

Abstract
Background
Hepatoerythropoietic porphyria (HEP) is a rare autosomal recessive disorder resulting from the markedly deficient, but not absent, activity of the heme biosynthetic enzyme, uroporphyrinogen decarboxylase (UROD). HEP typically manifests during infancy or early childhood with extreme photosensitivity, skin fragility in sun-exposed areas, hypertrichosis, erythrodontia, and pink urine.
Observations
We describe three siblings, offspring of parents of Puerto Rican and Dominican descent, who presented with excessive scarring on the face and dorsal aspect of the forearms, which initially led to the erroneous suspicion of child abuse. Although these lesions were photodistributed, overt photosensitivity had not been observed, with the exception of a single episode of blistering and onycholysis following intense sun exposure in one affected child. Mild facial hypertrichosis, chronic anemia, polyarticular arthritis, and developmental delay represented additional findings. Biochemical studies of urine, plasma, and erythrocyte porphyrins from the affected siblings established the diagnosis of HEP. Sequencing of the UROD gene revealed compound heterozygosity for a novel missense mutation, V166A, and a complex deletion/insertion, 645del1053ins10.
Conclusions
Our report expands the phenotypic and genotypic spectrum of HEP, highlighting mild cutaneous presentations that can occur without obvious photosensitivity and potentially masquerade as child abuse.
Introduction
Hepatoerythropoietic porphyria (HEP), a rare autosomal recessive disorder of heme biosynthesis, results from markedly deficient activity of uroporphyrinogen decarboxylase (UROD) due to mutations in the UROD gene.1-5 HEP is the recessive form of familial porphyria cutanea tarda (PCT), an autosomal dominant condition in which heterozygous UROD mutations predispose carriers to clinical manifestations.1,2 Since the initial description of HEP in 1969,6 there have been approximately 40 reported cases.2,7-34 Clinical manifestations usually develop during infancy or early childhood and include extreme photosensitivity, skin fragility (bullae, erosions, and scarring) in sun-exposed areas, hypertrichosis, erythrodontia, and pink-to-red urine. Sclerodermoid skin changes often become evident over time. Compared to PCT, the cutaneous features of HEP typically have earlier onset, increased severity leading to disfigurement, and closer resemblance to those in congenital erythropoietic porphyria (CEP; Günther disease).4,5 However, extracutaneous findings including hemolytic anemia are more frequent and severe in CEP than HEP.4,5,30
Here we describe three siblings with HEP who presented with mild cutaneous findings that led to a child abuse investigation. They also had chronic hemolytic anemia and other extracutaneous manifestations not previously recognized as features of HEP, including arthritis, developmental delay, and neonatal thrombocytopenia. Sequencing the UROD gene revealed a novel missense mutation and a complex deletion/insertion in the affected siblings. This kindred highlights the importance of recognizing mild cutaneous presentations of HEP, which may occur without obvious photosensitivity, and extends the phenotypic spectrum of the disease.
Report of a Case
The proband, a 7-year-old girl who was the third child of a Puerto Rican mother and a Dominican father, presented with a 2-month history of blistering, erosions, and scarring on the dorsal aspect of the hands and forearms as well as shedding of her fingernails. These findings developed after intense summer sun exposure. She took no medications and had not previously exhibited photosensitivity, although facial scarring had appeared after minor trauma. The patient had a history of severe neonatal thrombocytopenia requiring a platelet transfusion, chronic hemolytic anemia, and chronic reddish-brown urine. Her growth was normal, but she was developmentally delayed, including a receptive and expressive speech disorder and poor coordination resulting in an abnormal gait; she has been followed by a neurologist and received speech, physical, and occupational therapy since infancy.
Physical examination revealed linear-to-polygonal scars and mottled hyperpigmentation on the face, dorsal aspect of the hands, and extensor forearms. She had mild facial hypertrichosis, brownish teeth, and no evidence of hepatosplenomegaly. Two of the patient's four siblings had similar cutaneous and extracutaneous findings including chronic anemia and developmental delay (Table 1). The children's cutaneous findings are shown in Figure 1. Although both affected siblings were older (11-year-old brother, 10-year-old sister), neither had exhibited photosensitivity. However, multiple round (resembling cigarette burns), linear, and geometric scars in all three affected siblings had led to a year-long child abuse investigation by state authorities, which was initiated by teachers, school nurses, and emergency room physicians. The unaffected siblings (5-year-old brother, 3-year-old sister) had no history of photosensitivity, other skin findings, anemia, or developmental delay. With the exception of photosensitivity, these features had manifested in the affected siblings during the first years of life. The parents were non-consanguineous and unaffected, although the mother tended to sunburn easily.
Table 1. Summary of Clinical Findings for the Proband and her Family.
| Sib 1 – proband | Sib 2 – affected | Sib 3 – affected | Sib 4 – unaffected | Sib 5 – unaffected | Mother | Father | |
|---|---|---|---|---|---|---|---|
| Age (years), sex | 7, F | 11, M | 10, F | 5, M | 3, F | 29, F | 31, M |
| Acute photosensitivity (excessive erythema, blistering) | ++ | - | - | - | - | + | - |
| Erosions* | ++ | - | + | - | - | - | - |
| Linear to polygonal scars* | ++ | + | ++ | - | - | - | - |
| Sclerodactyly | + | - | + | - | - | - | - |
| Milia* | ++ | - | + | - | - | - | - |
| Mottled hyperpigmentation* | ++ | + | ++ | - | - | - | - |
| Facial hypertrichosis | + | ++ | ++ | - | - | - | - |
| Discolored teeth | + | + | - | - | - | - | - |
| Reddish urine due to porphyrinuria | + | + | + | - | - | - | - |
| Hemolytic anemia | ++ | + | + | - | - | - | - |
| Neonatal thrombocytopenia | ++ | - | - | - | - | - | - |
| Developmental delay | + | ++ | + | - | - | - | - |
| Abnormal gait | + | ++ | + | - | - | - | - |
| Polyarticular arthritis | + | ++ | - | - | - | - | - |
| Small nose with depressed bridge | + | ++ | ++ | - | - | - | - |
-, not present; +, mild; ++, moderate-to-severe; F, female; M, male; sib, sibling
In sun-exposed sites, primarily the face, extensor forearms, and dorsal aspect of the hands.
Figure 1.
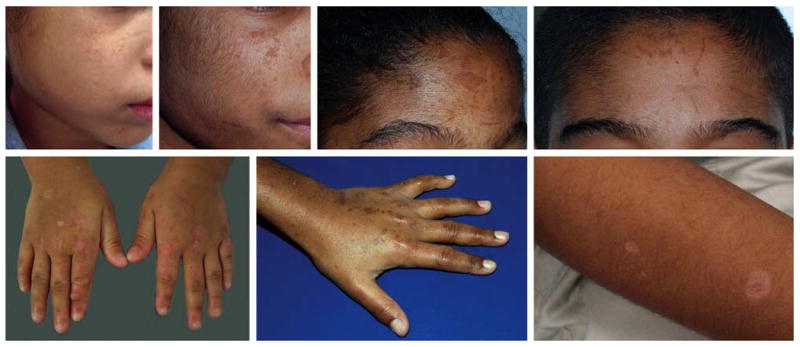
Cutaneous findings in the proband (A,E,G), 10-year-old sister (B,C,F), and 11-year-old brother (D). Linear and geometric hyperpigmented macules are evident on their faces (A-D) and the dorsal aspect of their hands (E,F). Polygonal to round hypopigmented scars are also present in these sites (A,E,F), and scars on the forearm resemble cigarette burns (G). Facial hypertrichosis is apparent (A-D).
Biochemical studies in the affected siblings were diagnostic of HEP. These included markedly elevated urine porphyrins (predominantly uroporphyrin-I/III and heptacarboxylporphyrins), a plasma porphyrin fluorescence peak at 620 nm, increased erythrocyte zinc protoporphyrin, and decreased erythrocyte UROD activities (Table 2). Sequencing the UROD gene in the affected siblings revealed compound heterozygosity for two UROD mutant alleles. A previously reported complex deletion/insertion, 645del1053ins10, inherited from their mother predicted truncation of the UROD enzyme's 367 amino acid sequence at residue 198. A novel missense mutation, a T-to-C transition at position 497, inherited from their father predicted a valine-to-alanine substitution at amino acid residue 166 (V166A). The two unaffected siblings were heterozygous for V166A. Neither of the mutations was identified in 100 normal individuals.
Table 2. Summary of Laboratory Findings for the Proband and her Family.
| Test, units (reference range) | Sib 1 – proband | Sib 2 – affected | Sib 3 – affected | Sib 4 – unaffected | Sib 5 – unaffected | Mother | Father |
|---|---|---|---|---|---|---|---|
| Hematocrit, % (34-48) | 29 | 33 | 32 | 36 | 32 | 38 | 41 |
| Serum ferritin, ng/mL (10-290) | 317 | 50 | 362 | 18 | 30 | 21 | 76 |
| Urine total porphyrins, nmol/24 hours (0-300) | 6,294 | 511 | 3,495 | 108 | 40 | 123 | 16 |
| Urine porphyrin pattern, %: | |||||||
| Uro (0-30) | 33 | 22 | 22 | N/A | N/A | N/A | N/A |
| Heptacarboxyl (0-5) | 25 | 29 | 27 | ||||
| Hexacarboxyl (0-5) | 10 | 13 | 12 | ||||
| Pentacarboxyl (0-5) | 9 | 11 | 13 | ||||
| Copro (50-100) | 23 | 25 | 26 | ||||
| Plasma total porphyrins, μg/dL (0-0.9) | 38.1 | 16.6 | 32.3 | 0.2 | 0.2 | 0.3 | 0.2 |
| Fluorescence peak, nm | 619 | 620 | 619 | No peak | No peak | No peak | No peak |
| Erythrocyte protoporphyrin, μg/dL (20-80) | 1,690* | 1,749* | 2,058* | 42 | 68 | 62 | 58 |
| Erythrocyte UROD activity, nmol/mL RBC/hr (35-60) | 16.2 | 13.8 | 15 | 45.8 | 53.6 | 38.1 | 43.8 |
| UROD mutation | del1053/ins10 V166A | del1053/ins10 V166A | del1053/ins10 V166A | V166A | V166A | del1053/ins10 | V166A |
| HFE mutation | H63D | - | H63D | N/A | N/A | - | N/A |
N/A, not available; sib, sibling; abnormal values are italicized
∼70% Zn-protoporphyrin.
HFE genotype analysis revealed heterozygosity for the H63D mutation in the proband and her affected sister, both of whom had elevated serum ferritin levels (see Table 2) and relatively pronounced skin fragility. Vitamin B12, folate, and liver chemistries were normal and viral hepatitis screens were negative in all family members. Brain MRIs in the affected children were normal.
A year after diagnosis, the proband and her older affected sister simultaneously developed pain, swelling, and limited range of motion in the interphalangeal and metacarpophalangeal joints and wrists of both hands; MRI confirmed the presence of synovitis. Mild cutaneous sclerosis and tapering of the fingertips was noted. Rheumatoid factor was negative in both girls and the older sister had antinuclear antibodies (titer 1:320) with a speckled pattern.
Comment
Most patients with HEP develop photosensitive eruptions during infancy or early childhood. Sun-induced erythema and blistering occurred by age 2 years in 75% of reported cases (25/33 patients with data available).2,6-9,11-16,19-24,26-34 Spontaneous improvement of acute photosensitivity during later childhood, but persistent skin fragility, has been described.8,11,29,33 Other patients have presented in the second or third decade of life with mild skin fragility or photodistributed annular plaques.26,28,30,33 Photomutilation can result in considerable morbidity in patients with HEP via impaired function of the hands and facial disfigurement, making photoprotection essential.4,5 Although helpful in PCT, phlebotomy and antimalarials are generally ineffective in HEP.5,35
The primary cutaneous manifestations in our kindred were fragility, scarring, and hyperpigmentation during childhood rather than acute photosensitivity. Round erosions and scars resembled cigarette burns, while linear and geometric lesions suggested forceful use of other instruments. The presence of multiple wounds in different stages of healing, the lack of an explanation for the injuries, and their occurrence in several siblings were additional features suspicious for child abuse.36 These skin findings had initially led to a long investigation by child protective services that resulted in considerable distress to the family.
Over 40 UROD mutations have been described, some occurring in both HEP and familial PCT.32,34 To date, 15 missense mutations, 2 deletions, and 1 nonsense mutation have been reported in HEP.3,5,17,18,21,25,26,29,30,32,33,34 Homozygosity for the F46L missense mutation causes relatively mild HEP,29,30 as may be true of the novel V166A mutation in our family. In contrast, mutations that abolish UROD activity, like the 645del1053ins10 lesion in our family, are only compatible with life when the individual's other UROD allele encodes residual enzymatic activity.5 Of note, 645del1053ins10 was previously described in an Argentinean kindred with PCT.37 Our three affected siblings had the same UROD mutations, but the severity of their clinical manifestations varied, underscoring the role of environmental and genetic modifiers. Our proband and her most severely affected sibling were heterozygous for the HFE H63D mutation and had increased serum ferritin levels, which may have contributed to their clinical presentations. Severe cutaneous findings, a HFE H63D mutation, and elevated ferritin levels were recently described in a 2-year-old boy with HEP.32
Sclerodactyly, osteolysis and shortening of the phalanges, and progressive joint deformities can occur as components of acral photomutilation in patients with HEP, CEP, and homozygous variegate porphyria.4,5,10,13,19,35,38 To our knowledge, arthritis has not been previously reported in patients with HEP. Whether our two sisters' painful polyarticular arthritis represents a typical (but heretofore unrecognized) inflammatory precedent of joint deformity in HEP or an idiosyncratic inflammatory process, perhaps triggered by porphyrin deposition together with exposure to ultraviolet light or another environmental insult, remains to be determined.
Anemia was present in >50% of HEP patients for whom hematologic status was reported (15/27),2,6-16,19-22,26,28,30,32,33 but severe anemia requiring transfusions or administration of epoetin-α has only been observed in a few individuals.28 The affected children in our family all had chronic anemia and were followed by a hematologist for years prior to diagnosis of HEP, emphasizing the importance of recognizing anemia as a feature of HEP. Thrombocytopenia, often due to secondary hypersplenism, has been described in patients with CEP (including neonates) but not in those with HEP.39,40,41
In contrast to the autosomal recessive form of variegate porphyria, which is characterized by developmental delay and seizures,38 neurologic abnormalities are not typically associated with HEP or CEP.4,5,39 Nevertheless, developmental delay and seizures have been previously reported in HEP.22,27 A 4-year-old boy had delayed speech and language skills then presented with focal seizures and acute left hemiparesis.22 Two young adults, ages 21 and 23 years, with severe HEP developed generalized seizures and had neuroimaging evidence of cerebral cortical atrophy and punctate calcifications in the frontal lobes, presumably related to hypoxic injury as in other porphyrias.27 These observations, together with our affected siblings' developmental delay, support neurologic assessment of HEP patients in order to better define this possible manifestation.
In summary, this report expands the clinical features of HEP to potentially include arthritis, neonatal thrombocytopenia, and developmental delay. The mutations identified in our kindred add to the UROD alleles that can cause HEP. We emphasize the importance of considering HEP in children who present with skin fragility and scarring in sun-exposed sites, even in the absence of acute photosensitivity. Increased awareness of the clinical manifestations of HEP will allow recognition of more affected individuals, delineation of the phenotypic spectrum, and evaluation of future therapies.42
Acknowledgments
Funding/Support: This study was supported in part by a grant from the Protect the Future Program of the American Porphyria Foundation to Dr Balwani, a NIH research grant (R37 DK34045) to Dr Desnick, and a Medical Dermatology Career Development Award from the Dermatology Foundation to Dr Schaffer.
Role of the Sponsors: The sponsors had no role in the design and conduct of the study; in the collection, analysis, and interpretation of data; or in the preparation, review, or approval of the manuscript.
Footnotes
Financial Disclosure: None reported.
Author Contributions: Dr Schaffer had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Cantatore-Francis, Balwani, Desnick, and Schaffer. Acquisition of data: Cantatore-Francis, Cohen, Balwani, Kahn, Lazarus, Desnick, and Schaffer. Analysis and interpretation of data: Cantatore-Francis, Cohen, Balwani, Kahn, Lazarus, Desnick, and Schaffer. Drafting of the manuscript: Cantatore and Schaffer. Critical revision of the manuscript for important intellectual content: Cohen, Balwani, Kahn, Lazarus, Desnick, and Schaffer. Obtained funding: Balwani, Desnick, and Schaffer. Administrative, technical, or material support: Balwani, Desnick, and Schaffer. Study supervision: Balwani, Desnick, and Schaffer.
Contributor Information
Dr Julie L. Cantatore-Francis, Email: julie.cantatore@gmail.com.
Dr Jessica Cohen, Email: jessica.cohen-pfeffer@mssm.edu.
Dr Manisha Balwani, Email: manisha.balwani@mssm.edu.
Dr Philip Kahn, Email: philip.kahn@nyumc.org.
Dr Herbert M. Lazarus, Email: herbert.lazarus@nyumc.org.
Dr Robert J. Desnick, Email: robert.desnick@mssm.edu.
References
- 1.Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CS, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th. New York, NY: McGraw-Hill; 2001. pp. 2961–3062. [Google Scholar]
- 2.de Verneuil H, Beaumont C, Deybach JC, Nordmann Y, Sfar Z, Kastally R. Enzymatic and immunological studies of uroporphyrinogen decarboxylase in familial porphyria cutanea tarda and hepatoerythropoietic porphyria. Am J Hum Genet. 1984;36(3):613–622. [PMC free article] [PubMed] [Google Scholar]
- 3.de Verneuil H, Grandchamp B, Beaumont C, Picat C, Nordmann Y. Uroporphyrinogen decarboxylase structural mutant (Gly281---Glu) in a case of porphyria. Science. 1986;234(4777):732–734. doi: 10.1126/science.3775362. [DOI] [PubMed] [Google Scholar]
- 4.Smith SG. Hepatoerythropoietic porphyria. Semin Dermatol. 1986;5(2):125–137. [Google Scholar]
- 5.Elder GH. Hepatic porphyrias in children. J Inher Metab Dis. 1997;20(2):237–246. doi: 10.1023/a:1005313024076. [DOI] [PubMed] [Google Scholar]
- 6.Piñol Aguadé J, Castells A, Indacochea A, Rodés J. A case of biochemically unclassifiable hepatic porphyria. Br J Dermatol. 1969;81(4):270–275. doi: 10.1111/j.1365-2133.1969.tb13979.x. [DOI] [PubMed] [Google Scholar]
- 7.Piñol Aguadé J, Herrero C, Almeida J, et al. Porphyrie hepato-érythrocytaire. Une nouvelle forme de porphyrie. Ann Dermatol Syphiligr (Paris) 1975;102(2):129–136. [PubMed] [Google Scholar]
- 8.Gunther WW. The porphyrias and erythropoietic protoporphyria: an unusual case. Australas J Dermatol. 1967;9(1):23–30. doi: 10.1111/j.1440-0960.1967.tb01147.x. [DOI] [PubMed] [Google Scholar]
- 9.Hofstad F, Seip M, Eriksen L. Congenital erythropoietic porphyria with a hitherto undescribed porphyrin pattern. Acta Paediatr Scand. 1973;62(4):380–384. doi: 10.1111/j.1651-2227.1973.tb08123.x. [DOI] [PubMed] [Google Scholar]
- 10.Simon N, Berkó G, Schneider I. Hepato-erythropoietic porphyria presenting as scleroderma and acrosclerosis in a sibling pair. Br J Dermatol. 1977;96(6):663–668. doi: 10.1111/j.1365-2133.1977.tb05212.x. [DOI] [PubMed] [Google Scholar]
- 11.Czarnecki DB. Hepatoerythropoietic porphyria. Arch Dermatol. 1980;116(3):307–311. [PubMed] [Google Scholar]
- 12.Ippen H, Fuchs T. Congenital porphyria. Clin Haematol. 1980;9(2):323–344. [PubMed] [Google Scholar]
- 13.Day RS, Strauss PC. Severe cutaneous porphyria in a 12-year-old boy: hepatoerythropoietic or symptomatic porphyria? Arch Dermatol. 1982;118(9):663–667. [PubMed] [Google Scholar]
- 14.Lim HW, Poh-Fitzpatrick MB. Hepatoerythropoietic porphyria: a variant of childhood-onset porphyria cutanea tarda. Porphyrin profiles and enzymatic studies of two cases in a family. J Am Acad Dermatol. 1984;11(6):1103–1111. doi: 10.1016/s0190-9622(84)70267-2. [DOI] [PubMed] [Google Scholar]
- 15.Lazaro P, de Salamanca RE, Elder GH, Villaseca ML, Chinarro S, Jaqueti G. Is hepatoerythropoietic porphyria a homozygous form of porphyria cutanea tarda? Inheritance of uroporphyrinogen decarboxylase deficiency in a Spanish family. Br J Dermatol. 1984;110(5):613–617. doi: 10.1111/j.1365-2133.1984.tb04687.x. [DOI] [PubMed] [Google Scholar]
- 16.Bundino S, Topi GC, Zina AM, D'Allessandro Gandolfo L. Hepatoerythropoietic porphyria. Pediatr Dermatol. 1987;4(3):229–233. doi: 10.1111/j.1525-1470.1987.tb00784.x. [DOI] [PubMed] [Google Scholar]
- 17.Romana M, Grandchamp B, Dubart A, et al. Identification of a new mutation responsible for hepatoerythropoietic porphyria. Eur J Clin Invest. 1991;21(2):225–229. doi: 10.1111/j.1365-2362.1991.tb01814.x. [DOI] [PubMed] [Google Scholar]
- 18.de Verneuil H, Bourgeois F, de Rooij F, et al. Characterization of a new mutation (R292G) and a deletion at the human uroporphyrinogen decarboxylase locus in two patients with hepatoerythropoietic porphyria. Hum Genet. 1992;89(5):548–552. doi: 10.1007/BF00219182. [DOI] [PubMed] [Google Scholar]
- 19.Fujimoto A, Brazil JL. Hepatoerythropoietic porphyria in a woman with short stature and deformed hands. Am J Med Genet. 1992;44(4):496–499. doi: 10.1002/ajmg.1320440423. [DOI] [PubMed] [Google Scholar]
- 20.Hift RJ, Meissner PN, Todd G. Hepatoerythropoietic porphyria precipitated by viral hepatitis. Gut. 1993;34(11):1632–1634. doi: 10.1136/gut.34.11.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meguro K, Fujita H, Ishida N, et al. Molecular defects of uroporphyrinogen decarboxylase in a patient with mild hepatoerythropoietic porphyria. J Invest Dermatol. 1994;102(5):681–685. doi: 10.1111/1523-1747.ep12374134. [DOI] [PubMed] [Google Scholar]
- 22.Parsons JL, Sahn EE, Holden KR, Pai GS. Neurologic disease in a child with hepatoerythropoietic porphyria. Pediatr Dermatol. 1994;11(3):216–221. doi: 10.1111/j.1525-1470.1994.tb00589.x. [DOI] [PubMed] [Google Scholar]
- 23.Boudghène-Stambouli O, Mérad-Boudia A. Hepato-erythropoietic porphyria. Ann Dermatol Venereol. 1995;122(9):615–617. [PubMed] [Google Scholar]
- 24.Castaño Suárez E, Zamarro Sanz O, Guerra Tapia A, Enríquez de Salamanca R. Hepatoerythropoietic porphyria: relationship with porphyria cutanea tarda. Dermatology. 1996;193(4):332–335. doi: 10.1159/000246284. [DOI] [PubMed] [Google Scholar]
- 25.McManus JF, Begley CG, Sassa S, Ratnaike S. Five new mutations in the uroporphyrinogen decarboxylase gene identified in families with cutaneous porphyria. Blood. 1996;88(9):3589–3600. [PubMed] [Google Scholar]
- 26.Moran-Jimenez MJ, Ged C, Romana M, et al. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet. 1996;58(4):712–721. [PMC free article] [PubMed] [Google Scholar]
- 27.Berenguer J, Blasco J, Cardenal C, et al. Hepatoerythropoietic porphyria: neuroimaging findings. AJNR Am J Neuroradiol. 1997;18(8):1557–1560. [PMC free article] [PubMed] [Google Scholar]
- 28.Horina JH, Wolf P. Epoetin for severe anemia in hepatoerythropoietic porphyria. N Engl J Med. 2000;342(17):1294–1295. doi: 10.1056/NEJM200004273421717. [DOI] [PubMed] [Google Scholar]
- 29.Ged C, Ozalla D, Herrero C, et al. Description of a new mutation in hepatoerythropoietic porphyria and prenatal exclusion of a homozygous fetus. Arch Dermatol. 2002;138(7):957–960. doi: 10.1001/archderm.138.7.957. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong DK, Sharpe PC, Chambers CR, Whatley SD, Roberts AG, Elder GH. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol. 2004;151(4):920–923. doi: 10.1111/j.1365-2133.2004.06101.x. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Bravo M, Segurado-Rodriguez MA, Moran-Jimenez MJ, et al. Successful treatment of hypertrichosis by high-intensity pulses of noncoherent light in a patient with hepatoerythropoietic porphyria. Arch Dermatol Res. 2004;296(3):139–140. doi: 10.1007/s00403-004-0490-3. [DOI] [PubMed] [Google Scholar]
- 32.Phillips JD, Whitby FG, Stadtmueller BM, Edwards CQ, Hill CP, Kushner JP. Two novel uroporphyrinogen decarboxylase (URO-D) mutations causing hepatoerythropoietic porphyria (HEP) Transl Res. 2007;149(2):85–91. doi: 10.1016/j.trsl.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Remenyik É, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol. 2008;33(5):602–605. doi: 10.1111/j.1365-2230.2008.02734.x. [DOI] [PubMed] [Google Scholar]
- 34.Granata BX, Parera VE, Melito VA, Teijo MJ, Batlle AM, Rossetti MV. The very first description of a patient with hepatoerythropoietic porphyria in Argentina. Biochemical and molecular studies. Cell Mol Biol (Noisy-le-grand) 2009;55(1):61–65. [PubMed] [Google Scholar]
- 35.Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135(3):281–292. doi: 10.1111/j.1365-2141.2006.06289.x. [DOI] [PubMed] [Google Scholar]
- 36.Swerdlin A, Berkowitz C, Craft N. Cutaneous signs of child abuse. J Am Acad Dermatol. 2007;57(3):371–392. doi: 10.1016/j.jaad.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Méndez M, Sorkin L, Rossetti MV, et al. Familial porphyria cutanea tarda: characterization of seven novel uroporphyrinogen decarboxylase mutations and frequency of common hemochromatosis alleles. Am J Hum Genet. 1998;63(5):1363–1375. doi: 10.1086/302119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer RA, Elder GH, Barrett DF, Keohane SG. Homozygous variegate porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene. Br J Dermatol. 2001;144(4):866–869. doi: 10.1046/j.1365-2133.2001.04147.x. [DOI] [PubMed] [Google Scholar]
- 39.Murphy GM. The cutaneous porphyrias: a review. The British Photodermatology Group. Br J Dermatol. 1999;140(4):573–581. doi: 10.1046/j.1365-2133.1999.02754.x. [DOI] [PubMed] [Google Scholar]
- 40.Kontos AP, Ozog D, Bichakjian C, Lim HW. Congenital erythropoietic porphyria associated with myelodysplasia presenting in a 72-year-old man: report of a case and review of the literature. Br J Dermatol. 2003;148(1):160–164. doi: 10.1046/j.1365-2133.2003.05040.x. [DOI] [PubMed] [Google Scholar]
- 41.Lazebnik N, Lazebnik RS. The prenatal presentation of congenital erythropoietic porphyria: report of two siblings with elevated maternal serum alpha-fetoprotein. Prenat Diagn. 2004;24(4):282–286. doi: 10.1002/pd.852. [DOI] [PubMed] [Google Scholar]
- 42.Fontanellas A, Mazurier F, Moreau-Gaudry F, Belloc F, Ged C, de Verneuil H. Correction of uroporphyrinogen decarboxylase deficiency (hepatoerythropoietic porphyria) in Epstein-Barr virus-transformed B-cell lines by retrovirus-mediated gene transfer: fluorescence-based selection of transduced cells. Blood. 1999;94(2):465–474. [PubMed] [Google Scholar]