

Abstract
The lung is one of the most common extra-muscular targets in idiopathic inflammatory myopathies (IIM) and interstitial lung disease (ILD) is a prevalent and often devastating manifestation of IIM. IIM-associated ILD (IIM-ILD) contributes to nearly 80% of the mortality in IIM with a reported prevalence of 65% of newly diagnosed IIM cases. Although ILD frequently accompanies clinical and laboratory findings of myositis, overt signs of muscle disease may be absent in the setting of significant lung disease. Understanding the varied scope of presentation of these diseases is essential to providing optimal patient care. This review will provide an in depth examination of ILD in IIM both from a rheumatologic and pulmonary perspective and will discuss the scope of disease, presenting features, genetic associations, pathogenesis, diagnosis, radiographic and histopathologic findings, along with biomarker assessment and a rationale for therapeutic intervention.
Keywords: Myopathy, polymyositis, dermatomyositis, myositis, interstitial lung disease
INTRODUCTION
The idiopathic inflammatory myopathies (IIM), commonly termed myositis, are a rare group of heterogeneous connective tissue diseases (CTD) affecting skeletal muscle and other organ systems. Subsets of IIM are clinically and pathologically assigned to one of three general subcategories: polymyositis (PM), dermatomyositis (DM) and inclusion body myositis (IBM). Some experts would include Amyopathic Dermatomyositis (ADM), skin findings consistent with DM but without muscle involvement, as a potentially distinct category. Juvenile dermatomyositis (JDM) occurs most commonly in the pediatric population, while IBM characteristically affects individuals older than 50 years of age and has not been reported in children.
Myositis is chronic and, if left untreated, is progressive and associated with significant morbidity and mortality. Physical disability, pulmonary compromise, cardiovascular disease and infection have the greatest impact on health and survival. Depending on the organ system(s) involved, myositis may present in many ways. Proximal muscle weakness predominates and may be accompanied by any one of several features including characteristic skin rashes (in DM), Raynaud’s phenomenon, polyarthritis, pulmonary involvement and dysphagia (pharyngeal involvement). Muscle enzymes are frequently elevated, including creatine kinase (CK), aldolase, aspartate aminotransferase (AST), alanine aminotransferase (ALT) and lactic acid dehydrogenase (LDH); however, some DM patients with active myositis present with normal muscle enzymes.
Other than skin involvement, the lung is the most common extra-muscular target in IIM. The first case of IIM-associated interstitial lung disease (ILD) was reported in 1956 in a DM patient [1]. Since this initial description, ILD has been increasingly recognized as an important and often devastating manifestation of IIM. IIM-associated ILD (IIM-ILD) has a reported prevalence of 78% in IIM [2], and is found in 65% of newly diagnosed IIM cases [3]. Because ILD appears to be restricted to subsets of DM and PM rather then IBM, this review will focus on the pulmonary manifestations of PM/DM--including the scope of disease, presenting features, pathogenesis, diagnosis, radiographic findings, biomarker assessment, and therapeutic rationale.
Of note, patients with mild to life-threatening ILD may have minimal, non-existent or late onset myositis or skin disease, and the lack of correlation between muscle disease and pulmonary symptoms often leads to delayed diagnosis and a compromised therapeutic response [4–7]. Yet, the coexistence of pulmonary and rheumatologic features in IIM warrants close collaboration between pulmonologists, rheumatologists and dermatologists. We therefore advocate that, even in the absence of overt rheumatologic disease, consultation with a rheumatology specialist might be considered in all patients with “idiopathic” ILD. By extension, a high index of suspicion for subclinical CTD amongst patients presenting with pneumonitis must be maintained among pulmonary specialists [8–11].
EPIDEMIOLOGY
The frequency of parenchymal pulmonary involvement has been reviewed in many studies, both retrospective and prospective [12]. These estimates have varied widely and range from 20–86% [13–15]. Prospective studies using sensitive screening techniques such as high resolution computed tomography (HRCT) frequently identify ILD as an early manifestation of PM/DM in which up to 78 % of patients may present with some degree of interstitial lung involvement (18% of which is occult) [2].
While ILD appears to occur with a similar incidence in both PM and DM, there has been a suggestion that DM-ILD and ADM-ILD have a more severe course and are more likely than PM-ILD to be associated with diffuse alveolar damage (DAD) [5, 16–21]. DM-ILD may also be more refractory to corticosteroid treatment, with a poorer prognosis and worse survival than PM-ILD [22]. In contrast to adult PM and DM, pulmonary disease in JDM is rare [23], though it may be under-recognized [24, 25].
In general, the epidemiology of ILD associated with CTD presents a number of complex questions. A significant potential confounder is that ILD may be caused by CTD as well as by the immunosuppressive medications used to treat it. This is also true of IIM-ILD, making it occasionally difficult to know the relative contribution of disease versus that of medications used for treatment. Screening with PFTs to assess baseline pulmonary status prior to initiation of immunosuppressive agents has therefore been suggested as a means to assess the potential contribution of these agents [26]. It is also important to recognize that a substantial percentage of IIM patients (up to 69%) can present with lung disease alone [7].
Beyond these issues, ILD occurring in concert with para-neoplastic inflammatory myopathy is of potential concern. Previous epidemiologic studies have convincingly demonstrated that incident malignancy occurs more frequently in DM than in the general population, though the link between PM and malignancy is less well established. IIM-ILD coincident malignancy is not a usual consideration and has been described as incidental [27–29] and malignancy is reportedly not associated with ILD in patients with anti-synthetase antibodies and IIM; however a 15% association was recently identified in a retrospective cohort with specific antibody combinations [30]. Screening for malignancy in IIM therefore remains appropriate regardless of the presence of associated ILD.
GENETIC ASSOCIATION
The prevalence of PM and DM is increased in subjects carrying particular HLA Class II haplotypes that in turn are associated with specific autoantibodies and phenotypic characteristics [31–33]. Trans-racial gene mapping has shown the greatest linkage to HLA-DQA1 [33]. Of particular interest are the linked alleles, DRB1*03-DQA1*05-DQB1*02, which form a haplotype in Caucasian populations that is strongly associated with the presence of anti-synthetase or anti-PM/Scl antibodies in both DM- and PM-ILD [32]. In patients with the anti-synthetase syndrome (see below), this haplotype is associated with poor outcomes [32]. This contrasts with the generally favorable outcome and lack of significant ILD in patients with anti-Mi-2 autoantibodies frequently associated with the haplotype DRB1*07-DQA1*02-DQB1*02 [32].
Based on previous studies in myositis that implicate infectious and non-infectious triggers and the known genetic predisposing factors discussed earlier in subsets of myositis, it is likely that IIM-ILD has environmental triggers. This may be more likely in certain autoantibody subsets such as patients with the anti-Jo-1 autoantibody as these individuals also possess unifying genetic characteristics [34, 35].
PATHOGENESIS
Polymyositis versus Dermatomyositis in Muscle and Lung Disease
Of striking interest, while both PM and DM can share the same repertoire of specific antibodies and possess similar clinical patterns of muscle involvement, their divergent muscle histopathology suggests a different immunopathogenesis. PM appears to be a CD8+ predominant T-cell mediated assault on the myofiber, suggesting that the targeted antigen lies within the muscle surface. This conclusion is supported by the demonstration of clonally restricted T cell populations invading non-necrotic muscle fibers [36].
In contrast to PM, DM appears to be humorally mediated [37], with B-cells and CD4+ T cells located in a perivascular pattern leading to a perifascicular atrophy and fibrosis. Although simple vascular insult may not fully explain the histopathologic abnormalities in DM muscle tissue, vascular abnormalities appear to contribute to the dermatologic features of the heliotrope rash and Gottron’s sign and papules. Additionally, DM is characterized by overexpression of alpha interferon - inducible genes [38]. The importance of these cytokine pathways is reinforced by the diminished expression of these genes in patients whose disease improves with immunomosuppressive therapy. Implicating the latter cytokine pathway in the pathogenesis of IIM-ILD remains speculative at this point. In contrast, T-cell activation appears to play a key role in initiating lung damage in the pathogenesis of both PM and DM-ILD. Bronchoalveolar lavage (BAL) has demonstrated a predominance of T lymphocytes with a decreased CD4+/CD8+ ratio [36]. Lung biopsy specimens reveal activated T-cells in the interstitium and within the alveolar walls of even normal appearing alveoli [39]. Interestingly, studies suggest that Th1- associated cytokines may mediate the immune response of IIM-ILD, which may distinguish this entity from IPF and scleroderma-related ILD wherein cytokines appear to be predominantly Th2 associated [40, 41].
Antibodies in IIM and IIM-ILD
Although the pathological mechanisms of muscle injury appear to be different between DM and PM, both share a repertoire of antibodies associated with unifying extramuscular manifestations. Numerous unsuccessful attempts have been made to link viral proteins with the development of IIM and antibody development. Though causative agents have been elusive, a common pattern in the variable region of T cell receptors has been identified in T cells found in both lung and muscle of IIM patients with ILD--suggesting shared auto-antigenic targets in both organs [36].
Providing a potential pathogenic clue, eight antibodies associated with IIM have each been identified as targeting different aminoacyl tRNA synthetases perhaps interfering with translation of messenger RNA and protein synthesis. Highly linked to ILD and the antisynthetase syndrome (see ‘clinical manifestations’ below), these autoantibodies are collectively referred to as antisynthetase autoantibodies (Table 1). Although different antisynthetase autoantibodies rarely coexist in the same individual, these antibodies can occur with other myositis associated autoantibodies that target other autoantigens. Anti-histidyl tRNA synthetase antibody, often referred to as anti-Jo-1 antibody, is the most frequently detected antisynthetase autoantibody and is strongly associated with the presence of ILD in both PM and DM. In one large cohort, ILD was identified in 86% of anti-Jo-1 antibody positive myositis patients [15]. Moreover, in an in vitro study, sera of anti-Jo-1 antibody positive patients with PM or DM induced activation of endothelial cells, suggesting that this may contribute to systemic organ involvement in the antisynthetase syndrome [42]. The presence of anti-Jo-1 antibodies and ILD have also been associated with elevated levels of C reactive protein as well as CXCL 9 and CXCL 10 [15], though the specific link to pulmonary manifestations of the antisynthetase syndrome remains speculative. Like anti-Jo-1, other antisynthetase antibodies such as anti-PL-12 (anti-alanyl tRNA synthetase) and anti-PL-7 (anti-threonyl tRNA synthetase) are strongly associated with ILD however, myositis and arthritis may be less frequent in these patients than in patients with anti-Jo-1 antibody [9, 11, 43].
Table 1.
Antibodies in Idiopathic Inflammatory Myopathy
| Autoantibodies in IIM | ||
|---|---|---|
| Antisynthetase Autoantibodies | ||
| Name | Antigen | Clinical Manifestation |
| Jo-1 | Histidyl tRNA synthetase | PM, DM + ILD |
| PL-7 | Threonyl tRNA synthetase | PM, DM + ILD |
| PL-12 | Alanyl tRNA synthetase | ILD>Myo |
| EJ | Glycyl tRNA synthetase | DM>PM +ILD |
| OJ | Isoleucyl tRNA synthetase | ILD+PM/DM |
| KS | Asparaginyl tRNA synthetase | ILD>Myo |
| Zo | Phenylalanyl tRNA synthetase | ILD+Myo |
| Ha | Tyrosyl tRNA synthetase | ILD+Myo |
| Non-synthetase Autoantibodies | ||
| Name | Antigen | Clinical Manifestation |
| SRP | Signal recognition particle | Severe, acute, resistant necrotizing myopathy |
| Mi-2 | DNA helicase | Dermatomyositis with rash>muscle symptoms, treatment responsive |
| P155/140 | Transcriptional Intermediary Factor 1- Gamma (TIF-γ) | Cancer in adult DM; Severe cutaneous JDM |
| Anti-CADM 140 | Melanoma Differentiation Associated Gene 5 (MDA5) | Amyopathic DM; Rapidly progressive lung disease |
| P140 kDa | Nuclear Matrix Protein NXP-2 | JDM |
| Anti-SAE | Small ubitquitin-like Modifier-Activating Enzyme | DM |
| Other Antibodies Associated with IIM | ||
| Name | Antigen | Clinical Manifestation |
| PM-Scl | Unidentified | PM or DM/SSc overlap |
| U1RNP | U1 small ribonuclear protein | MCTD (overlap syndrome) |
| Non-U1 snRNPs | U2, U4/6, U5, U3 snRNPs | PM or DM/SSc overlap or SSc |
| Ku | DNA binding proteins | Myo/SSc/SLE overlap |
| Ro (SSA); includes Ro60 and Ro52 | RNA protein | Myositis often with SS or SLE; may be associated with ILD (esp Ro-52) |
| 56 kD | Ribonuclear protein particle | Myositis, often with Jo-1 |
| KJ | Unidentified translation factor | PM, ILD, RP |
| Fer | Elongation factor 1a | Myositis |
| Mas | tRNASer binding protien | Myositis, rhabdomyolysis, chronic hepatitis |
| MJ | Unidentified nuclear pore | JDM |
| hPMS1 | DNA repair | Myositis |
Abbreviations KEY: PM=polymyositis; DM=dermatomysoitis; ILD= interstitial lung disease; Myo = myositis (may be either PM or DM); SLE= Systemic lupus erythematosus; SSc = Systemic sclerosis or scleroderma; RP= Raynaud phenomenon; JDM= juvenile dermatomyositis.
Anti-Mi-2
Anti-Mi-2 is an antinuclear antibody most closely, but not exclusively, associated with DM while anti-PM/Scl, an antinucleolar autoantibody, is associated with patients having an overlap syndrome of both myositis and systemic sclerosis. The autoantibody against signal recognition particle is associated with a predominantly necrotizing myopathy in PM and although the occurrence of ILD was greater than 20% in some studies [44], several series have noted the paucity of ILD associated with this antibody [45, 46]. Anti-CADM-140 has been identified as a specific antibody in ADM associated with a rapidly progressive ILD [47].
Antibody Concurrence
As suggested by phenotypic differences, antibodies coexisting in the sera of patients with DM or PM may play a role in disease pathogenesis. For example, anti-Ro52/SSA antibody frequently occurs with antisynthetase antibodies (Jo-1, PL-7, PL-12) and has been associated with particularly severe ILD [48, 49]. The frequency of these antibodies occurring together suggests that finding anti-Ro52 antibodies should prompt investigation for antisynthetase autoanti-bodies and ILD or other features of the antisynthetase syndrome [50] as this may impact treatment and prognosis [51].
CLINICAL PRESENTATIONS OF IIM-ILD
The breadth of presentation in IIM-ILD ranges from asymptomatic basilar lung fibrosis [2] to an acute, rapidly progressive process associated with adult respiratory distress syndrome (ARDS) and respiratory failure [16, 17, 19]. The most common presenting symptoms are cough and dyspnea; however, IIM-ILD may be associated with asymptomatic lung disease, as evidenced by the high frequency of radiographically documented ILD [15]. Of clinical importance, there is no correlation between severity of lung involvement and the degree of myositis disease activity [7, 19, 52–54], yet, the severity of respiratory symptoms at presentation appears to correlate with outcome in IIM-ILD [13, 55].
Asymptomatic or Occult ILD
Asymptomatic or occult ILD is identified on plain radiographs or computed tomography of the chest or with restrictive physiology on pulmonary function testing in patients with IIM [13]. Although these findings can occur at presentation, they highlight the importance of continued vigilant assessment to disclose silent but progressive disease. The clinical presentation of symptomatic IIM-ILD generally follows three patterns [56]:
Acute and rapidly progressive ILD - corresponding to ARDS with the histopathology of diffuse alveolar damage (DAD)
Subacute or chronic ILD - corresponding to organizing pneumonia (OP) or an overlap of OP and nonspecific interstitial pneumonia (NSIP) usually with a good response to corticosteroids
Chronic progressive fibrosing ILD - corresponding to fibrotic NSIP or usual interstitial pneumonia (UIP) which tends to respond poorly to steroids and other forms of immunosuppressive therapy
(See below for histopathologic definitions)
Perhaps the most common clinical pattern of IIM-ILD, however, is the gradual progression of non-productive cough, dyspnea, and decreased functional capacity. Thus, careful interview and physical examination at presentation are critical in detecting the presence of early, more treatable stages/forms of lung disease in IIM. At the same time, it is important to recognize that respiratory symptoms in the setting of IIM may represent non-parenchymal abnormalities such as infection, chest wall weakness, cardiac dysfunction, hemorrhage, pneumothorax or pneumomediastinum or pulmonary hypertension. Pleural effusion is generally not accepted to be associated with IIM and should prompt a search for another cause.
While many forms of IIM-ILD are associated with a subclinical presentation or relatively indolent progression, the worst outcomes are associated with an acute, rapidly progressive presentation characterized by diffuse parenchymal lung involvement in the presence or absence of active myopathy. The amyopathic form of dermatomyositis (ADM) described above certainly is associated with lung disease, as ILD has been seen in up to 24% of patients with this syndrome [52]. Moreover, ILD in ADM is often rapidly progressive [6, 18, 20, 21] and associated with poor survival [5, 18, 20]. Another potential scenario is the occurrence of overt muscle and/or skin disease several months after the pulmonary manifestations in IIM or overlap syndromes. Of note, the frequency of malignancy in ADM is similar to that of DM [53].
The antisynthetase syndrome is characterized by the presence of any one of several antisynthetase autoantibodies (perhaps occurring in up to one-third of DM and PM patients) in combination with fever, Raynaud’s phenomenon, polyarthritis, myositis, ILD and “mechanic hands” (thickened skin along sides of fingers with dry cracking and fissuring). ILD often dominates the clinical picture and may be the presenting feature [4, 7, 9, 54] of the syndrome with the acute onset of fever and respiratory distress in nearly 50% of patients [7]. Patients with anti-PM-Scl share similar features of the antisynthetase syndrome such as Raynaud phenomenon, ILD, myositis and even mechanic hands [57, 58].
Acute pulmonary decompensation can occur in any patient with IIM-ILD but is certainly common with the antisynthetase syndrome whether patients manifest complete forms of the syndrome or form frustes.
Complications of IIM-ILD
Beyond the underlying immune-mediated insult to the lung parenchyma of ILD, the following complications may appear in IIM in the presence or absence of ILD.
Chronic Respiratory Insufficiency
Chronic respiratory insufficiency may be the result of permanent damage to the lung parenchyma as a sequela to fibrosing ILD. Even after disease activity has been staved, patients may require life-long oxygen supplementation.
Opportunistic Infection
Opportunistic Infection is an important cause of morbidity and mortality (28% mortality in patients with opportunistic infection in IIM-ILD) [59]. Serious infections occur most frequently in the first year of diagnosis (62% of patients), but also immediately after IIM onset (89% of IIM patients) from pulmonary and gastrointestinal organisms. This complication results from immunosuppressive medications, myopathic hypoventilation with resultant atelectasis and diminished cough, or aspiration pneumonia secondary to pharyngeal involvement and dysphagia [59]. ILD, steroid use, and lymphopenia further increase the risk for opportunistic infections [59–61].
Hypoventilation
Hypoventilation due to decreased diaphragmatic, intercostal, and accessory muscle strength is seen in the setting of severe muscle weakness. Ventilatory insufficiency is a risk factor for serious pulmonary infection [59] and a mean inspiratory pressure of less than 40% of normal is associated with hypercapnia [62] leading to cognitive and further cardiopulmonary and muscular dysfunction.
Pneumomediastinum
Pneumomediastinum, a rare complication where free air collects around the structures of the mediastinum, occurs in 15% of patients with IIM-ILD [20, 63]. Pneumomediastinum may be an early feature of ILD in IIM and can respond to immunosuppressive therapy, but is a poor prognostic sign with a 25% mortality rate in the context of severe ILD in DM or ADM especially when associated with ARDS [64].
Aspiration
Aspiration results from the pharyngeal myopathy of IIM and results in chronic microaspiration of gastric fluids and aspiration pneumonia. On HRCT, aspiration pneumonia appears as consolidation in dependent areas of lung [65]. Importantly, chronic aspiration and gastro-esophageal reflux may exacerbate respiratory symptoms and incite parenchymal changes [66, 67].
Pulmonary arterial hypertension (PAH) is likely under-recognized, but a serious, manifestation of IIM that carries a dismal prognosis. It may be secondary to progressive pulmonary fibrosis with resultant hypoxemic vasoconstriction [68] or mechanistically similar to primary pulmonary hypertension secondary to a vasculopathy with intimal disruption [69, 70]. Thus, patients with IIM or IIM-ILD should have an echocardiogram done for screening purposes. An isolated reduction in the diffusion capacity of the lung for carbon monoxide (DLCO) without concomitant worsening of forced vital capacity (FVC) may herald the onset of pulmonary vascular disease. Ultimately, multiple abnormalities in ILD may lead to precapillary pulmonary hypertension, including vascular inflammation, perivascular fibrosis, and mechanical or fibrotic vascular destruction with subsequent compensatory hypertrophy of surrounding ‘healthy’ arterioles [68].
DIAGNOSIS AND DISEASE MONITORING
As previously discussed, establishing a diagnosis of myositis mandates careful assessment for underlying ILD. However, even though a large percentage of newly diagnosed IIM cases already present with abnormalities on HRCT or PFTs consistent with ILD and restrictive physiology, the significance of these findings in the absence of respiratory symptoms is unknown. As a result, there are no consensus screening guidelines for ILD in patients with IIM or accepted recommendations for treatment of the asymptomatic patient. Conversely, in the patient with IIM and symptoms of dyspnea, cough or decreased functional performance, it is important to obtain a chest radiograph, HRCT of the chest, and PFTs with a measurement of DLCO. BAL may also be necessary if infection is a consideration and open lung biopsy should be performed if there is diagnostic uncertainty.
Chest Radiographs
Chest radiographs, although sometimes helpful in screening and interval assessment of infection and overt ILD, have limited sensitivity and specificity in the diagnosis and follow-up of ILD. However, HRCT is sensitive and represents the method of choice for the detection and characterization of ILD. Some investigators believe that suggestive or typical features of UIP, NSIP or OP on HRCT may obviate the need for an invasive and potentially harmful surgical lung biopsy [65]. In IIM-ILD, HRCT parenchymal changes are fairly heterogeneous including basilar and posterior infiltrates, with various patterns that commonly include consolidation rather than honeycombing in non-septal, linear, plate-like, and sub-pleural patterns [71]. However, it is not uncommon for consolidation to progress to frank fibrosis [56].
Histopathologic Patterns
Several histopathologic patterns correlate with HRCT findings and have been described in IIM-ILD. Although initially described in idiopathic interstitial pneumonia (IIP), they are also observed in patients with CTD, specifically IIM.
The most common histology in PM/DM is NSIP with a prevalence of approximately 80% [27, 52, 72]. NSIP is characterized by temporally and geographically homogenous infiltration of lung interstitium by inflammatory cells, with moderate collagen deposition, preserved lung architecture, and scarcity of fibroblast foci and histologic honeycombing. HRCT findings are peripheral ground glass consolidation and non-coarse fibrosis with some reticularity also representative of fibrosis, and small septal thickening (Fig. 1). Such ground glass changes in conjunction with a significant reticular pattern may be more suggestive of fibrosing NSIP than cellular NSIP.
Fig. (1).

Nonspecific interstitial pneumonia in a 30-yr old man with dermatomyositis; A, high resolution computed tomography showing peripheral reticulation, irregular linear opacities, and mild ground glass opacity; B, C, microscopy of lung biopsy at low (B) and high (C) magnification showing uniform infiltration of alveolar septa by inflammatory cells and mild fibrosis (courtesy of Dr. L. Chalabreysse, Lyon).
UIP in contrast to NSIP is predominantly fibrotic, with spatially and temporally heterogenous collagen deposition within the lung, architectural destruction, fibroblastic foci, histologic honeycombing, and moderate inflammatory infiltrates. The corresponding HRCT pattern is that of peripheral and basilar honeycombing as well as diffuse septal thickening and traction bronchiectasis. UIP, among the idiopathic interstitial pneumonias, has a poorer prognosis and is less responsive to immunosuppressive treatment than NSIP. However, in CTD-ILD the outcome of UIP is more favorable, with NSIP and UIP in scleroderma having seemingly similar outcomes [73, 74], although bias in selecting patients requiring biopsy is likely. UIP is less common than NSIP in IIM-ILD, while fibrosing NSIP with peripheral honeycombing occurs more frequently.
DAD is the most rapidly progressive pathologic subset and carries the worst prognosis. DAD is diffuse interstitial inflammation with edema and hyaline membrane production ultimately leading to organizing fibrosis. On HRCT, it is diffuse in appearance with ground glass consolidations and in later stages traction bronchiectasis and honeycombing may appear. Any initial HRCT pattern can rapidly deteriorate to ARDS and DAD, often leading to patient demise [16, 75].
OP or Cryptogenic Organizing Pneumonia (COP) is defined by foci of granulation tissue in alveoli and their ducts which can progress to obstruct the smaller airways. The histopathologic pattern is somewhat different than that associated with non-IIM-ILD COP, as the consolidation in IIM-associated COP is more bronchocentric with a greater degree of fibrosis. The HRCT appearance (Fig. 2) corresponds more closely to a pattern of “fibrosing OP” with nodular or patchy bilateral infiltrates. This pattern has the most favorable outcome and is usually very responsive to treatment. Some overlap may occur between NSIP and OP.
Fig. (2).
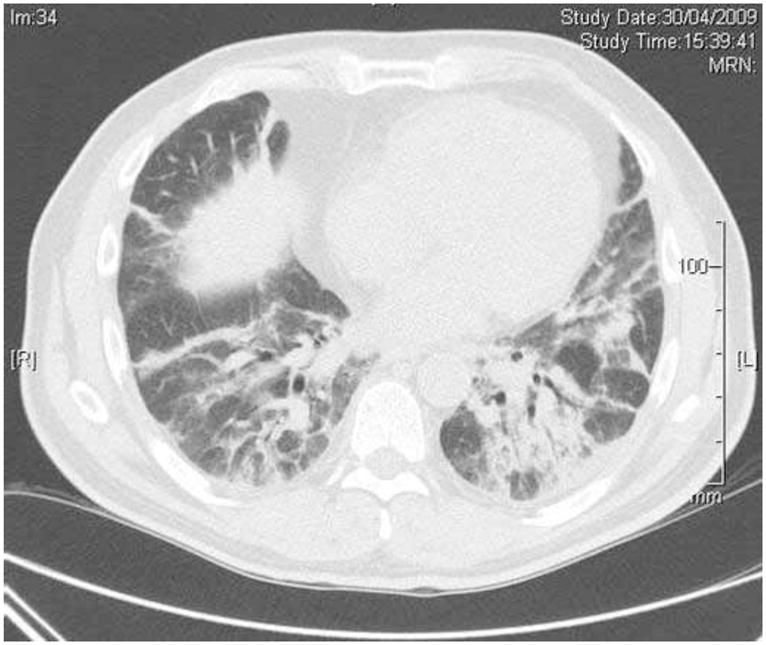
Organising pneumonia in a 55-year old patient with dermatomyositis and anti-Jo-1 antibodies. High resolution computed tomography of the chest demonstrates areas of alveolar consolidation with air bronchograms. Video-assisted thoracoscopic lung biopsy showed histologic pattern of predominant organising pneumonia, with associated features of nonspecific interstitial pneumonia.
Cellular and Tissue Diagnosis
Bronchoscopy
Bronchoscopy and BAL are important diagnostic tools to rule out infection, malignancy, or drug hypersensitivity as a cause for ILD. These diagnostic modalities should also be considered in patients with ILD who deteriorate on immunosuppressive therapy. On the other hand, the cellular profile of the BAL fluid is not helpful in elucidating parenchymal involvement due to lack of specificity, though findings may assist in guiding treatment and determining prognosis.
Surgical Lung Biopsy (SLB)
When HRCT, physiologic and clinical findings correlate with a pattern clearly suggestive of NSIP, SLB may not be necessary for diagnostic purposes. However, biopsy is an important consideration in patterns marked by considerable consolidation where malignant or infectious processes are possible.
Transbronchial Lung Biopsy (TBLB)
Transbronchial Lung Biopsy (TBLB) has a low yield but may be useful in identifying or ruling out infectious processes. The presence and type of intra-luminal fibrosis found on TBLB in IIM-ILD [76] may be helpful in classifying the severity, natural history, and therapeutic responsiveness in IIM-ILD. Two patterns of intraluminal fibrosis representing diverse histopathologic patterns have been identified [76]. The first type is more benign with mild epithelial damage superficial to the alveolar structure sparing the lung parenchyma with formation of isolated intra-luminal ‘polyps’ or ‘buds’ of granulation tissue occupying the alveolar space without compromise of the alveolar walls. Although intraalveolar granulation tissue is a key feature of OP that portends a favorable outcome, sampling error occurs and intraalveolar granulation tissue may represent an accompanying feature of many processes. The second type of intra-luminal fibrosis is defined by alveolar wall compromise and referred to as ‘mural incorporation’ which heralds a more severe course less responsive to corticosteroids and associated with fibrosing NSIP, and/or UIP.
Physiologic Testing
Pulmonary Function Tests (PFTs)
Pulmonary Function Tests (PFTs) are a relatively sensitive, but non-specific, diagnostic tool that remain an important component in the evaluation of respiratory symptoms, pulmonary disease severity, and therapeutic response. The typical pattern associated with ILD reflects restrictive physiology, with reductions in total lung capacity (TLC), forced vital capacity (FVC), DLCO (remembering to consider PAH), forced expiratory volume at 1 second (FEV1), functional residual capacity (FRC), and residual volume (RV). The FEV1:FVC ratio is typically elevated in IIM-ILD, depending on the relative severity of ILD. However, PFTs may be normal especially when confounded by the presence of a concomitant obstructive component due to pulmonary comorbidity related to smoking.
Distinguishing ILD from Respiratory Muscle Weakness (RMW)
Abnormalities in FVC, TLC, and FEV1 that suggest ILD can also be due to respiratory muscle weakness, adding to the challenge of assessing pulmonary disease activity. Some clues on PFTs suggesting RMW include an increase in RV with a normal FEV1:FVC ratio [62]. Evaluation of maximal inspiratory pressure at TLC and maximal expiratory pressure at RV will often distinguish between ILD and RMW [62].
Transdiaphragmatic Pressure Testing
Transdiaphragmatic pressure testing may distinguish between parenchymal lung disease and occult respiratory muscle weakness that can confound PFT or six minute walk testing. Another method to assess respiratory muscle weakness involves gauging positional differences in FVC or chest wall circumference measured by inspiratory supine and upright spiral CT images. Because the supine position requires more diaphragmatic strength for inspiration, large positional differences in these parameters indicate inspiratory muscle weakness [77].
Biomarkers: Krebs von den Lundgen-6 (KL- 6)
Biomarkers, Krebs von den Lundgen-6 (KL-6) expressed on alveolar cells and cells that constitute the bronchiole lining, may be a novel serologic marker for ILD. Several studies have identified KL-6 as a useful biomarker for assessment of disease activity, therapeutic response, and prognosis [78, 79]. In comparison to other experimental serum markers, such as surfactant proteins A and B, KL-6 correlates better with active ILD [80] but findings have not been validated in non-Asian populations. Cytokeratin 19 fragments (CK 19), a cellular component of the bronchial epithelium, are found in proportion to severity of respiratory disease in sera of patients with non-malignant respiratory diseases including IIM-ILD [78, 81].
CXCL9 and CXCL10
Chemokines induced by interferon-gamma (IFN-γ), and C reactive protein (CRP) were recently shown to be elevated in anti-Jo-1 antibody positive patients with ILD, distinguishing this cohort from both IPF and anti-signal recognition particle (anti-SRP) antibody-associated myositis [15]. However, none of these markers are commercially available.
In idiopathic ILD, a significant correlation between plasma concentrations of circulating vascular endothelial growth factor (VEGF) and changes in HRCT and PFTs were noted over a 6 month period [82]. Although not specifically linked to IIM-ILD, this correlation is provocative given that VEGF is highly expressed in the muscle tissue of PM/DM patients [83]. If future studies substantiate a correlation with IIM-ILD, targeting VEGF could have therapeutic implications.
Monitoring of Disease Activity
Longitudinal evaluation using PFTs, HRCT as well as clinical and functional assessments are critical in monitoring disease activity [2]. Six minute walk testing may not be reliable in patients with myopathy, but patient reported qualifiers, such as cough, dyspnea, and self report of functional status, may provide a better gauge for disease activity and sensitivity to change [84].
THERAPEUTICS
There are no approved agents in the treatment of IIM, and this lack of established treatment regimens extends to IIM-ILD. Nevertheless, corticosteroids remain the cornerstone of early treatment with initial doses at 1mg/kg of ideal body weight. In an effort to reduce treatment-related side effects, other immunosuppressive, steroid-sparing agents should be considered at the outset of therapy - particularly when treating the antisynthetase syndrome and other severe and progressive manifestations of ILD.
Cyclophosphamide
Cyclophosphamide administered intravenously has demonstrated efficacy in refractory IIM-ILD in a single, small open label trial [85] where mean FVC improvement was 15%. Although most patients discontinued oxygen supplementation, only 40% reported improvement in dyspnea. Cyclophosphamide has also been used in concert with other immunosuppressive agents in refractory disease [86, 87].
T-Cell Targeted Therapy May Become the Cornerstone
T-cell targeted therapy may become the cornerstone for the treatment of IIM-ILD based on clinical experience as well as the previously described histopathologic studies demonstrating the predominance of this cell type in BAL fluid and lung biopsy specimens. Calcineurin inhibitors such as cyclosporine and tacrolimus (FK506) have demonstrated consistent efficacy [88–92]. Cyclosporine inhibits interleukin-2 production and T-cell proliferation and may be an appropriate choice for early, slowly progressive, non-diffuse ILD. Tacrolimus, 100 fold more potent than cyclosporine in inhibiting T-cell activation, was efficacious in several case series of patients including those refractory to cyclosporine and in patients with ILD associated with antisynthetase autoantibodies with perhaps a more favorable safety profile [88, 90–92].
Mycophenolate Mofetil (MMF)
Mycophenolate Mofetil (MMF), an anti-metabolite that targets production of activated lymphocytes via inactivation of inosine monophosphate dehydrogenase, not only impacts T cells, but also interferes with fibroblast activity, proliferation, and release of profibrotic cytokines such as TGF-β [93]. Case series have revealed potential efficacy in reversing progression or stabilization of disease activity in CTD-ILD including IIM-ILD [94–96].
B-Cell Targeted Therapy
Rituximab has been helpful in both B- and T cell-mediated diseases and has shown efficacy in refractory IIM and IIM-ILD [97–101]. Ongoing studies of this agent may provide rationale for its use in the extramuscular complications of IIM including ILD. Further supporting the role of B cells as potential targets in IIM and IIM-ILD, has been the demonstration of elevated serum B-cell activating factor (BAFF), also known as B lymphocyte stimulator (BlyS). These cytokines and APRIL (a proliferation-inducing ligand) are necessary for B cell maturation and function. Because levels of BAFF and APRIL are higher in patients with Jo-1 positive IIM-ILD patients, therapeutic targeting of these B cell stimulating ligands represents a potential therapeutic approach not yet investigated in clinical trials [102].
Miscellaneous
Azathioprine is widely used in IIM and IIM-ILD, as is methotrexate despite possible concern for lung toxicity in underlying ILD, as immunosuppressive and steroid sparing agents [52]. Intravenous immunoglobulin (IVIG) is regularly employed in the treatment of refractory IIM and IIM-ILD [103, 104] and its empiric use includes the treatment of myositis and the skin manifestations of DM. A small retrospective report noted its potential efficacy in severe, refractory IIM-ILD as salvage therapy [105].
THERAPEUTIC HORIZONS
Anti-Endothelin Receptor Antagonists (ETA)
Endothelin is a potent vasoconstrictor, and mediator of inflammation and fibrosis. ETAs possess anti-fibrotic properties in lung and other organ tissues that appear to modulate the activity of other pro-fibrotic molecules such as transforming growth factor-beta (TGF-β), connective tissue growth factor, angiotensin II, and aldosterone [106, 107]. Some data suggest that ETAs may stimulate matrix metalloproteinase expression, potentially reversing early fibrosis [108, 109]. The first BUILD (Bosentan Use in Interstitial Lung Disease) trial, BUILD-1, examined the efficacy of bosentan in idiopathic pulmonary fibrosis. Favorable trends in dyspnea, health related quality of life, and delayed time to death, however, were only found to be significant in the patients who received surgical lung biopsy [108, 110]. While interesting, the applicability of these findings to IIM-ILD remains undefined.
SUPPORTIVE THERAPEUTICS
Supplemental Oxygen
Evaluation for home oxygen use is an important consideration in patients with IIM-ILD. Given that supplemental oxygen increases functional performance and may provide hemodynamic support in patients with active or stable disease associated with significant damage or compromise, patients should be regularly assessed for oxygen requirements throughout their disease course.
Control of Associated Exacerbating Conditions
Gastroesophageal reflux disease and post-nasal drip likely exacerbate symptoms of ILD and may lead to worsening parenchymal disease. In our experience, assessing the presence and severity of these two entities has proven useful in disease management including patient education on reflux hygiene.
Prophylaxis against Opportunistic Infections
Given the predisposition of patients with IIM to opportunistic infection, screening and prophylaxis are widely used to limit potentially devastating co-morbidity and mortality. The need for sulfamethoxazole - trimethoprim prophylaxis or its equivalent should be individualized for each patient on immunosuppressive regimens including moderate to high dose corticosteroids [59–61]. Due to anti-microbial properties of MMF, this agent may not require antibiotic prophylaxis. Screening for tuberculosis exposure with appropriate history and testing is generally recommended in all patients initiated on immunosuppressive therapy.
Vaccination
There is generally a lack of information on the efficacy and safety of vaccines in IIM patients. However, it is reasonable for IIM-ILD patients to receive the pneumococcal vaccine every five years as well as yearly influenza A vaccine and the non-live version of H1N1 as recommended by the Centers for Disease Control. Other non-live vaccines such as the meningococcal vaccine may also be considered, but live/attenuated vaccines should be avoided in patients receiving immunosuppressive agents [111].
Pulmonary Rehabilitation
Many patients with markedly improved or stable lung disease may benefit from pulmonary rehabilitation especially in regard to respiratory muscle strength. Pulmonary rehabilitation can be tailored to the patients’ performance level with resulting increases in energy and functional performance.
Transplantation
Preparation for transplantation should be considered in patients with progressive and fibrotic ILD. Frequently these patients are denied lung transplantation due to the presence of active muscle disease or other systemic manifestations. Because many of these patients may ultimately benefit from lung transplantation we propose that potential candidates not be excluded purely on the basis of extra-pulmonary manifestations.
References
- 1.Mills ES, Mathews WH. Interstitial pneumonitis in dermatomyositis. J Am Med Assoc. 1956;160(17):1467–70. doi: 10.1001/jama.1956.02960520029008b. [DOI] [PubMed] [Google Scholar]
- 2.Fathi M, Vikgren J, Boijsen M, et al. Interstitial lung disease in polymyositis and dermatomyositis: Longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 2008;59(5):677–85. doi: 10.1002/art.23571. [DOI] [PubMed] [Google Scholar]
- 3.Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis. 2004;63(3):297–301. doi: 10.1136/ard.2003.006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26:459–67. doi: 10.1016/s0049-0172(96)80026-6. [DOI] [PubMed] [Google Scholar]
- 5.Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford) 2005;44(10):1282–6. doi: 10.1093/rheumatology/keh723. [DOI] [PubMed] [Google Scholar]
- 6.Suda T, Fujisawa T, Enomoto N, et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur Respir J. 2006;28(5):1005–12. doi: 10.1183/09031936.06.00038806. [DOI] [PubMed] [Google Scholar]
- 7.Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax. 2008;63(1):53–9. doi: 10.1136/thx.2006.069237. [DOI] [PubMed] [Google Scholar]
- 8.Cottin V. Interstitial lung disease: are we missing formes frustes of connective tissue disease? Eur Respir J. 2006;28(5):893–6. doi: 10.1183/09031936.00101506. [DOI] [PubMed] [Google Scholar]
- 9.Fischer A, Swigris JJ, du Bois RM, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med. 2009;103(11):1719–24. doi: 10.1016/j.rmed.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittoo S, Gelber AC, Christopher-Stine L, Horton MR, Lechtzin N, Danoff SK. Ascertainment of collagen vascular disease in patients presenting with interstitial lung disease. Respir Med. 2009;103(8):1152–8. doi: 10.1016/j.rmed.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Kalluri M, Sahn SA, Oddis CV, et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest. 2009;135(6):1550–6. doi: 10.1378/chest.08-2233. [DOI] [PubMed] [Google Scholar]
- 12.Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. 2005;17(6):701–6. doi: 10.1097/01.bor.0000179949.65895.53. [DOI] [PubMed] [Google Scholar]
- 13.Marie I, Hachulla E, Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47(6):614–22. doi: 10.1002/art.10794. [DOI] [PubMed] [Google Scholar]
- 14.Chen IJ, Jan WYJ, Lin CW, et al. Interstitial lung disease in polymyositis and dermatomyositis. Clin Rheumatol. 2009;28(6):639–46. doi: 10.1007/s10067-009-1110-6. [DOI] [PubMed] [Google Scholar]
- 15.Richards TJ, Eggebeen A, Gibson K, et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum. 2009;60(7):2183–92. doi: 10.1002/art.24631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clawson K, Oddis CV. Adult respiratory distress syndrome in polymyositis patients with the anti-Jo-1 antibody. Arthritis Rheum. 1995;38(10):1519–23. doi: 10.1002/art.1780381020. [DOI] [PubMed] [Google Scholar]
- 17.Guglielmi S, Merz TM, Gugger M, Suter C, Nicod LP. Acute respiratory distress syndrome secondary to antisynthetase syndrome is reversible with tacrolimus. Eur Respir J. 2008;31(1):213–7. doi: 10.1183/09031936.00014707. [DOI] [PubMed] [Google Scholar]
- 18.Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136(5):1341–7. doi: 10.1378/chest.08-2740. [DOI] [PubMed] [Google Scholar]
- 19.Polosa R, Di Mauro C, Spampinato B, et al. A patient with antihistidyl-tRNA synthetase positive polymyositis presenting as acute respiratory distress syndrome. J Clin Rheumatol. 2008;14(4):219–21. doi: 10.1097/RHU.0b013e31817de0d4. [DOI] [PubMed] [Google Scholar]
- 20.Ye S, Chen XX, Lu XY, et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: A retrospective cohort study. Clin Rheumatol. 2007;26(10):1647–54. doi: 10.1007/s10067-007-0562-9. [DOI] [PubMed] [Google Scholar]
- 21.Tsuda T, Asanuma Y, Koyama S, Kawabata Y, Moriguchi M. A case of hypomyopathic dermatomyositis associated with rapid progressive interstitial pneumonia resistant to multi-immunosuppressive therapy. Am J Med Sci. 2007;333(3):185–90. doi: 10.1097/MAJ.0b013e318031b122. [DOI] [PubMed] [Google Scholar]
- 22.Fujisawa T, Suda T, Nakamura Y, et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol. 2005;32(1):58–64. [PubMed] [Google Scholar]
- 23.Gerami P, Walling HW, Lewis J, Doughty L, Sontheimer RD. A systematic review of juvenile-onset clinically amyopathic dermatomyositis. Br J Dermatol. 2007;157(4):637–44. doi: 10.1111/j.1365-2133.2007.08055.x. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi I, Yamada M, Takahashi Y, et al. Interstitial lung disease associated with juvenile dermatomyositis: Clinical features and efficacy of cyclosporin A. Rheumatology. 2003;43:371–4. doi: 10.1093/rheumatology/keg040. [DOI] [PubMed] [Google Scholar]
- 25.Morinishi Y, Oh-Ishi T, Kabuki T, Joh K. Juvenile dermatomyositis: Clinical characteristics and the relatively high risk of interstitial lung disease. Mod Rheumatol. 2007;17:413–7. doi: 10.1007/s10165-007-0610-y. [DOI] [PubMed] [Google Scholar]
- 26.Wilsher M, Wells AU. Pulmonary screening in patients with connective tissue disease prior to initiation of disease modifying drugs. Personal communication dated. Jun 6, 2009.
- 27.Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182–5. doi: 10.1164/ajrccm.164.7.2103110. [DOI] [PubMed] [Google Scholar]
- 28.Legault D, McDermott J, Crous-Tsanaclis AM, Boire G. Cancer-associated myositis in the presence of anti-Jo1 autoantibodies and the antisynthetase syndrome. J Rheumatol. 2008;35(1):169–71. [PubMed] [Google Scholar]
- 29.Rozelle A, Trieu S, Chung L. Malignancy in the setting of the anti-synthetase syndrome. J Clin Rheumatol. 2008;14(5):285–8. doi: 10.1097/RHU.0b013e31817d116f. [DOI] [PubMed] [Google Scholar]
- 30.Marie I, Lahaxe L, Benveniste O, et al. Long-term outcome of patients with polymyositis/dermatomyositis and anti-PM-Scl antibody. Br J Dermatol. 2010;162(2):337–44. doi: 10.1111/j.1365-2133.2009.09484.x. [DOI] [PubMed] [Google Scholar]
- 31.Chinoy H, Salway F, Fertig N, Oddis CV, Ollier WE, Cooper RG. Clinical, serological and HLA profiles in non-Caucasian UK idiopathic inflammatory myopathy. Rheumatology (Oxford) 2009;48(5):591–2. doi: 10.1093/rheumatology/kep035. [DOI] [PubMed] [Google Scholar]
- 32.Chinoy H, Salway F, Fertig N, et al. UK Adult Onset Myositis Immunogenetic Collaboration (AOMIC) In adult onset myositis, the presence of interstitial lung disease and myositis specific/associated antibodies are governed by HLA class II haplotype, rather than by myositis subtype. Arthritis Res Ther. 2006;8(1):R13. doi: 10.1186/ar1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Hanlon TP, Rider LG, Mamyrova G, et al. HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: Allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum. 2006;54:3670–81. doi: 10.1002/art.22205. [DOI] [PubMed] [Google Scholar]
- 34.Chinoy H, Payne D, Poulton KV, et al. UK Adult Onset Myositis Immunogenetic Collaboration & UK Juvenile Dermatomyositis Research Group. HLA-DPB1 associations differ between DRB1*03 positive anti-Jo-1 and anti-PM-Scl antibody positive idiopathic inflammatory myopathy. Rheumatology (Oxford) 2009;48(10):1213–7. doi: 10.1093/rheumatology/kep248. [DOI] [PubMed] [Google Scholar]
- 35.O’Hanlon TP, Rider LG, Schiffenbauer A, et al. Immunoglobulin gene polymorphisms are susceptibility factors in clinical and autoantibody subgroups of the idiopathic inflammatory myopathies. Arthritis Rheum. 2008;58(10):3239–46. doi: 10.1002/art.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Englund P, Wahlström J, Fathi M, et al. Restricted T cell receptor BV gene usage in the lungs and muscles of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 2007;56(1):372–83. doi: 10.1002/art.22293. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology. 2007;69:2008–19. doi: 10.1212/01.WNL.0000291619.17160.b8. [DOI] [PubMed] [Google Scholar]
- 38.Walsh RJ, Kong SW, Yao Y, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56(11):3784–92. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamadori I, Fujita J, Kajitani H, et al. Lymphocyte subsets in lung tissues of interstitial pneumonia associated with untreated polymyositis/dermatomyositis. Rheumatol Int. 2001;21(3):89–93. doi: 10.1007/s00296-001-0146-y. [DOI] [PubMed] [Google Scholar]
- 40.Kurasawa K, Nawata Y, Takabayashi K, et al. Activation of pulmonary T cells in corticosteroid-resistant and -sensitive interstitial pneumonitis in dermatomyositis/polymyositis. Clin Exp Immunol. 2002;129(3):541–8. doi: 10.1046/j.1365-2249.2002.01933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majumdar S, Li D, Ansari T, et al. Different cytokine profiles in cryptogenic fibrosing alveolitis and fibrosing alveolitis associated with systemic sclerosis: a quantitative study of open lung biopsies. Eur Respir J. 1999;14(2):251–7. doi: 10.1034/j.1399-3003.1999.14b03.x. [DOI] [PubMed] [Google Scholar]
- 42.Helmers SB, Englund P, Engström M, et al. Sera from anti-Jo-1-positive patients with polymyositis and interstitial lung disease induce expression of intercellular adhesion molecule 1 in human lung endothelial cells. Arthritis Rheum. 2009;60(8):2524–30. doi: 10.1002/art.24683. [DOI] [PubMed] [Google Scholar]
- 43.Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007;46(1):124–30. doi: 10.1093/rheumatology/kel112. [DOI] [PubMed] [Google Scholar]
- 44.Hengstman GJ, ter Laak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006;65(12):1635–8. doi: 10.1136/ard.2006.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50(1):209–15. doi: 10.1002/art.11484. [DOI] [PubMed] [Google Scholar]
- 46.Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70(6):360–74. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52(5):1571–6. doi: 10.1002/art.21023. [DOI] [PubMed] [Google Scholar]
- 48.Váncsa A, Csípo I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int. 2009;29(9):989–94. doi: 10.1007/s00296-009-0884-9. [DOI] [PubMed] [Google Scholar]
- 49.La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39(3):249–53. doi: 10.1080/08916930600623791. [DOI] [PubMed] [Google Scholar]
- 50.Limaye VS, Cassidy J, Scott G, Roberts-Thomson PJ, Gillis D. Anti-Ro52 antibodies, antisynthetase antibodies, and antisynthetase syndrome. Clin Rheumatol. 2008;27(4):521–3. doi: 10.1007/s10067-007-0762-3. [DOI] [PubMed] [Google Scholar]
- 51.Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senécal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. 2007;9(4):R78. doi: 10.1186/ar2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cottin V, Thivolet-Béjui F, Reynaud-Gaubert M, et al. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J. 2003;22(2):245–50. doi: 10.1183/09031936.03.00026703. [DOI] [PubMed] [Google Scholar]
- 53.Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597–613. doi: 10.1016/j.jaad.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt WA, Wetzel W, Friedländer R, et al. Clinical and serological aspects of patients with anti-Jo-1 antibodies--an evolving spectrum of disease manifestations. Clin Rheumatol. 2000;19(5):371–7. doi: 10.1007/s100670070030. [DOI] [PubMed] [Google Scholar]
- 55.Won HJ, Soon KD, Keun LC, et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med. 2007;101(8):1761–9. doi: 10.1016/j.rmed.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 56.Woodhead F, Wells AU, Desai SR. Pulmonary complications of connective tissue diseases. Clin Chest Med. 2008;29(1):149–64. vii. doi: 10.1016/j.ccm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Lega JC, Cottin V, Fabien N, Thivolet-Béjui F, Cordier JF. Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-tRNA synthetase autoantibodies: A similar condition? J Rheumatol. 2010 doi: 10.3899/jrheum.090652. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 58.Oddis CV, Okano Y, Rudert WA, Trucco M, Duquesnoy RJ, Medsger TA., Jr Serum antibody to the nucleolar antigen PM-Scl: Clinical and immunogenetic associations. Arthritis Rheum. 1992;25:1211–17. doi: 10.1002/art.1780351014. [DOI] [PubMed] [Google Scholar]
- 59.Marie I, Hachulla E, Chérin P, et al. Opportunistic infections in polymyositis and dermatomyositis. Arthritis Rheum. 2005;53(2):155–65. doi: 10.1002/art.21083. [DOI] [PubMed] [Google Scholar]
- 60.Kadoya A, Okada J, Iikuni Y, Kondo H. Risk factors for Pneumocystis carinii pneumonia in patients with polymyositis/dermatomyositis or systemic lupus erythematosus. J Rheumatol. 1996;23(7):1186–8. [PubMed] [Google Scholar]
- 61.Viguier M, Fouere S, de la Salmoniere P, et al. Peripheral blood lymphocyte subset counts in patient with dermatomyositis: Clinical correlations and changes following therapy. Medicine (Baltimore) 2003:82–6. doi: 10.1097/00005792-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 62.Braun NM, Arora NS, Rochester DF. Respiratory muscle and pulmonary function in polymyositis and other proximal myopathies. Thorax. 1983;38(8):616–23. doi: 10.1136/thx.38.8.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selva-O’Callaghan A, Labrador-Horrillo M, Muñoz-Gall X, et al. Polymyositis/dermatomyositis-associated lung disease: Analysis of a series of 81 patients. Lupus. 2005;14(7):534–42. doi: 10.1191/0961203305lu2158oa. [DOI] [PubMed] [Google Scholar]
- 64.Goff BL, Chérin P, Cantagrel A, et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum. 2009;61(1):108–18. doi: 10.1002/art.24372. [DOI] [PubMed] [Google Scholar]
- 65.Devaraj, Anand W, Hansell Athol U, David M. Computed tomographic imaging in connective tissue diseases. Semin Respir Crit Care Med. 2007;28:389–97. doi: 10.1055/s-2007-985611. [DOI] [PubMed] [Google Scholar]
- 66.Savarino E, Bazzica M, Zentilin P, et al. Gastroesophageal reflux and pulmonary fibrosis in scleroderma: A study using pH-impedance monitoring. Am J Respir Crit Care Med. 2009;179(5):408–13. doi: 10.1164/rccm.200808-1359OC. [DOI] [PubMed] [Google Scholar]
- 67.Tobin RW, Pope CE, 2nd, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158(6):1804–8. doi: 10.1164/ajrccm.158.6.9804105. [DOI] [PubMed] [Google Scholar]
- 68.Strange C, Highland KB. Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med. 2005;11:452. doi: 10.1097/01.mcp.0000174250.38188.6d. [DOI] [PubMed] [Google Scholar]
- 69.Minai OA. Pulmonary hypertension in polymyositis-dermatomyositis: Clinical and hemodynamic characteristics and response to vasoactive therapy. Lupus. 2009;18(11):1006–10. doi: 10.1177/0961203309102822. [DOI] [PubMed] [Google Scholar]
- 70.Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 71.Daimon T, Johkoh T, Honda O, et al. Nonspecific interstitial pneumonia associated with collagen vascular disease: Analysis of CT features to distinguish the various types. Intern Med. 2009;48(10):753–61. doi: 10.2169/internalmedicine.48.1714. [DOI] [PubMed] [Google Scholar]
- 72.Arakawa H, Yamada H, Kurihara Y, et al. Nonspecific interstitial pneumonia associated with polymyositis and dermatomyositis: Serial high-resolution CT findings and functional correlation. Chest. 2003;123(4):1096–103. doi: 10.1378/chest.123.4.1096. [DOI] [PubMed] [Google Scholar]
- 73.Flaherty KR, Colby TV, Travis WD, et al. Fibroblastic foci in usual interstitial pneumonia: Idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003;167:1410–15. doi: 10.1164/rccm.200204-373OC. [DOI] [PubMed] [Google Scholar]
- 74.Bouros D, Wells AU, Nicholson AG, et al. Histopathological subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:1581–86. doi: 10.1164/rccm.2106012. [DOI] [PubMed] [Google Scholar]
- 75.Bonnefoy O, Ferretti G, Calaque O, et al. Serial chest CT findings in interstitial lung disease associated with polymyositis-dermatomyositis. Eur J Radiol. 2004;49(3):235–44. doi: 10.1016/S0720-048X(03)00094-9. [DOI] [PubMed] [Google Scholar]
- 76.Mochimaru H, Kawamoto M, Enomoto T, et al. Transbronchial biopsy is clinically useful in classifying patients with interstitial pneumonia associated with polymyositis and dermatomyositis. Respirology. 2008;13(6):863–70. doi: 10.1111/j.1440-1843.2008.01363.x. [DOI] [PubMed] [Google Scholar]
- 77.Gregory SA. Evaluation and management of respiratory muscle dysfunction in ALS. NeuroRehabilitation. 2007;22(6):435–43. [PubMed] [Google Scholar]
- 78.Nakayama M, Satoh H, Ishikawa H, et al. Cytokeratin 19 fragment in patients with nonmalignant respiratory diseases. Chest. 2003;123(6):2001–6. doi: 10.1378/chest.123.6.2001. [DOI] [PubMed] [Google Scholar]
- 79.Satoh H, Kurishima K, Ishikawa H, Ohtsuka M. Increased levels of KL-6 and subsequent Mortality in patients with interstitial lung diseases. J Intern Med. 2006;260(5):429–34. doi: 10.1111/j.1365-2796.2006.01704.x. [DOI] [PubMed] [Google Scholar]
- 80.Ohnishi H, Yokoyama A, Kondo K, et al. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. 2002;165(3):378–81. doi: 10.1164/ajrccm.165.3.2107134. [DOI] [PubMed] [Google Scholar]
- 81.Fujita J, Dobashi N, Tokuda M, et al. Elevation of cytokeratin 19 fragment in patients with interstitial pneumonia associated with polymyositis/dermatomyositis. J Rheumatol. 1999;26:2377–82. [PubMed] [Google Scholar]
- 82.Simler NR, Brenchley PE, Horrocks AW, Greaves SM, Hasleton PS, Egan JJ. Angiogenic cytokines in patients with idiopathic interstitial pneumonia. Thorax. 2004;59(7):581–5. doi: 10.1136/thx.2003.009860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grundtman C, Tham E, Ulfgren AK, Lundberg IE. Vascular endothelial growth factor is highly expressed in muscle tissue of patients with polymyositis and patients with dermatomyositis. Arthritis Rheum. 2008;58(10):3224–38. doi: 10.1002/art.23884. [DOI] [PubMed] [Google Scholar]
- 84.Martinez FJ, Safrin S, Weycker D, et al. IPF Study Group. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963–7. doi: 10.7326/0003-4819-142-12_part_1-200506210-00005. [DOI] [PubMed] [Google Scholar]
- 85.Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum. 2006;54(6):2004–9. doi: 10.1002/art.21883. [DOI] [PubMed] [Google Scholar]
- 86.Kameda H, Nagasawa H, Ogawa H, et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol. 2005;32 (9):1719–26. [PubMed] [Google Scholar]
- 87.Tanaka F, Origuchi T, Migita K, et al. Successful combined therapy of cyclophosphamide and cyclosporine for acute exacerbated interstitial pneumonia associated with dermatomyositis. Intern Med. 2000;39 (5):428–30. doi: 10.2169/internalmedicine.39.428. [DOI] [PubMed] [Google Scholar]
- 88.Bongartz T, Ryu JH, Matteson EL. Is tacrolimus effective for treating antisynthetase-associated interstitial lung disease? Nat Clin Pract Rheumatol. 2005;1(2):80–1. doi: 10.1038/ncprheum0067. [DOI] [PubMed] [Google Scholar]
- 89.Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol. 1999;26(7):1527–33. [PubMed] [Google Scholar]
- 90.Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet. 1999;353(9166):1762–3. doi: 10.1016/S0140-6736(99)01927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: A new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity. 2005;38(5):383–92. doi: 10.1080/08916930500124023. [DOI] [PubMed] [Google Scholar]
- 92.Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52(8):2439–46. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- 93.Roos N, Poulalhon N, Farge D, Madelaine I, Mauviel A, Verrecchia F. In vitro evidence for a direct antifibrotic role of the immunosuppressive drug mycophenolate mofetil. J Pharmacol Exp Ther. 2007;321:583–9. doi: 10.1124/jpet.106.117051. [DOI] [PubMed] [Google Scholar]
- 94.Hervier B, Masseau A, Mussini JM, Audrain M, Hamidou MA. Long-term efficacy of mycophenolate mofetil in a case of refractory antisynthetase syndrome. Joint Bone Spine. 2009;76(5):575–6. doi: 10.1016/j.jbspin.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 95.Saketkoo LA, Espinoza LR. Experience of mycophenolate mofetil in 10 patients with autoimmune-related interstitial lung disease demonstrates promising effects. Am J Med Sci. 2009;337(5):329–35. doi: 10.1097/MAJ.0b013e31818d094b. [DOI] [PubMed] [Google Scholar]
- 96.Swigris JJ, Olson AL, Fischer A, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest. 2006;130(1):30–6. doi: 10.1378/chest.130.1.30. [DOI] [PubMed] [Google Scholar]
- 97.Brulhart L, Waldburger JM, Gabay C. Rituximab in the treatment of antisynthetase syndrome. Ann Rheum Dis. 2006;65(7):974–5. doi: 10.1136/ard.2005.045898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lambotte O, Kotb R, Maigne G, Blanc FX, Goujard C, Delfraissy JF. Efficacy of rituximab in refractory polymyositis. J Rheumatol. 2005;32(7):1369–70. [PubMed] [Google Scholar]
- 99.Levine TD. Rituximab in treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52(2):601–7. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 100.Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: A retrospective case series. Rheumatology (Oxford) 2009;48(8):968–71. doi: 10.1093/rheumatology/kep157. [DOI] [PubMed] [Google Scholar]
- 101.Vandenbroucke E, Grutters JC, Altenburg J, Boersma WG, ter Borg EJ, van den Bosch JM. Rituximab in life threatening antisynthetase syndrome. Rheumatol Int. 2009;29(12):1499–502. doi: 10.1007/s00296-009-0859-x. [DOI] [PubMed] [Google Scholar]
- 102.Krystufková O, Vallerskog T, Helmers SB, et al. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann Rheum Dis. 2009;68(6):836–43. doi: 10.1136/ard.2008.091405. [DOI] [PubMed] [Google Scholar]
- 103.Cherin P, Pelletier S, Teixeira A, et al. Results and longterm follow up of intravenous immunoglobulin infusions in chronic, refractory polymyositis: An open study with thirty-five adult patients. Arthritis Rheum. 2002;46:467–74. doi: 10.1002/art.10053. [DOI] [PubMed] [Google Scholar]
- 104.Donofrio PD, Berger A, Brannagan TH, 3, et al. Consensus statement: The use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the aanem. AD HOC committee. Muscle Nerve. 2009;40(5):890–900. doi: 10.1002/mus.21433. [DOI] [PubMed] [Google Scholar]
- 105.Suzuki Y, Hayakawa H, Miwa S, et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung. 2009;187(3):201–6. doi: 10.1007/s00408-009-9146-6. [DOI] [PubMed] [Google Scholar]
- 106.Park SH, Saleh D, Giaid A, Michel RP. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am J Respir Crit Care Med. 1997;156:600–8. doi: 10.1164/ajrccm.156.2.9607123. [DOI] [PubMed] [Google Scholar]
- 107.Clozel M, Salloukh H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann Med. 2005;37(1):2–12. doi: 10.1080/07853890410018925. [DOI] [PubMed] [Google Scholar]
- 108.Raghu G, King TE, Jr, Behr J, et al. Impact of bosentan on health-related quality of life and dyspnoea in idiopathic pulmonary fibrosis: The BUILD-1 trial. Eur Respir J. 2009 doi: 10.1183/09031936.00188108. Epub 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shi-Wen X, Denton CP, Dashwood MR, et al. Fibroblast matrix gene expression and connective tissue remodeling: Role of endothelin-1. J Invest Dermatol. 2001;116:417–25. doi: 10.1046/j.1523-1747.2001.01256.x. [DOI] [PubMed] [Google Scholar]
- 110.King TE, Jr, Behr J, Brown KK, et al. BUILD-1: A randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. doi: 10.1164/rccm.200705-732OC. [DOI] [PubMed] [Google Scholar]
- 111.Pickering LK, Baker CJ, Freed GL, et al. Infectious Diseases Society of America. Immunization programs for infants, children, adolescents, and adults: Clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49(6):817–40. doi: 10.1086/605430. [DOI] [PubMed] [Google Scholar]