

Abstract
A new technology has been developed by immunologix that allows human antibodies to be quickly generated against virtually any antigen. Using a novel process, naïve human B cells are isolated from tonsil tissue and transformed with efficiency up to 85%, thus utilizing a large portion of the human VDJ/VJ repertoire. Through ex vivo stimulation, the B cells class switch and may undergo somatic hypermutation, thus producing a human “library” of different IgG antibodies that can then be screened against any antigen. Since diversity is generated ex vivo, sampling immunized or previously exposed individuals is not necessary. Cells producing the antibody of interest can be isolated through limiting dilution cloning and the human antibody from the cells can be tested for biological activity. No humanization is necessary because the antibodies are produced from human B cells. By eliminating immunization and humanization steps and screening a broadly diverse library, this platform should reduce both the cost and time involved in producing therapeutic monoclonal antibody candidates.
Key words: human, antibody, monoclonal, novel platform, naïve, B cell, therapeutic
Introduction
The use of therapeutic monoclonal antibodies (mAbs) represents a second wave of ground-breaking innovations in the biotechnology industry that followed the success of recombinant proteins.1 After Kohler and Milstein developed a procedure for producing mAbs in 1975, scientists recognized that the agents could potentially be powerful tools for the treatment of many diseases.2 In 1986, the first mAb, OKT3 (Muromonab-CD3), was approved for human use.3 With more than 20 mAbs now in clinical use, mAbs have clearly come of age as therapeutics, generating an estimated $27 billion dollars globally in 2007 and continuing to grow annually.4,5
The first therapeutic antibodies were produced in immunized mice; however, when administered to humans, these foreign molecules were recognized and eliminated by the human immune system, sometimes with allergic responses ranging from a mild rash to renal failure.2 To circumvent this problem, researchers began engineering modified mouse antibodies to reduce immunogenicity. One approach resulted in the creation of chimeric antibodies in which murine variable domains responsible for the binding activity were fused to the human constant domains.6 These chimeric molecules are approximately 70% human and possess a human Fc sequence, which reduces the chances of immunogenicity, but does not eliminate it all together. As techniques to engineer antibodies progressed, it became possible to decrease the murine portion of the mAb even further by replacing the mouse hypervariable loops with those of human sequence using an approach called complementarity-determining region (CDR) grafting.7 The resulting “humanized” antibodies contain 85–90% human sequences, but the process of humanizing is more technically demanding than the mouse-human fusions used to generate chimeric antibodies.2 Furthermore, humanizing an antibody can result in a loss of activity, i.e., production of an antibody without any function. Most of the approved therapeutic mAbs on the market today are either chimeric or humanized and stimulate the human immune system to produce anti-drug antibodies (ADA).8,9
Therapeutic antibodies have also been successfully developed using display technologies in which human antibodies or antibody fragments are displayed on the surface of a simple organisms, such as phage, bacteria or yeast. These in vitro technologies, which we refer to as first generation technologies, have advanced the field of therapeutic antibody discovery, but they also have disadvantages. The largest of the phage display libraries comprises approximately 10 billion human antibodies, the sequences of which are derived from analysis of naturally occurring human gene sequences.10,11 Utilizing phage expression allows for many antibody variations to be screened at once and those that bind are identified and isolated with relative ease. The disadvantage of the phage display system is that the library does not consist of full-length antibodies; the sequences, are based on human sequences as opposed to being actual human sequences and the proteins are expressed by bacteria (prokaryotic organism) rather than human cells. Displaying antibodies or antibody fragments on a eukaryote such as yeast allows for some post-translational modifications to the antibodies that do not occur in prokaryotes; however, the modifications may differ from human.12 Phage and yeast display technologies have proved successful for identifying antibodies generated from human genes, but, due to differences in the expression and post-translational modification machinery in the bacterial and yeast cells, they may not be optimal.
Another common method of producing first generation therapeutic human antibodies utilizes transgenic animals that produce antibodies from human genes. When challenged with an antigen, these animals produce human antibodies, thus circumventing the laborious humanization steps.13 Six antibodies from transgenic animal platforms have been approved by the US Food and Drug Administration (FDA) and 32 more are in clinical trials.14 This technology, however, is limited to the production of antibodies that the immune system of the mouse will recognize.
Human antibodies can also be produced from B cells isolated from humans; however, this method involves screening large numbers of candidates to identify individuals with circulating antibodies to an antigen, which can be logistically daunting.15 As with transgenic animal models, target antigens are limited to what the human immune system will recognize.
A novel platform technology described here leverages the advantages of many of the first generation platforms and eliminates the disadvantages. The technology produces human IgG antibody libraries potentially comprising the entire human repertoire. Unlike the antibodies produced in phage or yeast, the antibodies produced from the Immunologix platform begin with naïve B cells isolated from human tonsil tissue. The antibodies are produced by human cells from human genes and are, therefore, 100% human antibodies. While the transgenic animal platforms are limited to a repertoire that arises in the animal, the Immunologix platform differentiates B cells ex vivo, thereby generating repertoire diversity potentially on par with phage or yeast display, but utilizing full-length human IgG rather than bacterial or yeast expressed Fv. The size and diversity of the library allows for the production of antibodies to potentially any and all antigens, including those that would otherwise be difficult or impossible to make in a transgenic mouse. To date, this novel technology is the only method that starts with naïve human B cells and produces full-length antibody libraries in an in vitro system. This brief report outlines our progress in the discovery of therapeutic mAbs and methods used in the production of these human antibodies derived from human naïve B cells.
Results
Enrichment of human B cells from tonsil tissue.
Searching for antibodies in an ex vivo system requires high numbers of B cells to maximize the inclusion of as many VDJ/VJ recombinants as possible, making human tonsils an ideal source of human B cells. Excised human tonsils from consenting patients were pressed through a sieve to obtain a single cell suspension. This suspension was then centrifuged over a Ficoll-Hypaque gradient to isolate mononuclear cells, yielding approximately 143 million mono-nuclear cells per gram of tonsil (Table 1). In comparison, peripheral blood typically yields less than two million mononuclear cells per milliliter (data not shown). The tonsil mononuclear cells were analyzed via flow cytometry and 60% were CD45+/CD19+/CD20+ B cells (Fig. 1A). Human B cells were enriched from the mononuclear cells using negative magnetic selection, in which all non-B cells are removed, leaving the B cells untouched. Before and after magnetic separation, hemacytometer counts were compared and around 50% of the total mononuclear cells were enriched as B cells (Table 1). When these enriched cells were analyzed by flow cytometry, they were found to be over 90% pure CD45+/CD19+/CD20+ B cells (Fig. 1D), consistent with the expected results from the manufacturer. The enriched cells were further analyzed to determine B-cell phenotype; over 90% of the enriched B cells were of the naïve B-cell phenotype, IgD+/IgM+ (Fig. 1E). Very few of the cells were positive for only IgM (Fig. 1B and E) or only IgG (Fig. 1C and F), which are indicative of memory B cells. Overall, human tonsils proved to be a readily available and robust source of naïve B cells.
Table 1.
Cellular yields from tonsil tissue
| Mean | 95% CI | ||||
| Mononuclear cell yield (cells/gram tonsil) | n = 28 | 1.43E + 08 | 1.08E + 08 | - | 1.78E + 08 |
| Total B-cell yield (% mononuclear cells) | n = 19 | 46.40% | 37.23% | - | 55.57% |
Figure 1.

Enrichment of naïve B cells from human tonsil tissue. Mononuclear cells (MC) prepared from a Ficoll-Hypaque gradient were enriched for B cells using negative magnetic separation. Samples of the mononuclear cells (A–C) and the enriched B-cells (D–F) were phenotyped by gating for CD45 positive cells and staining for CD19 and CD20 (A and D) and also by staining for surface IgD, IgM and/or IgG expression (B, C, E and F). Results from a representative sample are shown.
Transformation of human B cells.
The number of cells used to generate B-cell-derived antibodies limits the size and diversity of a B-cell library. Since primary B cells will not propagate readily ex vivo, transformation is necessary to maintain the cells in culture for longer periods of time; therefore, the number of cells that are transformed limits the size and diversity of a B-cell library. Prior to the method described in this brief report, transformation with supernatant from B95-8 cells, which contains high titers of Epstein-Barr virus (EBV), typically yields transformation efficiency of less than 10%.16–18 Using the novel patent-pending process of concentrating the B95-8 supernatant to 10× via ultrafiltration19 and introducing the supernatant to the B cells via centrifugation (“spinfection”),20 transformation efficiency of up to 86% was achieved (Table 2). Following transformation, B cells were suspended in DF media to induce class switching and somatic mutation ex vivo, and were distributed into 96-well plates at 105 cells per well. After 21 days, IgG secretion was analyzed via ELISpot and 14.6% of the B cells were secreting IgG, compared to low or undetectable levels for freshly isolated mononuclear cells or B cells (Fig. 2A). Additionally, all human IgG isotypes were found in the library supernatants (Fig. 2B) in expected proportions with IgG1 being the most abundant and IgG4 the least.21 Combining high-efficiency transformation and ex vivo differentiation increased the potential diversity of the human B-cell libraries by including more cells in each donor library and inducing class-switch recombination and somatic mutation, respectively.
Table 2.
B-cell transformation efficiency using spinfection with 10× concentrated EBV supernatant
| Mean | S.D. | S.E.M | 95% C.I. | |||
| n = 12 | 66.67% | 30.77% | 8.88% | 47.11% | - | 86.22% |
S.D., standard deviation; S.E.M, standard error of the mean;
C.I., confidence interval
Figure 2.

(A) Representative EliSpots displaying IgG secretion. (i) Freshly isolated naïve B cells from human tonsil [mean = 0.33 cells (0.0% positive)]. (ii) Total mononuclear cell population [mean = 17 cells (0.23% positive)]. (iii) B-cell population after undergoing Immunologix platform technology [mean = 438 cells (14.6% positive)]. All samples plated at 3 × 103 cells/well and done in triplicates. (B) Profile of IgG subclasses expressed by B-cell libraries. Duplicate experiment with two libraries.
Detecting cells secreting IgG reactive with desired antigen.
After at least 12 days in culture in Immunologix DF media, the supernatants from the transformed B cells were screened via ELISA to detect IgG binding to the antigen of choice. In order to determine the utility of the created human antibody libraries, three human antigens (a peptide of ∼15 amino acids, a peptide of ∼40 amino acids and an approximately 40 kDa protein) were each screened against supernatant from at least four B-cell libraries; 48 total 96-well plates were screened for each antigen. Regardless of the antigen (small peptide, medium peptide or large protein), supernatant from approximately 1% of the wells contained human IgG that bound the human antigen (Fig. 3). Cloning of the reactive cells from these wells by limiting dilution produced a monoclonal culture of human B cells secreting human IgG from which recombinant antibodies can be made. Since the antibodies are human and produced by human cells, it is anticipated any ADA response will be minimal or even eliminated.9,22
Figure 3.
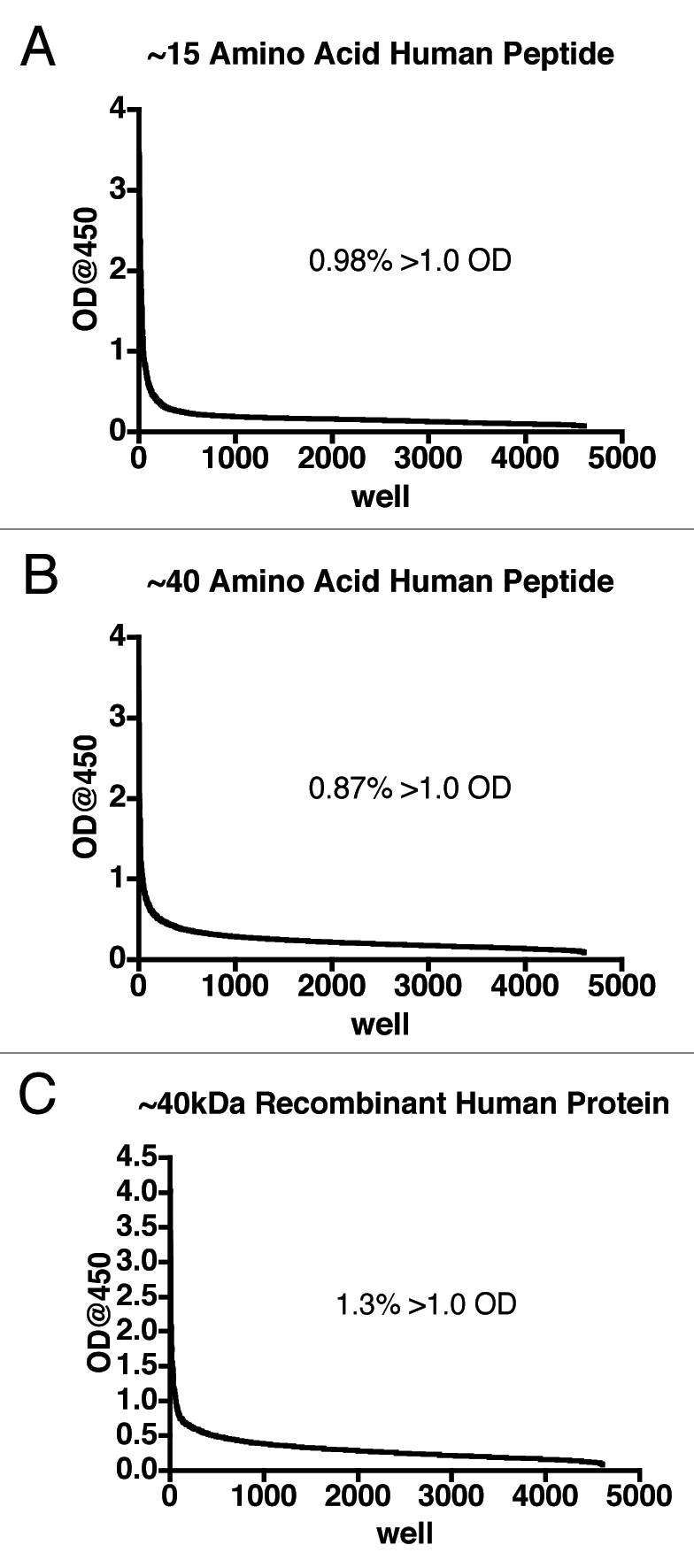
Primary screening of human B-cell supernatants. After at least 12 days, supernatant from each well of the 96-well plates containing transformed human B cells was added to microtiter plates on which antigen was immobilized and captured human IgG detected using an HRP conjugated anti-IgG-Fc antibody. The data from the wells were sorted in descending order and expressed as a line graph. Supernatants from at least four B-cell libraries (48 plates total) are represented for each antigen. The inset indicates the percentage of supernatants that returned an optical density (OD) of greater than 1.0.
Discussion
The FDA approved the first therapeutic antibody in 1986; since then, the production and uses of antibody therapeutics have dramatically changed. The emergence and evolution of antibody platforms designed to produce mouse chimeric, humanized and finally human antibodies was a result of the early recognition of the potential of monoclonal antibodies as targeted therapeutics. Over the next 25 years, numerous platform technologies intended to produce human antibodies have been developed. The most common classes of antibody platforms include phage display, transgenic animals and in vitro genetically modified cells. Muromonab-CD3 (OKT3) was an IgG2a murine antibody produced with hybridoma technology.3 OKT3 is produced in a mouse and causes varying levels of immunogenicity, so it is hypothesized that this adverse effect is due to the human immune system recognizing the murine antibody as foreign.23,24
Because of the immunogenicity associated with murine antibodies, the chimeric antibody was designed by removing the Fc region of the mouse antibody and replacing it with one from a human.25 The first antibodies were approximately 66% human and 34% mouse,26 and five (abciximab, rituximab, basiliximab, infliximab, cetuximab) of the currently FDA-approved mAbs are of this design. Grafting the CDRs of xenogenic antibodies onto human framework and constant regions to produce humanized antibodies may decrease the immunogenicity;24 however, CDR grafting may also result in the loss of some of the advantageous antibody-antigen binding properties.24
In an attempt to alleviate this issue, two major approaches have been used to yield effective CDR-grafted antibodies. The first involves database selection of human V regions with the greatest homology to a murine V region. Computer modeling greatly assists in the selection of the structurally important regions required to produce an antibody with the highest binding affinity and the lowest amount of immunogenicity. A second method involves grafting murine CDRs onto a human V region. If this method results in an antibody with reduced or no binding affinity, attempts can be made to produce new constructs to minimize the loss of affinity.24
Two examples of the successful use of this CDR approach include the humanization of rat anti-CDw52 CAMPATH-1 mAb and a murine mAb targeting the human respiratory syncytial virus, which ultimately resulted in the marketed products alemtuzumab (Campath®) and palivizumab (Synagis®) respectively.27,28 In the case of alemtuzumab, the anti-CDw52 Campath-1 antibody was grafted onto a human IgG1 and reshaped for serotherapy in human by including the six hyper-variable regions from the heavy- and light-chain variable domains of a rat antibody. This change led to an increase in cell-mediated lysis of human lymphocytes and allowed Campath to be indicated as a single agent for the treatment of B-cell chronic lymphocytic leukemia.29
The increase in efficacy achieved by increasing the amount of human sequence highlights the importance of native immunoglobin structure and reveals an unmet need for an efficient method to develop human antibody therapeutics. The novel platform technology described here produces human mAbs (IgG) against nearly any target. Previously, the only method that would produce 100% human antibodies involved selection of B cells from the blood of patients who had been exposed to the antigen, e.g., a viral antigen like influenza.30 While this screening method yields human antibodies, the time and expense of screening potentially thousands of blood samples can become impractical. In addition, searching human blood for a previously produced antibody may yield results for antigens that have been identified by the human immune system, but not for antigens that have eluded the body's defenses. Therefore, a technology that produces a diverse, screenable library of human antibodies appears to be well suited for producing therapeutics.
Platforms utilizing phage or yeast display of antibodies or antibody fragments enable screening against a large library, yet the antibody proteins are not expressed by mammalian cells and may differ in folding or post-translational modifications. Conversely, recombinant animal models are capable of producing full-length human IgG from mammalian cells, but the library is limited to antigens that elicit an immune response by the mouse. The Immunologix platform combines the best characteristics of each technology. By enriching naïve B cells from a robust source and transforming at high efficiency, large and diverse libraries of human antibody variable regions are created. Stimulating differentiation ex vivo further increases the potential diversity, which ongoing research is more precisely measuring. For example, an antibody reactive with Ricin B chain was also previously isolated,31 indicating that the process elicits antibodies against antigens to which the subjects were not previously exposed. Since the antibodies generated are full-length human IgG expressed from human cells, the native conformation is retained. With this novel technology, human antibodies can be generated against virtually any antigen, making advances in antibody therapeutics attainable.
Materials and Methods
Tonsil sample collection.
All samples were collected at Charleston Ear, Nose and Throat using an Institutional Review Board (IRB)-approved protocol and patient consent form. Tonsils were removed by a surgeon and aseptically transferred in a sterile container. Cold storage solution (RPMI-1640, HyClone; antibiotic/Antimycotic Solution 100x, HyClone) was added to the specimen to cover the tonsil during transport to the lab for processing.
Mononuclear cell isolation.
Tonsil tissue was cut into pieces using dissecting scissors and ground through an autoclaved Cellector® (Bellco, Vineland, NJ) 100 uM mesh screen with the sterile end of a 10 cc syringe. Cell suspension was put in 50 ml RPMI-1640, split into 25 ml aliquots and layered over 13 ml of Ficoll-Paque PLUS (GE Healthcare) for a final volume of 50 ml. Tubes were centrifuged (Sorvall) at 1,500 rpm for 25 min at room temperature with break and acceleration set at setting 6. Following centrifugation, the mononuclear cell layer was removed, washed three times in DPBS and resuspended for B-cell enrichment.
B-cell enrichment.
CD19+ cells were isolated using the StemSep B Cell Negative Isolation System according to the manufacturer's instructions (StemCell Technologies, Vancouver, BC Canada).32 Briefly, mononuclear cells were resuspended at 5 × 10e7 cells/ml in DPBS containing 2% fetal bovine serum. EasySep Human B Cell Enrichment Cocktail and EasySep D Magnetic Particles were added to the cells and placed into the Big Easy Magnet (StemCell Technologies). Enriched cells were counted by trypan blue exclusion to calculate the yield.
B-cell immortalization.
Using a process exclusively licensed from the Foundation for Research Development in Charleston, SC and patented by Immunologix,31 purified and enriched B cells were resuspended at 1 × 10e7 cell/ml in Complete Medium (RPMI-1640, 10% Heat-Inactivated FBS, L-Glutamine, Penicillin/Streptomycin). Supernatant from B95-8 cells was concentrated by ultra-filtration using Centricon-Plus 70 100 kDa filter units (Millipore) to one-tenth the original volume (10×). Equal amounts (1:1) of cells and 10× ultra-filtration concentration of EBV supernatant were added to one well of a six-well plate and spun at 900 g for 1 h at room temperature. Infected cells were resuspended to a concentration of 5 × 10e5 cells/ml in differentiation (DF) Media [Complete Media and Human BAFF (Peprotech)], Human CD40L, anti-Human IgM Fc5 µF(ab')2 fragment and other proprietary items and seeded into 96-well plates at 200 µl per well.31 Cells were incubated at 37°C with 5% CO2, feeding as necessary and replacing media every seven days. After 21 days in DF Media, cells were cultured in Complete Media.
Antibodies.
Antibodies used in the flow cytometry include Anti-Human CD19 Alexa Fluor 488 (eBioscience), Anti-Human CD20 APC (eBioscience), Anti-Human CD45 PE-Cy7 (eBioscience), Anti-Human IgM APC (BD Pharmingen), Anti-Human IgD APC (BD Pharmingen) and Anti-Human IgG FITC (BD Pharmingen). The secondary antibody used in the ELISA assays was Goat Anti-Human IgG (γ chain specific) (SouthernBiotech).
Flow cytometry.
All flow cytometry methods were performed using standard techniques.33 Briefly, cells were counted and resuspended at 2 × 10e7 cell/ml in flow buffer (1% FCS, 2 mM EDTA, DPBS) and 100 ul aliquots were taken per sample. Samples were washed three times by centrifugation at 1,500 rpm for 5 min in 1 ml flow buffer at 4°C, antibodies were then added to each sample and incubated on ice for 30 min. After incubation, samples were again washed three times in flow buffer and resuspended in 200 ml of 4% paraformaldehyde for 30 min on ice. Samples were finally washed two times in 1 ml PBS, resuspended in 200 ul PBS and stored at 4°C until read. Samples were read on a BD FACSCalibur™ (BD Biosystems, Rockville, MD) at wavelengths specific to each labeled antibody and analyzed using FlowJo, Flow Cytometry Analysis Software (Ashland, OR).
ELISpot.
The ELISpot IgG assay was performed according to manufacturer's instructions (Mabtech, Inc., Mariemont, OH). Briefly, capture antibody was immobilized and 3.0 × 10e3 naïve B cells/well, Tonsil mononuclear cells or day 21 B cells were added in triplicates overnight. After washing, biotinylated detection antibody was added, followed by Avidin-HRP and AEC substrate. Plates were read on an AID EliSpot Reader (AID Diagnostika, Germany).
Isotype ELISA.
Human IgG isotype was determined by sandwich ELISA according to manufacturer's instructions (Invitrogen Corp., Camarillo, CA). Briefly, B-cell supernatant was mixed with subclass specific capture antibody and added to pre-coated capture strips. Human IgG was detected using a peroxidase conjugated antibody and TMB substrate. Concentrations were calculated using a standard curve generated with a standard serum sample of known isotype concentrations.
Indirect ELISA.
An Indirect Enzyme Linked Immunosorbent Assay (ELISA) was used to detect the positive clones to each target using standard techniques.34 Briefly, 96-well microtiter plates (Corning) were coated with 100 µl of antigen in 0.05 M bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. Plates were washed five times in PBS-T and blocked in (0.1%) non-fat milk blocking buffer from concentrate (KPL, Inc.) at room temperature for 1 h. Following the blocking, the buffer was removed and 100 µl/well supernatants from an antibody library (produced using the Immunologix method above) was added and incubated at room temperature for 1 h. After rinsing three times with PBST, Goat Anti-Human IgG with horseradish peroxidase (HRP) was added and incubated for 1 h in the dark. SureBlue HRP Substrate (KPL, Inc.) was added to each well and incubated for 15 min at room temperature in the dark. Color development was stopped with 100 µl 1 N HCl and optical density (OD) was read at 450 nm in an Epoch plate reader (BioTek).
Acknowledgements
The authors thank W. Guy Bradley for graciously assisting with the flow cytometry and ELISpot experiments contained in this manuscript. We would also like to thank the members of our Scientific Advisory Board for assisting in the preparation of this manuscript.
Abbreviations
- EMA
European Medicines Agency
- FDA
US Food and Drug Administration
- ADA
anti-drug antibody
- mAb
monoclonal antibody
- OKT3
orthoclone
- PBMC
peripheral blood mononuclear cell
- TNF
tumor necrosis factor
- CDR
complementarity-determining region
- IRB
institutional review board
- DF
differentiation
- HRP
horseradish peroxidase
- EBV
Epstein-Barr virus
- CHO
Chinese hamster ovary cells
Conflict of Interest
All three authors are full-time employees of Immunologix, Inc.
References
- 1.Frost & Sullivan, author. Antibody Technology Developments (Technical Insights) 2005:30. [Google Scholar]
- 2.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith SL. Ten years of Orthoclone OKT3 (muromonab-CD3): a review. J Transpl Coord. 1996;6:109–119. doi: 10.7182/prtr.1.6.3.8145l3u185493182. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal S. What's fueling the biotech engine—2009–2010. Nat Biotech. 2010;28:1165–1171. doi: 10.1038/nbt1110-1165. [DOI] [PubMed] [Google Scholar]
- 5.Scolnik PA. mAbs: a business perspective. mAbs. 2009;1:179–184. doi: 10.4161/mabs.1.2.7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuberger MS, Williams GT, Mitchell EB, Jouhal SS, Flanagan JG, Rabbitts TH. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature. 1985;314:268–270. doi: 10.1038/314268a0. [DOI] [PubMed] [Google Scholar]
- 7.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321:522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 8.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 9.Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Deliv Rev. 2006;58:640–656. doi: 10.1016/j.addr.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 10.Hoet RM, Cohen EH, Kent RB, Rookey K, Schoonbroodt S, Hogan S, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23:344–348. doi: 10.1038/nbt1067. [DOI] [PubMed] [Google Scholar]
- 11.Rothe C, Urlinger S, Lohning C, Prassler J, Stark Y, Jager U, et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol. 2008;376:1182–1200. doi: 10.1016/j.jmb.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 13.Lonberg N. Human monoclonal antibodies from transgenic mice. Handb Exp Pharmacol. 2008:69–97. doi: 10.1007/978-3-540-73259-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 15.Crowe JE., Jr Recent advances in the study of human antibody responses to influenza virus using optimized human hybridoma approaches. Vaccine. 2009;27:47–51. doi: 10.1016/j.vaccine.2009.10.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, Delecluse HJ. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J Gen Virol. 2005;86:3009–3019. doi: 10.1099/vir.0.81153-0. [DOI] [PubMed] [Google Scholar]
- 17.Aman P, Ehlin-Henriksson B, Klein G. Epstein-Barr virus susceptibility of normal human B lymphocyte populations. J Exp Med. 1984;159:208–220. doi: 10.1084/jem.159.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller G, Lipman M. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc Natl Acad Sci USA. 1973;70:190–194. doi: 10.1073/pnas.70.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanbe E, Zhang DE. A simple and quick method to concentrate MSCV retrovirus. Blood Cells Mol Dis. 2004;33:64–67. doi: 10.1016/j.bcmd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 20.O'Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74:10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7:1703–1710. doi: 10.1111/j.1538-7836.2009.03568.x. [DOI] [PubMed] [Google Scholar]
- 22.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies. mAbs. 2010;2:256–265. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards J, Auger J, Peace D, Gale D, Michel J, Koons A, et al. Phase I evaluation of humanized OKT3: toxicity and immunomodulatory effects of hOKT3gamma4. Cancer Res. 1999;59:2096–2101. [PubMed] [Google Scholar]
- 24.Kipriyanov SM, Little M. Generation of recombinant antibodies. Mol Biotechnol. 1999;12:173–201. doi: 10.1385/MB:12:2:173. [DOI] [PubMed] [Google Scholar]
- 25.Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA. 1984;81:6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chadd HE, Chamow SM. Therapeutic antibody expression technology. Curr Opin Biotechnol. 2001;12:188–194. doi: 10.1016/s0958-1669(00)00198-1. [DOI] [PubMed] [Google Scholar]
- 27.Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332:323–327. doi: 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- 28.Tempest PR, Bremner P, Lambert M, Taylor G, Furze JM, Carr FJ, et al. Reshaping a human monoclonal antibody to inhibit human respiratory syncytial virus infection in vivo. Biotechnology (NY) 1991;9:266–271. doi: 10.1038/nbt0391-266. [DOI] [PubMed] [Google Scholar]
- 29.Hillmen P, Skotnicki AB, Robak T, Jaksic B, Dmoszynska A, Wu J, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol. 2007;25:5616–5623. doi: 10.1200/JCO.2007.12.9098. [DOI] [PubMed] [Google Scholar]
- 30.Collarini EJ, Lee FE, Foord O, Park M, Sperinde G, Wu H, et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J Immunol. 2009;183:6338–6345. doi: 10.4049/jimmunol.0901373. [DOI] [PubMed] [Google Scholar]
- 31.Sutkowski N, Rubinchik S. Human monoclonal antibodies and methods for producing the same. 2007 inventor; PCT/US2008/072124. [Google Scholar]
- 32.Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- 33.Siemer D, Kurth J, Lang S, Lehnerdt G, Stanelle J, Kuppers R. EBV transformation overrides gene expression patterns of B cell differentiation stages. Mol Immunol. 2008;45:3133–3141. doi: 10.1016/j.molimm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Sun SQ, Guo HC, Sun DH, Yin SH, Shang YJ, Cai XP, et al. Development and validation of an ELISA using a protein encoded by ORF2 antigenic domain of porcine circovirus type 2. Virol J. 2010;7:274. doi: 10.1186/1743-422X-7-274. [DOI] [PMC free article] [PubMed] [Google Scholar]