

Abstract
Mesangial cells are critical for glomerular filtration. Mesangial cell dysfunction, the hallmark of diabetic nephropathy, results from disordered mesangial growth induced by cytokines, abnormal hemodynamic influence, and metabolic factors associated with chronic hyperglycemia. Insulin-like growth factors (IGFs) and their high affinity binding proteins (IGFBPs) exert major actions on mesangial cell survival, but their underlying mechanisms remain unclear. In light of emerging IGF-independent roles for IGFBP-3, we investigated IGFBP-3 actions during mesangial cell apoptosis induced by cytokine or high glucose concentration.
Quantified by DNA fragmentation ELISA and Annexin V flow cytometry, apoptosis occurred in rat mesangial cells (RMC) exposed to 2 μg/mL IGFBP-3 for 24 h under high ambient or standard glucose. Anti-sense IGFBP-3 oligo at 10 μg/mL significantly inhibited apoptosis induced by 100 ng/mL TNF-α, serum-free conditions, or high (25 mM) glucose. Increased IGFBP-3 release associated with high ambient glucose or TNF-α was inhibited by pre-treatment with anti-sense oligo. Under serum-free conditions, recombinant human IGFBP-3 blocked Akt phosphorylation at threonine 308 (pThr308), whereas anti-sense oligo treatment was associated with enhanced pThr308 activity. In summary, these data support a novel mechanism for TNF-α-induced mesangial cell apoptosis mediated by IGFBP-3 and present regulation of pThr308 activity as a novel mechanism underlying IGFBP-3 action.
Keywords: Mesangial cell, Diabetic nephropathy, Insulin-like growth factor binding protein, Apoptosis, Inflammation
1. Introduction
Glomerular injury represents the earliest stage in the etiopathophysiology of diabetic nephropathy [1], the leading cause of end-stage renal failure for North American adults since at least 1978 [2]. Mesangial cells occupy central anatomic positions within the glomeruli and comprise critical elements of the renal filtration mechanism. In addition to metabolic and hemodynamic factors, inflammation and growth factors significantly remodel the mesangium in vivo [3].
Hyperglycemia induces stable phenotypic and genotypic changes in mesangial cells, resulting in cellular hypertrophy [4]. Mesangial cells isolated from non-obese diabetic (NOD) mice after the onset of diabetes display increased expression of insulin-like growth factor-I (IGF-I) signaling and enhanced production of extracellular matrix [5,6]. A member of the relaxin/insulin superfamily of peptide hormones, IGF-I exerts major actions in carbohydrate, lipid, and protein metabolism, predominantly through activation of type I IGF receptors [7]. High affinity IGF binding proteins (IGFBPs) modulate IGF actions, are secreted by diverse cell types, and are found in most biological fluids [8]. Site-specific and time-dependent expressions of IGFs and IGFBPs regulate mesangial growth in vivo during renal development and injury [9-12]. Although high ambient glucose has been shown to induce mesangial IGF signaling and cellular hypertrophy, high glucose has also been shown in vitro to induce programmed mesangial cell death by apoptosis [13,14]. No known mechanism underlies this paradoxical apoptosis in the face of increased IGF-I signaling, and the predominant cellular effect of high ambient glucose in clinical circumstances remains unknown.
Navarro et al. [15] reported clinical data suggesting that, in addition to traditional metabolic and hemodynamic factors, inflammation participates in the etiopathophysiology of diabetic nephropathy. Serum levels of pro-inflammatory cytokines, particularly tumor necrosis factor (TNF)-α, are significantly higher in patients with type 2 diabetes and correlate to progression of DN [16-19]. Urinary TNF-α levels were higher in type 2 diabetic patients with respect to normal controls; furthermore, urinary TNF-α excretion directly increased as DN progressed [18,20]. Hasegawa et al. [21] implicated TNF-α as a major contributor to the early development of DN in rats with diabetes induced by streptozotocin. As a potential mechanism, TNF-α has been associated with sodium retention and renal hypertrophy during the early stages of DN in such diabetic rats [22].
These data support a recent paradigm shift proposing that chronic inflammation contributes to the etiopathophysiology of DN via the paracrine actions exerted upon mesangium by pro-inflammatory cytokines. Recently, several groups have reported in vitro studies linking IGFs to inflammatory cytokines in the process of mesangial cell dysfunction [34]. IGF-I has been associated with diabetic proliferation of mesangial cells in vivo[23,24]; paradoxically, end stage progression to uremia appears to reduce IGF production and paracrine action, contributing to mesangial cell death [24].
IGFBP-3 accumulates in serum obtained from children with chronic renal failure as its free, low molecular weight form [25,26]. Uremic diabetic adults excrete IGFBP-3, as do type 1 diabetic adolescents with microalbuminuria, patients whose urinary levels of IGFBP-3 correlate directly to urinary albumin excretion [27]. Elevated levels of IGFBP fragments have been identified in urine from non-diabetic children with acute renal failure due to increased IGFBP-3 protease activity [28,29]. This may reflect damage to the filtration mechanism in the face of higher IGFBP levels in serum from uremic children; a damaged filter due to apoptosis results, in part, from decreased IGF action during ESRD [30]. Other investigators have not observed IGFBP protease activity in urine obtained from non-diabetic children with chronic renal failure [31,32].
IGFBP-3 has recently been shown to mediate apoptosis in human mesangial cells under starvation conditions [33]. Although human mesangial cells express all six IGFBPs in vitro [34], IGFBP-3 (the most abundant IGFBP species in human serum) is normally expressed in vivo only in the mature ureteric duct and epithelia of the collecting ducts and pelvicalyceal system. Integrating all current data, we hypothesize that high ambient glucose and associated inflammatory cytokines induce mesangial apoptosis via the IGFBP-3 signaling pathway. Using the rat mesangial cell model, we assessed the role of IGFBP-3 during apoptosis induced in the presence of high ambient glucose or TNF-α, a major cytokine implicated in the diabetic milieu.
2. Materials and methods
2.1. Cell culture and reagents
Rat mesangial cells (RMC) were obtained from ATCC (Manassas, VA, USA). Derived from the eighth passage of Sprague–Dawley rat mesangium and immortalized with pSV3-Neo [35,36], cells were propagated at 37 °C under 5% ambient CO2 in Dulbecco’s modified Eagle’s medium with 4 mM l-glutamine, adjusted to contain 1.5 g/L sodium bicarbonate, 1.5 g/L glucose, 0.4 mg/mL G418, and 15% fetal bovine serum. Growth media was changed every third day.
Recombinant rat (rr) TNF-α, rrIL-1β, and recombinant human (rh) IGF-I were purchased from R&DSystems (Minneapolis, MN, USA). RhIGFBP-3 was purchased from Sigma (St. Louis, MO, USA). Polyclonal anti-rabbit IGFBP-3 IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-sense phosphorothioate oligodeoxynucleotide encompassing the initiation codon of rat IGFBP-3 (5′-CGCGGGATGCATGGCGCTGGCGGAGGGCTC) and the complementary sequence as the sense oligo were purchased from Sigma-Genosys, Ltd. (The Woodlands, TX, USA). Thioester bonds linked the first three and final three residues of each oligo. The concentrations applied were: 100 ng/mL TNF-α, 2 μg/mL rhIGFBP-3, 1 μg/mL rhIGF-I, and 10 μg/mL oligo.
2.2. Fluorescent activated cell sorting (FACS) by annexin V
Cells were grown in 75 cm2 flasks to 80% confluence, then harvested with trypsin and plated in 12-well plates (Carrollton, TX, USA) at approximate density of 100,000 cells/well (1 mL medium/well). Precisely 24 h later, growth media was removed, and cells were washed thrice with serum-free media (SFM). Media was replaced with equal volumes of SFM containing experimental conditions. To terminate the experiment, cells were gently separated by trypsin, collected by centrifugation, then immediately analyzed with fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (BioVision, Mountain View, CA, USA), performed according to manufacturer’s instructions. FITC-labeled Annexin V antibody binding was quantified by flow cytometry with excitation by the 488 nm line of an argon laser and emission at 530 nm using FITC signal detector (FL1). Propidium iodide (PI) stain was detected on the phycoerythrin emission signal detector (FL2).
2.3. DNA fragmentation ELISA
Cells were plated in 96-well plates (Falcon, Franklin Lakes, NJ, USA) at concentration of 104 cells/well in 200 μL complete media containing standard glucose (5 mM) or high glucose (25 mM). After overnight attachment, cells were washed thrice with serum-free medium (SFM). Then, 200 μL SFM with experimental conditions were added to each well. To terminate each experiment, plates were centrifuged at 200g in Beckman-Coulter Allegra 6 Series or Spinchron R centrifuges for 10 min at 25 °C. After centrifugation, conditioned media were removed by gentle vacuum filtration.
Mono- and oligo-nucleosomes specific to apoptotic cells comprise the histone-associated DNA fragments quantified by Roche Cell Death Detection ELISAplus (Mannheim Germany), performed according to the manufacturer’s instructions. Briefly, cells re-suspended in 200 μL 1X Lysis Buffer were incubated 30 min at 25 °C. Lysates were centrifuged at 200g for 10 min, and 20 μL from each supernatant were transferred carefully into streptavidin-coated 96-well plates. Immunoreagent 80 μL was added to each well, and plates were covered with adhesive cover foil under gentle shaking (300 rpm) for 2 h at 15 °C. Solution was removed thoroughly by gentle suction, then each well was rinsed thrice with 300 μL 1X incubation buffer. ABTS solution 100 μL/well developed the color signal, detected at 405 nm against ABTS solution as blank (reference 490 nm).
2.4. Western blot
Mesangial cells were grown to 80–90% confluence in 60 mm2 dishes. Media were replaced to SFM (with 5 or 25 mM glucose) containing experimental conditions for 24 h. Protein was extracted by lysis buffer, and 50 lg/lane was loaded on 15% SDS–PAGE. After transferring protein to nitrocellulose (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA), the membrane was incubated in 5% milk-TBST (20 mM Tris–HCl, pH 7.6, 137 mM NaCl, 0.1% Tween-20), then immunoblotted with anti-IGFBP-3 antibody. The membrane was stripped and re-blotted with tubulin antibody to confirm loading was equal in each lane.
We used 1:500 dilution of primary rabbit polyclonal IGFBP-3 IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), followed by horseradish peroxidase-conjugated secondary IgG (Jackson Immunologicals, West Grove, PA, USA). Bands were visualized by ECL™ Western Blotting Analysis System (Amersham Biosciences, Little Chalfont of Buckinghamshire, UK).
2.5. Akt kinase assay
Total Akt and Akt phosphorylated at threonine 308 were quantified by Akt [pThr308] phosphoELISA™ (BioSource International, Inc., Camarillo, CA, USA), performed according to the manufacturer’s instructions. Mesangial cells were grown to 80–90% confluence in 60 mm2 dishes, then changed to SFM with experimental conditions for 24 h. Cells were collected in PBS by scraping culture flasks, then washed twice with cold PBS. The supernatant was discarded, and cell pellets were lysed in Cell Extraction Buffer (BioSource International, Inc., Camarillo, CA, USA) for 30 min at 4 °C with vortexing every 10 min. Extracts transferred to microcentrifuge tubes were centrifuged at 13,000 rpm for 10 min at 4 °C. Clear lysate was aliquoted into clean microfuge tubes. Akt [pThr308] standard and samples diluted 1:10 in Standard Diluent Buffer were added, 100 μL/well, and incubated 2 h at 25 °C. Incubation solution was aspirated, then wells were washed four times with working wash buffer. Detection antibody 100 μL/well was added, followed by incubation for 1 h at 25 °C. After four washes, cells were incubated with anti-rabbit IgG-HRP working solution 100 μL/well for 30 min, then incubated with stabilized chromogen 100 μL/well for 30 min under darkness. Finally, stop solution was added prior to reading optical density at 450 nm on Ultramark plate reader (Bio-Rad, Hercules, CA, USA). Data were analyzed with Scion Corporation Imaging software (Frederick, MD, USA).
2.6. Statistical analysis
Data from a minimum of three experiments were expressed as means ± SE and analyzed by ANOVA and post-hoc Kruskal-Wallis test for ordinary, unmatched measures using InStat (version 2.00; GraphPad Software Inc., San Diego, CA, USA). Analyses were run on G3 Macintosh PowerBook (Apple Computer, Cupertino, CA, USA). Probability values (p) below 5% were considered significant.
3. Results
3.1. RhIGFBP-3 induced apoptosis in RMC
To quantify the percentage of cells undergoing apoptosis, RMC cultures were analyzed by FACS after exposure to 2 μg/mL rhIGFBP-3 for 2 h or 24 h under 5 mM glucose. RhIGFBP-3 rapidly induced apoptosis (Fig. 1(a) and (b)). Detected by ELISA, 2 μg/mL IGFBP-3 for 24 h significantly increased DNA fragmentation under both 5 and 25 mM glucose conditions (Fig. 1(c), n = 8, p < 0.05, IGFBP-3 vs. SF). DNA fragmentation results are consistent with FACS data. High ambient glucose increased apoptosis compared to standard glucose (Fig. 1(c), n = 8, p < 0.01, 25 mM vs. 5 mM).
Fig. 1.

RhIGFBP-3 induced apoptosis in RMC. FACS analysis: The cells were treated with or without 2 μg/mL IGFBP-3 in SFM containing 5 mM glucose for 2 h (a) or 24 h (b). Black lines represent rhIGFBP-3 treatment, and blue lines represent SF control. Annexin (upper panel) reflects apoptosis, and propidium iodide stain (lower panel) depicts necrosis. DNA fragmentation ELISA: (c) cells were treated with or without 2 μg/mL rhIGFBP-3 under 5 or 25 mM glucose-containing media for 24 h. Data were represented as means ± SE (n = 8; *p < 0.05 vs. SFM control by ANOVA).
3.2. Anti-sense IGFBP-3 oligo inhibited apoptosis induced by SFM, TNF-α, or high glucose
Pre-treatment for 30 min with anti-sense IGFBP-3 oligo significantly prevented DNA fragmentation induced under serum-free conditions at all concentrations tested (5–25 μg/mL) (Fig. 2, n = 8, p < 0.05 or p < 0.0001 oligo vs. SFM control). As expected, sense oligo exerted no significant effect on RMC apoptosis.
Fig. 2.
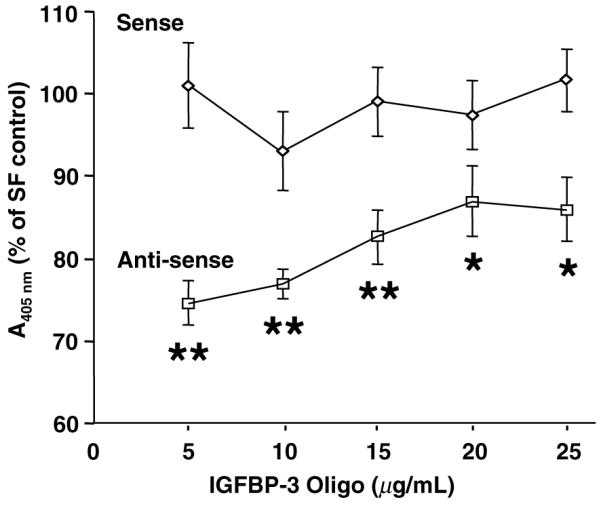
Anti-sense IGFBP-3 oligo inhibited SFM-induced apoptosis by DNA fragmentation ELISA. Cells were incubated with dose ranges of oligos for 24 h under SFM containing 5 mM glucose. Data are presented as percentage of SFM control. Histogram depicts means ± SE (n = 8, *p < 0.05, **p < 0.0001 vs. SF control by ANOVA).
TNF-α has been implicated in the etiopathophysiology of DN in patients [16-19]. To model this action in vitro, cells were treated with 100 ng/mL TNF-α for 24 h under SFM containing 5 mM glucose, which induced apoptosis as detected by FACS (Fig. 3(a)). To further study the action of IGFBP-3 on TNF-α-induced apoptosis, cells were pre-treated with anti-sense IGFBP-3 oligo (10 μg/mL) for 30 min prior to treatment with TNF-α. FACS revealed significantly increased apoptosis after 2 h or 24 h TNF-α treatment (Fig. 3(b) and (c), n = 3, p < 0.05, TNF-α vs. SFM control). Anti-sense, but not sense, oligo prevented apoptosis induced at both time points (Fig. 3(b) and (c), n = 3, p < 0.05, TNF-α + AS vs. TNF-α alone).
Fig. 3.

Anti-sense IGFBP-3 oligo inhibited TNF-α-induced apoptosis by FACS. (a) Cells were incubated with 100 ng/mL TNF-α (black lines) or SFM control (blue lines) containing 5 mM glucose for 24 h. Annexin (upper panel) reflects apoptosis, and PI (lower panel) reflects necrosis. Cells pre-treated with or without sense or anti-sense oligo for 30 min, then 100 ng/mL TNF-α for 2 h (b) or 24 h (c). Data presented in the histograms are expressed as percentage of SF control with means ± SE (n = 3; *p < 0.05 vs. SF control; #p < 0.05 vs. TNF-α). (d) DNA fragmentation ELISA: cells pre-treated with or without oligo before 24 h of TNF-α under SFM with 5 or 25 mM glucose. Data represented percentage of SF control, with histogram depicting means ± SE (n = 8; *p < 0.05 vs. SF control; #p < 0.05 vs. TNF-α by ANOVA).
TNF-α increased DNA fragmentation (apoptosis) under both 5 and 25 mM glucose conditions (Fig. 3(d), n = 8, p < 0.05, TNF-α vs. SFM control), apoptosis was blocked by anti-sense oligo but not sense oligo (Fig. 3(d), n = 8, p < 0.05, TNF-α + AS vs. TNF-α alone). Anti-sense oligo, but not sense oligo, prevented DNA fragmentation induced by SFM under both 5 and 25 mM glucose (Fig. 3(d), n = 8, p < 0.01, AS oligo vs. SF).
We further evaluated the effects of AS oligo on apoptosis induced by SFM or high ambient glucose. The cells were treated with or without sense or anti-sense IGFBP-3 oligo under 5 mM glucose or 25 mM glucose for 2 or 24 h. Twenty-four hour serum withdrawal significantly increased DNA fragmentation compared to 2 h treatment (Fig. 4, n = 8, p < 0.001, 24-h under 5 mM glucose vs. 2-h under 5 mM glucose). This increase was partially inhibited by AS oligo at the 24-h time point (Fig. 4, n = 8, p < 0.01, AS under 5 mM glucose vs. SFM under 5 mM glucose). Twenty four hour high glucose treatment increased apoptosis compared to 2-h high glucose treatment (Fig. 4, n = 8, p < 0.001, 24-h high glucose vs. 2-h high glucose). Anti-sense oligo partially inhibited SFM-induced or high glucose induced apoptosis (Fig. 4, n = 8, p < 0.001, AS with 24-h high glucose treatment vs. SF with 24-h high glucose treatment). High glucose treatment also induced more apoptosis than low glucose at 24 h (Fig. 4, n = 8, p < 0.05, SF with 24-h high glucose vs. SFM with 24-h low glucose).
Fig. 4.
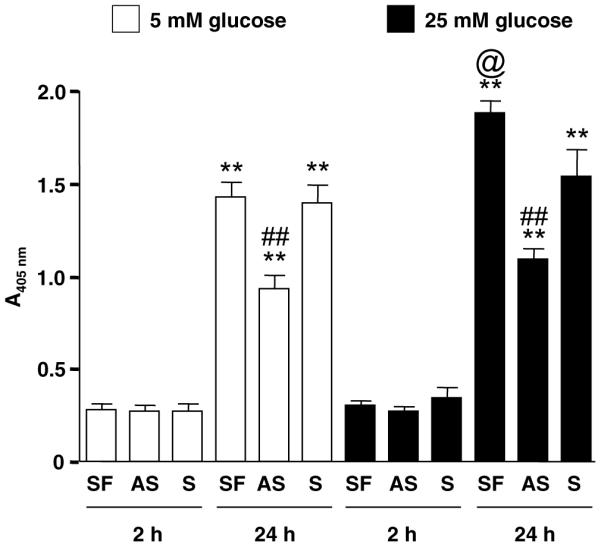
DNA fragmentation ELISA. Data represent means ± SE (n = 8; **p < 0.01 vs. 2 h under identical glucose; ##p < 0.01 vs. 24 h SFM under identical glucose; ©p < 0.01 vs. 24 h SFM under 5 mM glucose, by ANOVA).
3.3. Anti-sense IGFBP-3 oligo inhibited TNF-α-induced IGFBP-3 release
IGFBP-3-specific Western blot revealed two bands in cell lysates that correspond to the predicted 46 and 44 kDa forms of glycosylated IGFBP-3 (Fig. 5). TNF-α treatment was associated with increased IGFBP-3 synthesis. This effect was blocked by anti-sense IGFBP-3 oligo pre-treatment but enhanced in the presence of high ambient glucose.
Fig. 5.

Anti-sense oligo inhibit TNF-α-induced IGFBP-3 synthesis by Western blot of cell lysates. Cells were pre-treated with or without oligo (10 μg/mL) for 30 min, before incubated with TNF-α under 5 mM or 25 mM glucose for 24 h. Representative blot from three independent experiments.
3.4. RhIGFBP-3 and AS IGFBP-3 oligo regulate Akt phosphorylation at Thr308
RhIGFBP-3 treatment was associated with reduced phosphorylation of Akt at Thr308 (Fig. 6, n = 7, p < 0.01). By contrast, pre-treatment with anti-sense IGFBP-3 oligo significantly increased Thr308 phosphorylation (n = 7, p < 0.01).
Fig. 6.
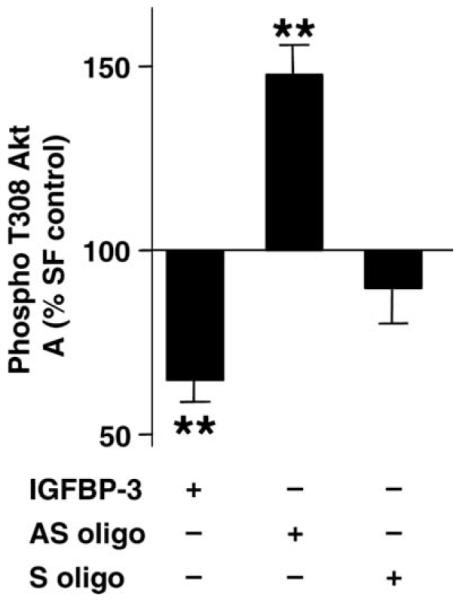
Thr-308 Akt phosphorylation ELISA. Data represent percentage of SF control expressed as mean ± SE (n = 7; **p < 0.01 vs. SF control by ANOVA).
4. Discussion
We have presented the first evidence that IGFBP-3 mediates mesangial apoptosis via intracellular signal transduction in the context of high ambient glucose or TNF-α. Classically, somatotropin is recognized as the primary stimulator in vivo for endocrine IGF release [37] and endocrine IGFBP-3 release [8]. Pharmacologic antagonism against somatotropin action has been reported to prevent early DN in vivo [38]. It is plausible these clinical observations derive from reduced paracrine or autocrine actions of IGFBP-3 delivered at the mesangium.
Related to its proposed pathologic role in diabetic mesangial hypertrophy [5,6], IGF-I has been shown to promote mesangial survival in the face of hyperglycemia [13]. The early stage of DN is characterized by mesangial cell proliferation and expansion of extracellular matrix. The balance between local delivery of unbound, free IGF-I vs. unbound IGFBP-3 appears to determine the mesangial fate between survival or death.
We observed that rhIGF-I rescued RMC from apoptosis, but this action was eliminated in the presence of high glucose concentration. We observed consistent, anti-apoptotic effects from anti-sense IGFBP-3 oligo in the presence of high ambient glucose or the cytokine TNF-α. Our data support paracrine IGFBP-3 action as a mechanism to account for these observations, most likely through local sequestration of IGF. Yet IGFBP-3 is emerging as a novel intracellular regulator of signal transduction via major tyrosine kinase-mediated pathways that control cell growth [39-41]. For the first time, we have presented inhibition of threonine phosphorylation of cytosolic Akt as a novel, non-genomic, intracellular mechanism for IGFBP-3-mediated apoptosis in the mesangial cell.
Whereas IGFBP-3 reduced Akt Thr308 phosphorylation, anti-sense IGFBP-3 oligo was associated with an increase in Thr308 phosphorylation. These observations suggest that regulating the Akt signaling pathway is a mechanism underlying IGFBP-3-induced apoptosis. Although anti-sense IGFBP-3 oligo pre-treatment blocked TNF-induced apoptosis, anti-sense oligo did not prevent TNF-induced inhibition of Thr-308 Akt phosphorylation (data not shown). These observations cannot exclude the existence of an Akt-independent pathway for IGFBP-3-mediated apoptosis. Delineation of the IGFBP-3 signaling pathway(s) warrants future investigation. Use of a cell model limits the conclusions to be drawn about IGFBP-3 action on mesangial cells in vivo. In conclusion, we have shown that IGFBP-3 represents a novel intracellular mechanism for mesangial cell apoptosis, which offers novel therapeutic targets for intervention during the early, inflammatory progression of DN.
Acknowledgments
This work was Supported in part by NIH Grant K08 DK02876 and the Genentech Clinical Scholar Award of the Lawson Wilkins Pediatric Endocrine Society (to R.F.). Portions of the work were presented in abstract form at The 2004 Southern Regional Meeting of the American Federation for Medical Research held February 14, 2004 (New Orleans, LA). The opinions herein are solely those of the authors and do not represent an official position of The State of Texas, the Army nor their subordinate agencies. The authors gratefully thank Maria Bunegin for her technical support.
References
- [1].Flyvbjerg A, Landau D, Domene H, Hernandez L, Gronbaek H, LeRoith D. The role of growth hormone, insulin-like growth factors (IGFs), and IGF-binding proteins in experimental diabetic kidney disease. Metabolism. 1995;44:67–71. doi: 10.1016/0026-0495(95)90223-6. [DOI] [PubMed] [Google Scholar]
- [2].Atlas of ESRD in the US. Available from: < http://www.usrds.org/atlas.htm>.
- [3].Brosius FC., 3rd Trophic factors and cytokines in early diabetic glomerulopathy. Exp. Diabesity Res. 2003;4:225–233. doi: 10.1155/EDR.2003.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fornoni A, Lenz O, Striker LJ, Striker GE. Glucose induces clonal selection and reversible dinucleotide repeat expansion in mesangial cells isolated from glomerulosclerosis-prone mice. Diabetes. 2003;52:2594–2602. doi: 10.2337/diabetes.52.10.2594. [DOI] [PubMed] [Google Scholar]
- [5].Tack I, Elliot SJ, Potier M, Rivera A, Striker GE, Striker LJ. Autocrine activation of the IGF-I signaling pathway in mesangial cells isolated from diabetic NOD mice. Diabetes. 2002;51:182–188. doi: 10.2337/diabetes.51.1.182. [DOI] [PubMed] [Google Scholar]
- [6].Pugliese G, Pricci F, Locuratolo N, Romeo G, Romano G, Giannini S. Increased activity of the insulin-like growth factor system in mesangial cells cultured in high glucose conditions. Relation to glucose-enhanced extracellular matrix production. Diabetologia. 1996;39:775–784. doi: 10.1007/s001250050510. [DOI] [PubMed] [Google Scholar]
- [7].Mauras N, Martinez V, Rini A, Guevara-Aguirre J. Recombinant human insulin-like growth factor I has significant anabolic effects in adults with growth hormone receptor deficiency: studies on protein, glucose, and lipid metabolism. J. Clin. Endocrinol. Metab. 2000;85:3036–3042. doi: 10.1210/jcem.85.9.6772. [DOI] [PubMed] [Google Scholar]
- [8].Ferry RJ, Jr., Cohen P. The insulin-like growth factor axis in pediatrics. Clin. Pediatr. Endocrinol. 1999;8:1–10. [Google Scholar]
- [9].Matsell DG, Delhanty PJ, Stepaniuk O, Goodyear C, Han VK. Expression of insulin-like growth factor and binding protein genes during nephrogenesis. Kidney Int. 1994;46:1031–1042. doi: 10.1038/ki.1994.364. [DOI] [PubMed] [Google Scholar]
- [10].Lindenbergh-Kortleve DJ, Rosato RR, van Neck JW, Nauta J, van Kleffens M, Groffen C, et al. Gene expression of the insulin-like growth factor system during mouse kidney development. Mol. Cell. Endocrinol. 1997;132:81–91. doi: 10.1016/s0303-7207(97)00123-8. [DOI] [PubMed] [Google Scholar]
- [11].Price GJ, Berka JL, Edmondson SR, Werther GA, Bach LA. Localization of mRNAs for insulin-like growth factor binding proteins 1 to 6 in rat kidney. Kidney Int. 1995;48:402–411. doi: 10.1038/ki.1995.308. [DOI] [PubMed] [Google Scholar]
- [12].Hise MK, Li L, Mantzouris N, Rohan RM. Differential mRNA expression of insulin-like growth factor system during renal injury and hypertrophy. Am. J. Physiol. 1995;269:F817–F824. doi: 10.1152/ajprenal.1995.269.6.F817. [DOI] [PubMed] [Google Scholar]
- [13].Kang BP, Urbonas A, Baddoo A, Baskin S, Malhotra A, Meggs LG. IGF-1 inhibits the mitochondrial apoptosis program in mesangial cells exposed to high glucose. Am. J. Physiol. Renal Physiol. 2003;285:F1013–F1024. doi: 10.1152/ajprenal.00209.2003. [DOI] [PubMed] [Google Scholar]
- [14].Kang BP, Frencher S, Reddy V, Kessler A, Malhotra A, Meggs LG. High glucose promotes mesangial cell apoptosis by oxidant-dependent mechanism. Am. J. Physiol. Renal Physiol. 2003;284:F455–F466. doi: 10.1152/ajprenal.00137.2002. [DOI] [PubMed] [Google Scholar]
- [15].Navarro JF, Mora C, Maca M, Garca J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am. J. Kidney Dis. 2003;42:53–61. doi: 10.1016/s0272-6386(03)00408-6. [DOI] [PubMed] [Google Scholar]
- [16].Desfaits A-C, Serri O, Renier G. Normalization of plasma lipid peroxides, monocyte adhesion, and tumor necrosis factor-α production in NIDDM patients after gliclazide treatment. Diabetes Care. 1998;21:487–493. doi: 10.2337/diacare.21.4.487. [DOI] [PubMed] [Google Scholar]
- [17].Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumor necrosis factor α and blood cytokine production in type 2 diabetes. Life Sci. 2000;67:291–300. doi: 10.1016/s0024-3205(00)00622-6. [DOI] [PubMed] [Google Scholar]
- [18].Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 2003;52:605–608. doi: 10.1053/meta.2003.50096. [DOI] [PubMed] [Google Scholar]
- [19].Shikano M, Sobajima H, Yoshikawa H, Toba T, Kushimoto H, Katsumata H, et al. Usefulness of a highly sensitive urinary and serum IL-6 assay in patients with diabetic nephropathy. Nephron. 2000;85:81–85. doi: 10.1159/000045634. [DOI] [PubMed] [Google Scholar]
- [20].Mora C, Navarro JF. Inflammation and pathogenesis of diabetic nephropathy. Metabolism. 2004;53:265–266. doi: 10.1016/j.metabol.2003.11.005. [DOI] [PubMed] [Google Scholar]
- [21].Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, Ienaga K, et al. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991;40:1007–1012. doi: 10.1038/ki.1991.308. [DOI] [PubMed] [Google Scholar]
- [22].DiPetrillo K, Coutermarsh B, Gesek FA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am. J. Physiol. Renal Physiol. 2003;28:F113–F121. doi: 10.1152/ajprenal.00026.2002. [DOI] [PubMed] [Google Scholar]
- [23].Segev Y, Landau D, Marbach M, Shehadeh N, Flyvbjerg A, Phillip M. Renal hypertrophy in hyperglycemic non-obese diabetic mice is associated with persistent renal accumulation of insulin-like growth factor I. J. Am. Soc. Nephrol. 1997;8:436–444. doi: 10.1681/ASN.V83436. [DOI] [PubMed] [Google Scholar]
- [24].Blum WF. Insulin-like growth factors (IGFs) and IGF binding proteins in chronic renal failure: evidence for reduced secretion of IGFs. Acta Paediatr. Scand. 1991;379:24–31. doi: 10.1111/j.1651-2227.1991.tb12039.x. [DOI] [PubMed] [Google Scholar]
- [25].Powell DR, Liu F, Baker B, Lee PD, Belsha CW, Brewer ED, et al. Characterization of insulin-like growth factor binding protein-3 in chronic renal failure serum. Pediatr. Res. 1993;33:136–143. doi: 10.1203/00006450-199302000-00011. [DOI] [PubMed] [Google Scholar]
- [26].Durham SK, Mohan S, Liu F, Baker BK, Lee PD, Hintz RL, et al. Bioactivity of a 29-kilodalton insulin-like growth factor binding protein-3 fragment present in excess in chronic renal failure serum. Pediatr. Res. 1997;42:335–341. doi: 10.1203/00006450-199709000-00014. [DOI] [PubMed] [Google Scholar]
- [27].Spagnoli A, Chiarelli F, Vorwerk P, Boscherini B, Rosenfeld RG. Evaluation of the components of insulin-like growth factor (IGF)-IGF binding protein (IGFBP) system in adolescents with type 1 diabetes and persistent microalbuminuria: relationship with increased urinary excretion of IGFBP-3 18 kD N-terminal fragment. Clin. Endocrinol. 1999;51:587–596. doi: 10.1046/j.1365-2265.1999.00842.x. [DOI] [PubMed] [Google Scholar]
- [28].Lee DY, Cohen P, Krensky AM, Rosenfeld RG, Yorgin PD. Insulin-like growth factor binding protein-3 protease activity in the urine of children with chronic renal failure. Pediatr. Nephrol. 1993;7:416–423. doi: 10.1007/BF00857555. [DOI] [PubMed] [Google Scholar]
- [29].Lee DY, Park SK, Yorgin PD, Cohen P, Oh Y, Rosenfeld RG. Alteration in insulin-like growth factor-binding proteins (IGFBPs) and IGFBP-3 protease activity in serum and urine from acute and chronic renal failure. J. Clin. Endocrinol. Metab. 1994;79:1376–1382. doi: 10.1210/jcem.79.5.7525635. [DOI] [PubMed] [Google Scholar]
- [30].Tonshoff B, Blum WF, Mehls O. Derangements of the somatotropic hormone axis in chronic renal failure. Kidney Int. 1997;58:S106–S113. [PubMed] [Google Scholar]
- [31].Blum WF, Ranke MB, Kietzmann K, Tonshoff B, Mehls O. Growth hormone resistance and inhibition of somatomedin activity by excess of insulin-like growth factor binding protein in uraemia. Pediatr. Nephrol. 1991;5:539–544. doi: 10.1007/BF01453697. [DOI] [PubMed] [Google Scholar]
- [32].Hodson EM, Brown AS, Roy LP, Rosenberg AR. Insulin-like growth factor-1, growth hormone-dependent insulin-like growth factor-binding protein and growth in children with chronic renal failure. Pediatr. Nephrol. 1992;6:433–438. doi: 10.1007/BF00874005. [DOI] [PubMed] [Google Scholar]
- [33].Verzola D, Villaggio B, Berruti V, Gandolfo MT, Deferrari G, Garibotto G. Apoptosis induced by serum withdrawal in human mesangial cells. Role of IGFBP-3. Exp. Nephrol. 2001;9:366–371. doi: 10.1159/000052634. [DOI] [PubMed] [Google Scholar]
- [34].Grellier P, Sabbah M, Fouqueray B, Woodruff K, Yee D, Abboud HE, et al. Characterization of insulin-like growth factor binding proteins and regulation of IGFBP3 in human mesangial cells. Kidney Int. 1996;49:1071–1078. doi: 10.1038/ki.1996.156. [DOI] [PubMed] [Google Scholar]
- [35].Zent R, Ailenberg M, Waddell TK, Downey GP, Silverman M. Puromycin aminonucleoside inhibits mesangial cell-induced contraction of collagen gels by stimulating production of reactive oxygen species. Kidney Int. 1995;47:811–817. doi: 10.1038/ki.1995.123. [DOI] [PubMed] [Google Scholar]
- [36].Zent R, Ailenberg M, Silverman M. Tyrosine kinase cell signaling pathways of rat mesangial cells in 3-dimensional cultures: response to fetal bovine serum and platelet-derived growth factor-BB. Exp. Cell Res. 1998;240:134–143. doi: 10.1006/excr.1998.4008. [DOI] [PubMed] [Google Scholar]
- [37].Kaplan SA. Somatomedin hypothesis: time for reexamination. The Endocrinologist. 2001;11:470–473. [Google Scholar]
- [38].Segev Y, Landau D, Rasch R, Flyvbjerg A, Phillip M. Growth hormone receptor antagonism prevents early renal changes in nonobese diabetic mice. J. Am. Soc. Nephrol. 1999;10:2374–2381. doi: 10.1681/ASN.V10112374. [DOI] [PubMed] [Google Scholar]
- [39].Franklin SL, Ferry RJ, Jr., Cohen P. Rapid insulin-like growth factor (IGF)-independent effects of IGF binding protein-3 on endothelial cell survival. J. Clin. Endocrinol. Metab. 2003;88:900–907. doi: 10.1210/jc.2002-020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Martin JL, Weenink SM, Baxter RC. Insulin-like growth factor-binding protein-3 potentiates epidermal growth factor action in MCF-10A mammary epithelial cells. Involvement of p44/42 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2003;278:2969–2976. doi: 10.1074/jbc.M210739200. [DOI] [PubMed] [Google Scholar]
- [41].Grill CJ, Sivaprasad U, Cohick WS. Constitutive expression of IGF-binding protein-3 by mammary epithelial cells alters signaling through Akt and p70S6 kinase. J. Mol. Endocrinol. 2002;29:153–162. doi: 10.1677/jme.0.0290153. [DOI] [PubMed] [Google Scholar]