

Abstract

Synthesis of a series of novel, broad spectrum anticancer agents containing the tricyclic 5:7:5-fused diimidazo[4,5-d:4′,5′-f][1,3]diazepine ring system is reported. Compounds 1, 2, 8, 11, and 12 in the series show promising in vitro antitumor activity with low micromolar IC50 values against prostate, lung, breast, and ovarian cancer cell lines. Some notions about structure−activity relationships and a possible mechanism of biological activity are presented. Also presented are preliminary in vivo toxicity studies of 1 using SCID mice.
Keywords: Synthesis; 5:7:5-fused tricycles; diimidazo[4,5-d:4′,5′- f][1,3]diazepine ring system; in vitro screening; broad spectrum anticancer activity; prostate, lung, breast, and ovarian cancers; preliminary mechanistic studies; effects on G1 and S phases of the cell cycle
A class of diseases involving groups of cells displaying uncontrolled growth (proliferation), intrusion and destruction of adjacent tissues (invasion), and spreading to other parts of the body via lymph or blood (metastasis) are generally termed cancer.1 Damaged genes are ultimately responsible for the loss of growth control mechanisms leading to out of control cell proliferation.2 When cells are damaged beyond repair, they are normally eliminated by apoptosis, but cancer cells avoid apoptosis and continue to undergo unregulated cell division.3 While proto-oncogenes promote such cell growth and mitosis,4 tumor suppressor genes discourage it or temporarily halt it to perform gene repair.5
Cancer is the second leading cause of death worldwide after heart disease, claiming more than 7 million deaths in 2007, the year for which the most recent global cancer statistics are available.6 Out of the many cancer deaths, those related to lung (30%), prostate (9%), colorectal (9%), and pancreatic cancers are predominant among men, while lung (26%), breast (15%), colorectal (9%), pancreatic (6%), and ovarian (5%) cancers are predominant among women.7,8 Deaths from cancer worldwide are projected to continue rising with an estimated 12 million deaths in the year 2030.8 While surgery, radiation, and chemotherapy are the principal modes of cancer treatment,9 the targeted chemotherapy acting specifically on detectable molecular abnormalities in certain tumors, and which minimizes damage to normal cells, is becoming increasingly promising and popular.10
We have recently reported the synthesis and physical as well as chemical properties of a novel tricyclic, 5:7:5-fused heterocyclic ring system containing the diimidazo[4,5-d:4′,5′-f][1,3]diazepine (1) nucleus.11 We also speculated its possible medicinal use in cancer chemotherapy.11 We now report here the potent, broad spectrum anticancer activity of not only 1 but also some of its analogues against six different tumor cell lines belonging to lung (A-549 and H-460), prostate (PC-3), breast (MCF-7 and MDA-MB-231), and ovarian (OVCAR-3) cancers. In addition, we have performed some preliminary in vitro mechanistic and in vivo toxicity studies on 1. Limited structure−activity relationship (SAR) studies were also performed by synthesizing a few analogues of 1 incorporating systematic changes in the R1, R2, and R3 substitutions at 7-, 5-, and 3-positions, respectively.

The analogue synthesis for SAR studies was initially focused on replacing the H atom at position 5 of compound 1 with various aryl and alkyl substitutions. Thus, compounds 2−7 were synthesized12 by varying the R2 substitutions, while retaining the R1 and R3 groups same as that of 1 (Scheme 1). While compound 2 contained an electron-withdrawing NO2 substituent at the p-position of an aromatic ring, compound 3 with no aromatic substitution served as a reference. Compound 4 with multiple carboxylic acid groups was designed to enhance the electron-withdrawing inductive effect, while also increasing its aqueous solubility. Compound 5, with a p-methyl substituent, was to probe the effect of the electron-donating inductive effect as well as the enhanced hydrophobicity of the aromatic ring. Compounds 6 and 7 were to explore the effect of replacing an aromatic ring with an aliphatic chain containing hydrophilic carboxylic acid groups. Compounds 8−13 (Scheme 2) were designed to throw light on the consequence of replacing the p-methoxybenzyl substitution of 1 at position 3 with various substituted or unsubstituted benzyl (n = 1) or phenyl (n = 0) groups. Thus, compounds 8−13 were synthesized12 by varying the R3 substitutions, while retaining the R1 and R2 groups the same as those of 1. Compound 14 (Scheme 3) was synthesized12 to explore the effect of replacing the p-methoxybenzyl group at position 7 of 1 with an o-chlorobenzyl substituent.
Scheme 1.

Scheme 2.

Scheme 3.

Compounds 2 and 3 (Scheme 1) were synthesized,12 in 40−41% yield, by the reaction of 15, an intermediate that we used in the synthesis of 1,11 with p-nitro- and parent benzaldehyde, respectively, at 80 °C, catalyzed by concentrated sulfuric acid. Compound 4 was obtained12 in 46% yield using a similar procedure as described for 2 and 3 above but using pyromellitic dianhydride. Compound 5 was prepared12 in 81% yield by the reaction of 15 with toluoyl chloride in pyridine at room temperature. Compounds 6 and 7 were synthesized12 by reacting 15 with succinic and glutaric anhydrides, respectively, at 60 °C in DMF, catalyzed by concentrated sulfuric acid, in 37 and 25% yields, respectively. Compounds 8−13 (Scheme 2) were synthesized12 through a series of sequential reactions starting from another intermediate 16 that was also employed in the synthesis of 1.11 Compound 16 was condensed with variously substituted benzyl isocyanates (17−21) or phenyl isocyanate (22) in acetonitrile at room temperature to obtain the corresponding ureido intermediates (not shown), which, without isolation, were subjected to further ring closure, mediated by DBU, to obtain the respective iminoimidazolone intermediates, 23−28, respectively, in 24−63% yields. The desired final 5:7:5-fused tricyclic heterocycles 8−13 were obtained12 in 53−81% yields by ring closure of the intermediates 23−28 by heating at reflux with triethyl orthoformate, catalyzed by concentrated sulfuric acid. Compound 14 was synthesized12 starting from N′-[(Z)-2-amino-1,2-dicyanovinyl]-N-(2-chlorobenzyl)imidoformamide (29),13 which in turn was prepared by reaction of ethyl (Z)-N-(2-amino-1,2-dicyanovinyl)formimidate14 with o-chlorobenzyl-amine.13 Ring closure of 29, catalyzed by DBU to obtain 30 (69%), followed by condensation with p-methoxybenzylisocyanate, gave a mixture (74%) of 32 and its ring-closed product 33. The latter was also obtained (67%) by treatment of the mixture with catalytic amount of DBU. Finally, the target 14 was obtained (85%) by acid-catalyzed ring closure of 33 with triethyl orthoformate. All new compounds were fully characterized by 1H and 13C NMR, mass spectral, and elemental microanalytical and/or high-resolution mass spectral (HRMS) data.
Compounds 1−14 were screened in vitro against six cancer cell lines, including A-549 and H-460 (lung cancer), MCF-7 and MDA-MB-231 (breast cancer), OVCAR-3 (ovarian cancer), and PC-3 (prostate cancer).12 The compounds that showed the most promising in vitro broad spectrum anticancer activity are collected in Table 1.
Table 1. In Vitro Broad Spectrum Anticancer Activity of Leading Analogues of 1a.
| μM |
||||||
|---|---|---|---|---|---|---|
| compd ID | A-549 (lung) | H-460 (lung) | MCF-7 (breast) | MDA-MB-231 (breast) | OVCAR-3 (ovarian) | PC-3 (prostate) |
| 1 | 2.5 ± 0.06 | 2.8 ± 0.06 | 7.5 ± 0.09 | 4.01 ± 0.03 | 14.5 ± 0.0 | 4.8 ± 0.1 |
| 2 | 2.7 ± 0.0 | 2.5 ± 0.5 | 5.7 | 3.3 ± 0.1 | 5.20 ± 0.6 | 4.3 ± 0.1 |
| 8 | 1.7 ± 0.9 | 1.8 ± 0.5 | 7.0 ± 0.9 | 2.3 ± 0.2 | 5.00 ± 0.7 | 4.05 ± 0.2 |
| 11 | 1.5 ± 0.4 | 1.6 ± 0.5 | 6.0 ± 5 | 2.6 ± 0.6 | 5.1 ± 0.6 | 2.5 ± 0.3 |
| 12 | 1.6 ± 0.6 | 1.5 ± 0.4 | 4.0 ± 0.9 | 2.5 ± 0.3 | 9.0 ± 0.0 | 2.1 ± 0.1 |
Average IC50 values are shown. Each compound was tested at nine different concentrations, and each drug dilution was repeated four times. Cells treated with DMSO (equivalent volume) were used as a vehicle control.
Some important, albeit limited, SARs emerged from these studies: (a) replacement of H at the 5-position of 1 (R2 = H) with a phenyl ring containing an electron-withdrawing group either enhances or retains activity as in 2 (R2 = p-NO2PhCH2−), whereas (b) an unsubstituted phenyl ring as in 3 (R2 = Ph) or that containing an electron-donating group as in 5 (R2 = p-CH3PhCH2−) leads to loss of activity, (c) multiple substitutions on the aromatic ring as in 4 [R2 = 2,4,5-(CO2H)3-Ph] also lead to loss of biological activity despite enhancement in solubility properties, (d) replacement of H at position 5 with an aliphatic chain as in 6 and 7 [R2 = −CH2−(CH2)n-CO2H] also leads to loss of biological activity despite enhancement in solubility properties, (e) replacement of the p-methoxybenzyl substituent of 1 at position 3 with an unsubstituted benzyl group as in 8 (R3 = −CH2Ph) enhanced the anticancer potency in all six cell lines tested, whereas (f) substitution of the same benzyl group with a single chloro or methoxy group in ortho position as in 9 or 10 (R3 = o-Cl-PhCH2− or o-OMe-Ph−CH2−) leads to significant loss of activity, (g) movement of the ortho-methoxy group of 10 to the meta position as in 11 (R3 = m-OMe-Ph−CH2−) considerably enhances the anticancer activity, (h) replacement of the para-methoxybenzyl substituent at position 3 of 1 with a para-fluorobenzyl group as in 12 (R3 = p-F-Ph−CH2−) gave closely comparable anticancer activity as that of 11, (i) replacement of the p-methoxybenzyl group at position 3 of 1 with a directly attached phenyl group with multiple fluoro substituents at ortho and meta positions as in 13 [R3 = 2,4-(F)2-Ph] also leads to considerable loss of activity, and finally, (j) replacement of the para-methoxy substituent at position 7 of 1 with an ortho-chlorobenzyl group as in 14 also leads to considerable loss of activity.
Our preliminary mechanistic studies on anticancer activity focused on exploring the stage(s) of the cell cycle being affected by compound 1. To this end, we performed flow cytometry on MCF-7 cells treated with a 3 μM concentration of 1 for 24 h, and the results are collected in Figure 1. As shown, the compound exerted a significant effect on the G1 (Gap1) and S (synthesis) phases of the cell cycle, while G2 (Gap 2) and M (mitosis) phases were practically unaffected. The G1 phase of the cell cycle concerns RNA production and protein synthesis, as well as the activation of an important cell cycle control mechanism, called G1 Checkpoint, which ensures that everything is ready for the subsequent S phase that involves DNA synthesis. Apparently, MCF-7 cells treated with 1 accumulate in the G1 phase of the cell cycle, indicating a G1 arrest (79.15% cells in treated vs 65.05% in untreated control, P = 0.003). Moreover, the treatment with 1 causes a large reduction of cells in the S phase (5.87 vs 18.1%, P = 0.002). On the other hand, the G2/M phase of the cell cycle exhibited only a minimal effect upon treatment with 1 (14.5% cells in treated vs 16.15 ± 1.48% in untreated control, P = 0.26) of the cell cycle. This indicates the G1 cell cycle arrest is one of the possible mechanisms of action of compound 1. More studies are currently underway to explore further details on the mechanism of action. One of the prime candidates based on what we have discovered so far is the effect of 1 on regulation of expression of DDX3, a member of the DEAD-box RNA helicase family.15,16 Members of this family not only act as molecules that unwind RNA but also play important roles in virtually all aspects of RNA synthesis and function as well as in helicase-independent transcriptional regulations.16−20 Not surprisingly, the DDX family of proteins are important targets for potential anticancer as well antiviral therapy in many laboratories,21−25 including our own.26
Figure 1.
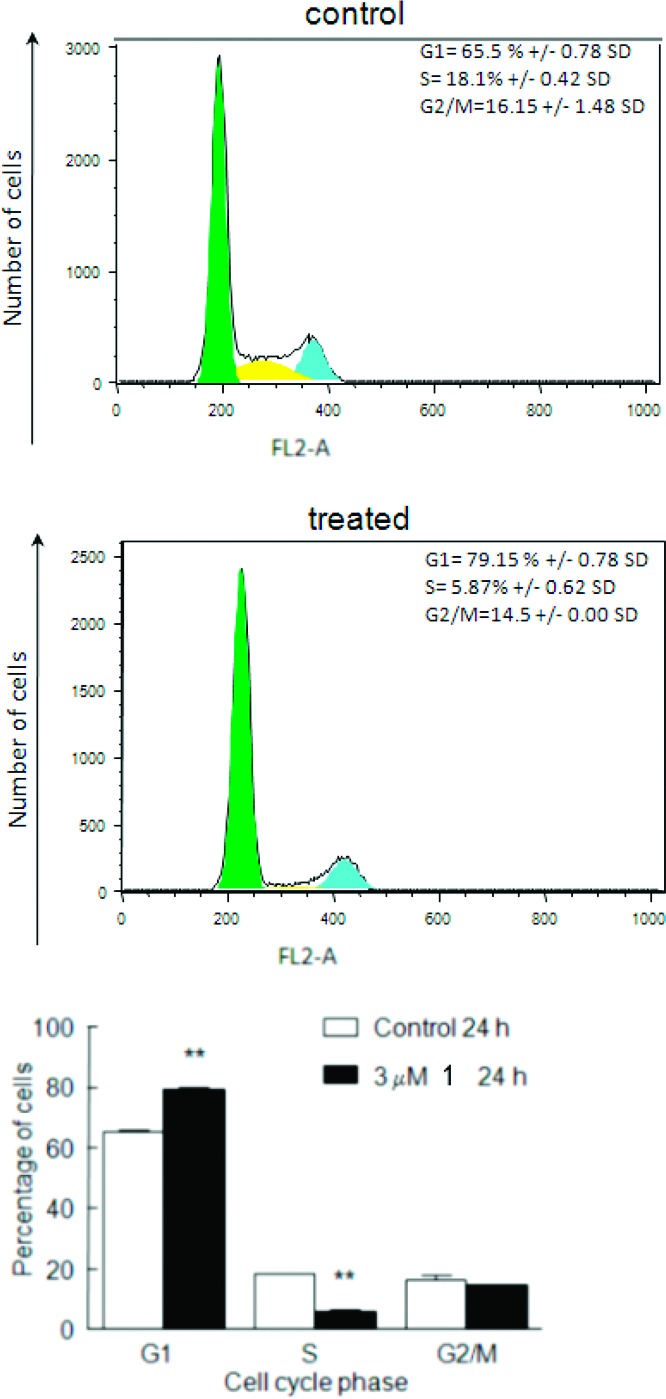
Flow cytometric analysis of 1-treated cells. The top panel shows a cell cycle histogram of DMSO-treated MCF-7 cells, and the middle panel shows the effect of 1 (3 μM) for 24 h on the cell cycle. Percentages of cells in the various phases of the cell cycle are indicated with standard deviations (SDs). The experiment was repeated four times, and the results are representative of the changes seen in reduction. The bottom panel shows statistical analysis of data of changes in the G1 and S phases of the cell cycle upon treatment with 1. Data were analyzed by Student's t test. P < 0.005 (**).
Finally, limited in vivo toxicity studies of 1 were conducted employing five SCID mice. Following injection of 1 up to 500 μM (≈20 mg/kg), twice a week for 7 weeks, we did not observe any toxicity in SCID mice. An extensive pathological examination of tissues was performed following necropsy, which indicated no tissue damage. Identical patterns were observed in kidney (glomerulus and tubules), liver (centrilobular area), spleen (red and white pulp), and cerebellum-brain (molecular and granular cell layers, purkinje cells, and white matter) cells in both control and treated mice. Furthermore, blood analysis (Figure 2) did not exhibit any variance suggesting 1 was nontoxic. Therefore, given that 1 had no apparent toxicity in mice even at concentrations far greater than that showed anticancer activity in vitro suggests its potential clinical utility for treating various neoplastic conditions.
Figure 2.
Blood analysis of SCID mice before and after treatment with 1.
In conclusion, we have discovered some novel, broad spectrum antitumor agents that show promising in vitro activity with low micromolar IC50 values against six cancer cell lines tested. Compounds 2, 8, 11, and 12 listed in Table 1, besides 1, are candidates for further explorations of biological activity as anticancer agents. Our preliminary mechanistic studies of biological activity of 1 suggest its effect on the G1 and S phases of the cell cycle. Our limited toxicity studies of 1 using SCID mice show no apparent tissue damage of kidney, liver, spleen, and brain cells nor variance in blood pattern at concentrations far greater than what is necessary for exerting biological activity. Further detailed investigations of SARs as well as mechanistic explorations of antitumor activity are currently in progress.
Author Status
∥ Recently retired.
This research was supported in part by a grant (#1R01 GM087738-01A1) from the National Institute of General Medical Sciences of the National Institutes of Health. A grant from the Flight Attendant Medical Research Institute to V.R. is also acknowledged.
Supporting Information Available
Synthetic procedures; 1H, 13C, and mass spectral data; and elemental microanalytical and/or high-resolution mass spectral data (HRMS), as well as experimental procedures used for biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vincenzi B. Biology in Anticancer Treatment. Curr. Cancer Drug Targets 2010, 1011–2. [DOI] [PubMed] [Google Scholar]
- Yang X. H.; Zou L. DNA damage: Sensing. Wiley Encycl. Chem. Biol. 2009, 1, 535–541. [Google Scholar]
- Vucic D. Apoptotic Pathways as Targets for Therapeutic Intervention. Curr. Cancer Drug Targets 2008, 8286. [DOI] [PubMed] [Google Scholar]
- Lleonart M. E. A new generation of proto-oncogenes: Cold-inducible RNA binding proteins. Biochim. Biophys. Acta, Rev. Cancer 2010, 1805, 43–52. [DOI] [PubMed] [Google Scholar]
- Yokota J.; Akiyama T.; Eds. Frontiers of Oncosuppressor Genes; 1995; 160 pp.
- American Cancer Society. Cancer Statistics: Global Facts & Figures 2007; 2009; http://www.cancer.org/downloads/STT/Global_Facts_and_Figures_2007_rev2.pdf.
- American Chemical Society. Cancer Statistics 2009 Presentation; 2009; http://www.cancer.org/docroot/PRO/content/PRO_1_1_Cancer_Statistics_2009_presentation.asp.
- World Health Organization (WHO). Factsheets, Cancer; 2009; http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
- Lee A.Options for Cancer Treatment: Surgery, Chemotherapy, and Radiation; 2007; http://cancer.suite101.com/article.cfm/options_for_cancer_treatment.
- Targeted Chemotherapy--Fighting Cancer without the Side Effects; 2009; http://www.gizmag.com/isoflow-isolate-tumour-chemotherapy/12820/.
- Kumar R.; Ujjinamatada R. K.; Hosmane R. S. The first synthesis of a novel 5:7:5-fused diimidazodiazepine ring system and some of its chemical properties. Org. Lett. 2008, 10, 4681–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Experimental: See the Supporting Information.
- Yahya-Zadeh A.; Booth B. L. Synthesis of aryl(or benzyl)-(Z)-N-[2-amino-1,2-dicyanovinyl] formamidines. Synth. Commun. 2002, 32, 3241–3246. [Google Scholar]
- Sun Z.; Hosmane R. S. An improved synthesis of 9-benzyladenine: A model for adenosine and its analogues. Synth. Commun. 2001, 31, 549–554. [Google Scholar]
- Botlagunta M.; Vesuna F.; Mironchik Y.; Raman A.; Lisok A.; Winnard P. Jr.; Mukadam S.; Van Diest P.; Chen J. H.; Farabaugh P.; Patel A. H.; Raman V. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 2008, 27, 3912–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem. Pharmacol. 2010, 79, 297–306. [DOI] [PubMed] [Google Scholar]
- Abdelhaleem M. Over-expression of RNA helicases in cancer. Anticancer Res. 2004, 24, 3951–3953. [PubMed] [Google Scholar]
- Abdelhaleem M. Do human RNA helicases have a role in cancer?. Biochim. Biophys. Acta, Rev. Cancer 2004, 1704, 37–46. [DOI] [PubMed] [Google Scholar]
- Fuller-Pace F. V. DExD/H box RNA helicases: Multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006, 34, 4206–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myohanen S.; Baylin S. B. Sequence-specific DNA binding activity of RNA helicase A to the p16INK4a promoter. J. Biol. Chem. 2001, 276, 1634–1642. [DOI] [PubMed] [Google Scholar]
- Shin S.; Rossow K. L.; Grande J. P.; Janknecht R. Involvement of RNA Helicases p68 and p72 in Colon Cancer. Cancer Res. 2007, 67, 7572–7578. [DOI] [PubMed] [Google Scholar]
- Toretsky Jeffrey A.; Erkizan V.; Levenson A.; Abaan Ogan D.; Parvin Jeffrey D.; Cripe Timothy P.; Rice Anna M.; Lee Sean B.; Uren A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–81. [DOI] [PubMed] [Google Scholar]
- Wei X.; Pacyna-Gengelbach M.; Schluens K.; An Q.; Gao Y.; Cheng S.; Petersen I. Analysis of the RNA helicase A gene in human lung cancer. Oncol. Rep. 2004, 11, 253–258. [PubMed] [Google Scholar]
- Yang L.; Lin C.; Liu Z.-R. Phosphorylations of DEAD box p68 RNA helicase are associated with cancer development and cell proliferation. Mol. Cancer Res. 2005, 3, 355–363. [DOI] [PubMed] [Google Scholar]
- Yang L.; Lin C.; Zhao S.; Wang H.; Liu Z.-R. Phosphorylation of p68 RNA helicase plays a role in platelet-derived growth factor-induced cell proliferation by up-regulating cyclin D1 and c-Myc expression. J. Biol. Chem. 2007, 282, 16811–16819. [DOI] [PubMed] [Google Scholar]
- Yedavalli V. S. R. K.; Zhang N.; Cai H.; Zhang P.; Starost M. F.; Hosmane R. S.; Jeang K.-T. Ring expanded nucleoside analogues inhibit RNA helicase and intracellular human immunodeficiency virus type 1 replication. J. Med. Chem. 2008, 51, 5043–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.