

Abstract
The LDL receptor (LDLR) is an endocytic receptor that plays a major role in the clearance of atherogenic lipoproteins from the circulation. During the endocytic process, the LDLR first binds lipoprotein at the cell surface and then traffics to endosomes, where the receptor releases bound lipoprotein. Release is acid-dependent and correlates with the formation of an intramolecular contact within the receptor. Human mutations at residues that form the contact are associated with familial hypercholesterolemia (FH) and the goal of the present study was to determine the role of contact residues on LDLR function. We show that mutations at nine contact residues reduce the ability of the LDLR to support lipoprotein uptake. Unexpectedly, only four of the mutations (W515A, W541A, H562Y and H586Y) impaired acid-dependent lipoprotein release. The remaining mutations decreased the lipoprotein-binding capacity of the LDLR through either reduction in the number of surface receptors (H190Y, K560W, H562Y and K582W) or reduction in the fraction of surface receptors that were competent to bind lipoprotein (W144A and W193A). We also examined three residues, distal to the contact, which were predicted to be necessary for the LDLR to adopt the acidic conformation. Of the three mutations we tested (G293S, F362A and G375S), one mutation (F362A) reduced lipoprotein uptake. Together, these data suggest that the intramolecular interface plays multiple roles in LDLR function.
1. Introduction
The LDL receptor (LDLR) removes all major classes of atherogenic lipoproteins from the circulation including chylomicrons, chylomicron remnants, VLDL, VLDL remnants and LDL. LDLR-mediated uptake involves four principal steps: receptor binding to lipoproteins at the cell surface, internalization of bound lipoproteins through clathrin coated pits, release of lipoproteins in endosomes and recycling of receptors back to the cell surface for further rounds of uptake. Hereditary defects that impair any step in the uptake process result in familial hypercholesterolemia (FH) and heightened risk of coronary artery disease [1].
Different domains of the LDLR are responsible for different steps during lipoprotein uptake. The LDLR is composed of 7 LDLR type A repeats (LA repeats 1–7), two EGF-like repeats (EGF-A and EGF-B), six YWTD repeats that form a six-bladed β-propeller, a third EGF-like repeat (EGF-C), a domain that is heavily glycosylated, a single transmembrane domain and a short cytoplasmic tail (Fig. 1). The LDLR uses LA repeats 4 and 5 (LA4/5) to bind to apolipoprotein E (apoE) on chylomicrons, VLDL and remnant particles [2–4]. LDL lacks apoE, and the LDLR instead binds to apolipoprotein B100 (apoB100) on LDL using LA repeats 3–7 and the EGF-A module of the receptor [2, 3]. Both LA repeats and EGF-like repeats bind calcium and calcium is required for lipoprotein binding activity [5–9]. Lipoprotein uptake occurs through clathrin-coated pits and requires interaction of the cytoplasmic domain of the LDLR with adaptor proteins that couple LDLR-lipoprotein complexes to the endocytic machinery [10–12]. Lipoprotein release in endosomes requires acidic pH and may also require efflux of calcium from the endosomal lumen [9, 13–17]. How acidic pH drives release is not fully understood; however, acidic pH has been shown to drive a conformational change in the LDLR from an extended conformation at neutral pH to a compact conformation at acidic pH [18, 19]. In the acidic state, the β-propeller module makes an intramolecular contact with LA4/5 [19]. LA4/5 is required for all lipoprotein binding, suggesting that the intramolecular contact may participate in acid-dependent release. Consistent with this possibility, deletion of the β-propeller prevents lipoprotein release at endosomal pH (pH6-6.5) and cripples LDL uptake [17, 20].
Fig. 1. Structure of the LDLR ectodomain and the location of mutations.

In the upper left is a diagram of the domain structure of the LDLR with seven LA repeats (1–7), three EGF-like repeats (A, B, C), the β-propeller, a region of heavy glycosylation (wavy lines), a single transmembrane domain and a short cytoplasmic domain. Centrally located in the figure is the structure of the LDLR ectodomain at acidic pH (pdb 1N7D). The LA repeats, EGF repeats and the β-propeller have the same color as in the diagram in the upper left. The red balls in the structure indicate bound calcium ions. The three insets at the right show the contact residues, the region around G293 and the region around F362 and G375. Residues that have been mutated are underlined.
The intramolecular contact between the β-propeller and LA4/5 can be divided into several groups of interactions (Fig. 1). The largest hydrophobic (van der Waals) contact group is composed of P141 and W144 on LA4 and W515 and W541 on the β-propeller. W193, E581 and K582 form a second, smaller van der Waals contact grouping between LA5 and the β-propeller. H562 and H586 of the β-propeller interact with D149 of LA4 with what are likely ionic contacts at acidic pH. Ionic contacts are also made by K560 and K582 with acidic residues that chelate calcium in LA4 and LA5, respectively. H190 of LA5 packs against the main chain of LA4 between N148 and D149 and the side chain of H562. H190 may make ionic contact with D149 depending upon the rotomer of the D149 side chain.
The role of these contacts in lipoprotein uptake is not clear; however, mutations at contact residues correlate with FH, suggesting that the contact plays an important role in lipoprotein uptake. Known FH mutations at contact residues include P141L, H190Y, H190L and H562Y [21–25]. Prior studies have focused on the role of H190, H562 and H586 on lipoprotein uptake. These histidines are implicated in uptake because the pKa of histidine is similar to the pH necessary to drive LDL release. Surprisingly, replacement of all three histidines with alanine has no effect on the acid-dependent conformational change [26]; however, replacement of the three histidines with either alanine or tyrosine reduces the acid sensitivity of LDL release [26, 27]. Recently, Huang and colleagues made mutations at several additional interface residues and tested for the effect of these mutations on acid-dependent conformational change and LDL release [28]. They chose to examine the W144A, H190Y, W193A, W515A, W541A, K560W, H562Y, K582W and H586Y mutations. None of these mutations had a measurable effect on acid-dependent conformational change; however, several mutations had effects on acid-dependent LDL release in in vitro assays using purified ectodomains [28]. Whether these effects correlated with impaired lipoprotein uptake was not examined. Because the mutations did not influence conformational change, Huang and colleagues also examined the G293S, F362A and G375S mutations [28]. G293 and G375 were proposed to function as pivots during conformational change because these residues sit at the boundaries between LA7/EGF-A and EGF-B/β-propeller, respectively. FH mutations have also been identified at these positions [23, 29]. F362 provides van der Waals contacts that hold the EGF-B module against the β-propeller and may influence the ability of the β-propeller to sample conformational space. As with the other nine mutations, the G293S, F362A and G375S mutations had no effect on receptor conformational change [28]. Again, the role of these residues in lipoprotein uptake was not tested.
The goal of this study is to characterize how the contact between the β-propeller and LA4/5 participates in lipoprotein uptake. Because of the prior biochemical work done with the W144A, H190Y, W193A, G293S, F362A, G375S, W515A, W541A, K560W, H562Y, K582W and H586Y mutations, we chose to introduce these mutations into full-length LDLRs, express these receptors in LDLR deficient fibroblasts and compare the ability of these cells to support uptake of LDL and the VLDL remnant, β-VLDL. Our results show that all but the G293S and G375S mutations impair lipoprotein uptake. We investigated how each mutation impaired lipoprotein uptake, using assays for receptor surface expression, lipoprotein binding and acid-dependent release. Our results show that different mutations produce different defects in LDLR-dependent lipoprotein uptake, suggesting that contact residues play multiple roles in LDLR function. We present a model for how these residues may function during lipoprotein uptake.
2. Methods
2.1 Materials
Human LDL and rabbit β-VLDL were provided by Michael Brown and Joseph Goldstein. Rabbit polyclonal antibody against the LDLR was provided by Joachim Herz. The C7 mouse monoclonal antibody against the LDLR was purchased from Santa Cruz. The CD44 antibody was purchased from Chemicon. Cell culture media was from Invitrogen. All other chemicals and buffers were purchased from Sigma.
2.2 Cell lines
LDLR−/− fibroblasts were infected with recombinant retroviruses that encode a bicistronic mRNA in which both an LDLR variant and GFP were linked by an internal ribosomal entry site (IRES). Because both the LDLR variant and GFP are translated from the same mRNA, GFP expression is proportional to LDLR synthesis [30]. This feature allows LDLR expressing cells to be selected by fluorescence activated cell sorting (FACS) and allows selection of cells expressing similar levels of LDLR. To facilitate homogeneous LDLR expression, the multiplicity of infection (MOI) was kept low to limit viral integration events to a single genomic insertion. This approach produced cells that showed a variance in LDLR surface expression that was similar to that of endogenous LDLR expression in normal human fibroblasts (data not shown). The 5′ UTR of the virus acts as the promoter for mRNA transcription and the bicistronic mRNA yields approximately twice as much LDLR as the combined production from both copies of the LDLR gene in normal human fibroblasts [26]. Mean GFP expression was comparable for all variants (Fig. S1), indicating that synthesis of LDLR protein was similar for all variants. GFP expression was determined by flow cytometry and data in Figure S1 is reported as the mean ± one standard deviation from three independent determinations.
2.3 Surface expression assays
Surface expression was monitored by flow cytometry using the C7 monoclonal antibody to the LDLR as previously described [26]. Briefly, cells were fixed with 3% paraformaldehyde, washed with phosphate buffered saline (PBS) and blocked with PBS containing 0.1% bovine serum albumin. Cells were then incubated in the same buffer with 10 μg/ml C7 antibody for 1 hr at room temperature. Cells were then washed and incubated for 1 hr at room temperature with a secondary antibody coupled to APC. Cells were then washed, lifted from dishes and processed by flow cytometry. The reported data is the average ± one standard deviation from three independent experiments.
2.4 Uptake assays
LDL and β-VLDL uptake assays used previously published protocols [26]. Briefly, cells were grown in normal growth medium (low glucose D-MEM supplemented with 10% fetal bovine serum) for 5 days. The medium was then replaced with LPPS medium (low glucose D-MEM supplemented with 10% lipoprotein poor serum) for two days. On the day of the experiment, cells were incubated with Alexa546 labeled LDL (10 μg/ml) or Alexa546 labeled β-VLDL (5 μg/ml) in LPPS medium for 1–4 hrs. LDL and β-VLDL were labeled using Alexa546 succinimidyl ester as previously described [26]. Cells were then washed with PBS, fixed with 3% paraformaldehyde, washed with PBS again and subject to flow cytometry. LDLR-dependent lipoprotein uptake in this assay shows a linear increase in cellular fluorescence with respect to time allowing rate constants to be determined by linear regression analysis. As a negative control, all assays included trials with cells infected with a retrovirus that does not encode an LDLR. These vector control cells have a constant fluorescence over the 4 hr time course and this fluorescence has been subtracted from the reported data. Some of the reported data has been normalized to surface receptor number.
2.5 Lipoprotein binding assays
Binding assays were performed as previously described [31, 32]. LDL and β-VLDL were labeled with 125I using the Bolton-Hunter protocol [33]. Saturation binding assays were performed over a range of 0.25 to 16 μg/ml 125I-lipoprotein. To facilitate comparison between LDLR variants, the saturation data has been plotted using the method of Scatchard (ng lipoprotein bound vs ng bound per μg/ml unbound). This method allows linear regression analysis to be used to determine the binding affinity (−1/slope). The X-intercept in the Scatchard plot also provides an estimate of the binding capacity of surface receptors. Individual Scatchard plots for the data in Figure 6 are provided in the supplement (Fig. S2 and S3). Affinity measurements given in Figure 6C and 6F are the means ± standard deviation from at least three independent experiments.
Fig. 6. Comparison of the ability of LDLR variants to bind LDL and β-VLDL.

Saturation binding assays were performed using 125I-LDL and 125I-β-VLDL for each of the variants. Representative data from three independent experiments is presented in Scatchard form for LDL (Panel A) and β-VLDL (Panel B) binding. The Scatchard plot data has been divided into three panels for clarity. WT data is the same in all three panels. Panels C and D show the average affinities from multiple determinations of LDL and β-VLDL binding. Error bars correspond to one standard deviation. W144A and W193A had significantly reduced affinity for LDL (*, p<0.05), but not β-VLDL. No other variant had a statistically significant difference in affinity for either lipoprotein as compared to cells expressing normal LDLR.
2.6 Lipoprotein release assays
Release assays were performed as previously described [26]. Briefly, cells were incubated with 10 μg/ml 125I-LDL in bicarbonate-free minimal essential media supplemented with 20 mM HEPES, 20 mM maleate, 0.45 M sucrose and 10% lipoprotein poor FBS at pH 7.5 and then incubated with media at pH 5.5–7.5 for 30 min. Cells were then washed on ice and 125I-LDL that remained bound was solubilized with 0.1N NaOH. All assays were done in triplicate and are reported as means ± one standard deviation.
2.7 Structural alignment
Residues P4-C39 of LA5 from the crystal structure determined at neutral pH (PDB: 1AJJ) were aligned with residues P175-C210 of LA5 region from the crystal structure of the LDLR ectodomain at acidic pH (PDB 1N7D) using DeepView/Swiss-PdbViewer (http://www.expasy.org/spdbv/). This program was also used to produce the geometric primitives sets used to render the images of the LDLR shown in Figure 1 and 7. Rendering was performed using MegaPov v1.2.1 (http://megapov.inetart.net/index.html).
Fig. 7. LA5 has structural differences with and without the β-propeller bound.

Alpha carbons of LA5 alone at neutral pH (Green, PDB: 1AJJ) and LA5 as part of the ectodomain at acidic pH (Red, PDB 1N7D) were aligned using SwissPDB viewer (Panel A). Overall rms deviation over residues P175-C210 was 1.78 Å. The C-terminal half (W193-C210) aligned better than the N-terminal half (P175-W193) with an rms deviation of 1.38Å vs 1.97Å. Side chain differences of note include a twist in W193 (Panel B), a 1.5 Å movement of H190 (Panel C) and a flip in E187 (Panel D). The image in panel D is from the opposite side as that depicted in panels A–C.
2.8 Statistical analysis
Significance was determined by one-way ANOVA.
3. Results
3.1 Generation of fibroblast lines expressing LDLR variants
Fibroblasts stably expressing different LDLR variants were generated by infection of immortalized LDLR−/− fibroblasts (549T cells) with retroviruses expressing no receptor (Vector), normal LDLR (WT), LDLR-W144A, LDLR-H190Y, LDLR-W193A, LDLR-G293S, LDLR-F362A, LDLR-G375S, LDLR-W515A, LDLR-W541A, LDLR-K560W, LDLR-H562Y, LDLR-K582W or LDLR-H586Y. After genomic integration, the viral promoter drives expression of a bicistronic mRNA that encodes both the receptor variant and GFP. GFP expression thus correlates with LDLR expression [30]. All cells expressed similar GFP levels (Fig. 1S), indicating that LDLR synthesis was similar for each variant. Steady state receptor expression levels were similar for all variants with the exception of LDLR-H190Y, which was somewhat less (Fig. 2A). Surface expression was quantified by flow cytometry using the C7 monoclonal antibody to the LDLR ectodomain. Most LDLR variants had similar surface expression with some notable exceptions. The W144A and F362A variants both had elevated surface expression, while the K560W, H562Y and K582W variants each had reduced surface expression (Fig. 2B). Surface expression of H190Y also trended lower, though this reduction did not reach statistical significance. Normally, approximately half of the total receptor in fibroblasts is on the cell surface at steady state, while the remaining half is cycling through the endosomal system. Thus, the presence of more surface receptors on cells expressing the W144A and F362A variants suggests that these receptor variants may spend more time on the cell surface, while the reduced surface expression on cells expressing the K560W, H562Y and K582W variants suggest that these variants may spend more time in the endosomal system.
Fig. 2. Total and surface LDLR levels of cell lines.
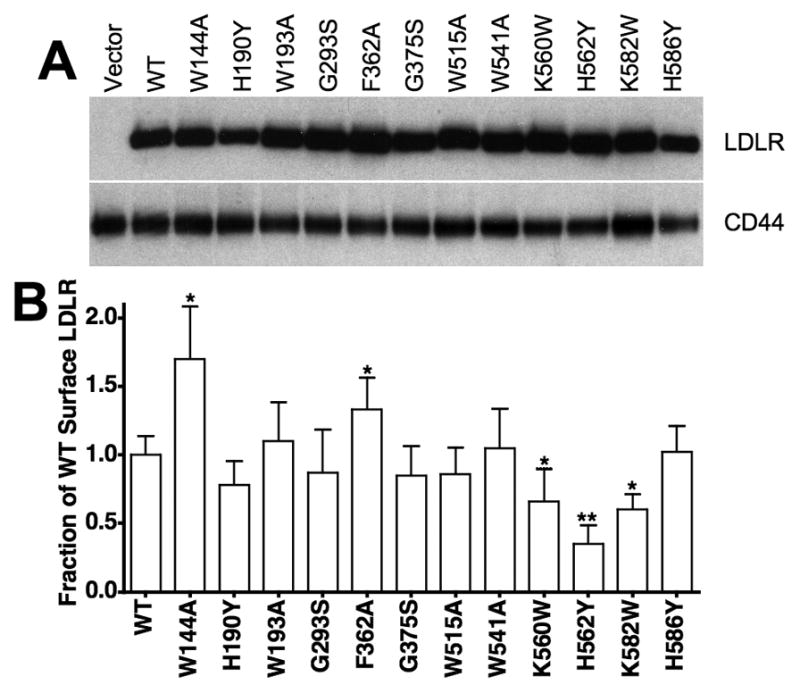
LDLR−/− fibroblasts were infected with retroviruses encoding no LDLR (Vector), normal LDLR (WT) or LDLR variants W144A, H190Y, W193A, G293S, F362A, G375S, W515A, W541A, K560W, H562Y, K582W and H586Y. Panel A contains immunoblots of whole cell lysates showing relative total expression of LDLR with CD44 as a loading control. Relative surface expression (panel B) was determined by flow cytometry using the C7 monoclonal antibody to the LDLR ectodomain. Data presented in panel B are means ± one standard deviation of three independent determinations. In panel B, *indicates p<0.05 and **indicates p<0.001 relative to WT. H190Y trended lower, but did not reach 95% confidence (p=0.064).
3.2 Cells expressing variants with mutations at contact residues show impaired lipoprotein uptake
We compared the ability of different variants to support lipoprotein uptake using a cell cytometry assay with fluorescently labeled LDL and β-VLDL. Assays were performed over a four-hour time course, and this assay provides a measure of the amount of lipoprotein that reaches lysosomes [26]. LDL uptake data from the 12 variants showed that only cells expressing the G293S and G375S variants had normal LDL uptake (Fig. 3A–B), indicating that all of the residues tested at the interface are required for normal LDL uptake. Because uptake for all variants was linear with respect to time, we calculated rate constants by linear regression. Averaged rates from three independent experiments of each variant showed that rates of LDL uptake were reduced for all variants with the exception of the G293S and G375S variants (Fig. 3C). Because several mutations reduced the number of surface receptors (Fig. 2), we normalized LDL uptake to the relative number of surface receptors to test whether reduced surface expression correlated with reduced uptake (Fig. 3D–F). This normalization showed that reduced surface expression was likely responsible for the reduced LDL uptake observed with cells expressing the H190Y, K560W, H562Y and K582W variants. By contrast, cells expressing variants W144A, W193A, W515A, W541A and H586Y had defective uptake that could not be explained by surface expression. Normalization of the uptake data for the F362 variant suggests that this variant also impairs LDL uptake, though the effect of the F362A mutation was modest as compared to the W144A, W193A, W515A, W541A and H586Y mutations. LDL uptake by these latter variants was particularly poor with rates that approached that of the LDLR-ΔBC, which is a deletion variant that lacks the EGF-B, β-propeller and EGF-C modules.
Fig. 3. LDL uptake by cells expressing LDLR variants.

Cells expressing LDLR variants were incubated with 10 μg/ml Alexa546 labeled LDL for the indicated times. Cellular fluorescence was determined by flow cytometry. LDL uptake was assayed in three independent experiments and is shown as means ± one standard deviation. For clarity, the data is divided into two panels (A, B) and is shown as a fraction of WT uptake at 4 hrs. Data for WT is the same in both panels. Cells expressing an LDLR variant lacking the EGF-B, β-propeller and EGF-C modules (ΔBC) were used as a negative control. In panel C, rate constants were determined from each experiment separately and then averaged. Error bars correspond to one standard deviation. Panels D, E, and F show data from panels A, B and C that has been normalized to surface expression using the data presented in figure 2. In panels C and F, *indicates p<0.05 and **indicates p<0.001 relative to WT.
3.3 The W515A, W541A and H586Y mutations impair acid-dependent release
We performed two types of assays to assess whether defects in LDL uptake were caused by failure of acid-dependent release. First, we tested whether cells defective in LDL uptake also had defective β-VLDL uptake. We previously showed that β-VLDL uptake does not require the β-propeller and that endosomes may instead drive release of β-VLDL by extracting calcium from LA4/5 through acidic pH and low free calcium [17]. Thus, if a mutation only impairs β-propeller-dependent release, then β-VLDL uptake should be normal. The second type of assay was to directly measure the pH-dependence of LDL release.
The β-VLDL uptake experiments showed that the W144A, H190Y, W193A, F362A, K560W, H562Y and K582W variants had reduced ability to support β-VLDL uptake (Fig. 4A–B). With the exception of the W193A variant, all reductions in the rates of β-VLDL uptake (Fig. 4C) were modest as compared to the rate reductions observed in LDL uptake experiments (Fig. 3C). After normalization to surface expression, only three variants, the W144A, W193A and F362A variants, displayed a significant reduction in β-VLDL uptake (Fig. 4D–F). The normal ability of the H190Y, K560W, H562Y and K582W variants to support β-VLDL uptake relative to surface receptor number supports the conclusion that these four mutations impair lipoprotein uptake by reducing the number of surface receptors. The ability of the W515A, W541A and H586Y variants to support normal rates of β-VLDL uptake, but not LDL uptake, suggests that these three mutations impair acid-dependent release.
Fig. 4. β-VLDL uptake by cells expressing LDLR variants.

Cells expressing LDLR variants were incubated with 5 μg/ml Alexa546 labeled β-VLDL for the indicated times and analyzed as described in Figure 3. Panels A and B show the time courses of β-VLDL uptake over 4 hrs. Panel C shows the relative rate constants derived from the time courses in Panels A and B. Panels D, E and F show data from panels A, B and C that has been normalized to surface expression. All data is reported as the average of three determinations ± one standard deviation. In panels C and F, *indicates p<0.05 and **indicates p<0.001 relative to WT.
To confirm that the W515A, W541A and H586Y mutations impair acid-dependent release of LDL, we compared the pH-dependence of LDL release from cell surface receptors (Fig. 5). These assays revealed that the W515A, W541A, H562Y and H586Y mutations decreased by 0.3–0.4 pH units the pH at which half of the surface bound LDL released. A smaller effect was also observed with the F362A variant. These results support the conclusion that the reduction in LDL uptake observed with the W515A, W541A and H586Y variants results from a defect in acid-dependent LDL release. We were unable to generate meaningful data with the W144A and W193A variants because LDL binding was greatly reduced with these variants.
Fig. 5. Comparison of acid-dependent lipoprotein release.

Saturating amounts of 125I-LDL were bound to surface receptors of each variant and then cells were exposed to media of the indicated pH. The plots show the fraction of 125I-LDL that remained bound after the incubation. Assays using cells expressing the LDLR-ΔBC are shown as a negative control. Experiments were not conducted with cells expressing W144A and W193A because these cells did not bind LDL with normal affinity. Data presented is the mean ± one standard deviation of triplicate trials and is representative of three independent experiments.
3.4 The W144A and W193A mutations impair lipoprotein binding
Because of the reduction in LDL binding observed with W144A and W193A in LDL release assays, we compared the ability of the 12 variants to bind LDL and β-VLDL using saturation binding assays (Fig. 6). These assays showed that the W144A variant had a 5-fold reduction in LDL binding affinity, while the W193A variant had a 3-fold reduction in LDL binding affinity. The W515A, W541A and H586Y variants also trended lower (Fig. 6A), but did not show a significant reduction in affinity when averaged over three independent experiments (Fig. 6C). The effects on LDL binding are similar to prior observations made with recombinant ectodomains bearing these mutations in in vitro LDL binding assays [28]. In contrast to the observations with LDL binding, we did not observe reduced binding affinity for β-VLDL for any of the 12 variants (Fig. 6B and 6D). Where we did observe a substantial difference was in the lipoprotein binding capacities. Comparison of the binding capacities showed that the W144A and W193A greatly reduced the amount of both LDL and β-VLDL that was bound. The W144A mutation decreased β-VLDL binding by 75%, while the W193A mutation reduced both LDL and β-VLDL binding by more than 90%. The failure to bind lipoprotein is the likely cause of the impairment in LDL and β-VLDL uptake observed in cells expressing the W144A and W193A variants.
4. Discussion
The results of this study show that residues, which form the intramolecular contact between the β-propeller and LA4/5, play critical roles in lipoprotein uptake by the LDLR (Table 1). We tested nine residues at the interface and three residues in the EGF-A/EGF-B modules. All nine interface mutations and one mutation in EGF-B resulted in defective lipoprotein uptake. Most defects fell into one of three categories. Mutations H190Y, K560W, H562Y and K582W impaired both LDL and β-VLDL uptake by reducing the number of surface receptors. The W144A and W193A mutations reduced lipoprotein uptake by impairing lipoprotein binding. This defect manifested as a reduction in the affinity of LDL binding and as reductions in the fraction of surface receptors that were competent to bind LDL and β-VLDL. The W515A, W541A, H562Y and H586Y mutations reduced the ability of the LDLR to release LDL in response to acidic pH. These mutations crippled LDL uptake, but not β-VLDL uptake. The G293S and G375S mutations had no effect on lipoprotein uptake and we observed no defects in receptor distribution, lipoprotein binding, or acid-dependent release with cells expressing LDLRs with these mutations.
Table 1.
Summary of LDLR variants
| LDLR Variant | Location of Mutation | Surface LDLRa | LDL Bindinga | VLDL Bindinga | Acid-dep Release | Normalized LDL Uptakeb | Normalized VLDL uptakeb |
|---|---|---|---|---|---|---|---|
| WT | N/A | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| W144A | LA4 | ▲ |
|
|
N/D |
|
|
| H190Y | LA5 | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| W193A | LA5 | ⚊ |
|
|
N/D |
|
|
| G293S | EGF-A | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| F362A | EGF-B | ▲ | ⚊ | ⚊ |
|
⚊ |
|
| G375S | EGF-B | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| W515A | β-propeller | ⚊ | ⚊ | ⚊ |
|
|
⚊ |
| W541A | β-propeller | ⚊ | ⚊ | ⚊ |
|
|
⚊ |
| K560W | β-propeller |
|
⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| H562Y | β-propeller |
|
⚊ | ⚊ |
|
⚊ | ▲ |
| K582W | β-propeller |
|
⚊ | ⚊ | ⚊ | ⚊ | ⚊ |
| H586Y | β-propeller | ⚊ | ⚊ | ⚊ |
|
|
⚊ |
| ΔBC | deletion | ⚊ | ⚊ | ⚊ |
|
|
⚊ |
Differences with p<0.05;
Differences with p<0.001;
N/D Not Done
The H190Y, G375S and H562Y mutations were originally identified in individuals with FH and our data provide a mechanistic basis for loss-of-function for the H190Y and H562Y mutations. Fibroblasts expressing LDLR variants with these mutations had reduced LDL and β-VLDL uptake that correlated well with the relative number of surface receptors, suggesting that these mutations impair uptake by reducing the surface expression of the LDLR. In the case of H190Y mutation, the effect on surface expression was small and did not quite reach statistical significance (Fig. 2). Synthesis of the H190Y variant appeared to be normal because expression of GFP, which is translated from the same message as the LDLR-H190Y, was normal (Fig. 1S); however, somewhat less LDLR-H190Y protein was observed in fibroblasts at steady state (Fig. 2). The H190Y mutation may slightly reduce the fraction of receptors that successfully transit through the ER-Golgi system. This type of defect (Class 2 FH mutation) reduces the total number of receptors through ER-mediated degradation of receptors that fail to achieve a native state. Class 2 FH mutations are the most common type of FH mutation observed in LA5 [34] and biochemical studies have identified several mutations near H190 that hinder refolding of LA5 [7, 35]. Any effect of H190Y on folding is minor, however, because we do not observe immature, poorly-glycosylated receptors by immunoblot (Fig. 2), a feature observed with severe Class 2 FH mutations. Cells expressing LDLR-H190Y had normal LDL and β-VLDL binding affinity (Fig. 6), indicating that the LDLR-H190Y, which successfully arrives at the cell surface, is natively folded. Consistent with a minor effect of H190Y on LDLR function, individuals bearing an H190Y LDLR allele do not always exhibit hypercholesterolemia [22]. In contrast to the H190Y mutation, the H562Y mutation significantly reduced surface expression (Fig. 2). Because total expression was normal, the LDLR-H562Y may spend more time in the endosomal system than normal. The H562Y variant had acid-sensitivity that was as poor as the W515A, W541A and H586Y variants (Fig. 5); however, normalization of LDL uptake to surface receptor number indicates that the abridgement in release was not responsible for the lipoprotein uptake defect observed in cells expressing the H562Y variant (Fig. 3). An extended time in endosomes may give the H562Y variant additional time to release its LDL cargo.
Interestingly, the K560W and K582W variants also had reduced surface expression and the region centered on H562 may play a role in LDLR recycling. As with lipoprotein release, recycling of the LDLR is inhibited when endosomes are unable to acidify [13]. Recycling is also regulated by the presence of bound lipoprotein in a manner that is dependent upon the EGF-homology region, which encompasses the EGF-A, EGF-B, β-propeller and EGF-C modules [17, 20, 36]. Point mutations that impair LDLR recycling have been identified throughout the EGF-A, EGF-B and β-propeller modules [34], suggesting that control of receptor recycling involves coordinated actions of multiple sites. The K560W, H562Y and K582W mutations may highlight one such site, and the decrease in acid-sensitivity observed with the H562Y variant suggests that this site may play a role in coupling lipoprotein release with receptor recycling.
F362 may also participate in receptor recycling. The F362A variant had slightly reduced sensitivity to acidic pH (Fig. 5); however, the release defect does not appear to be causal in uptake impairment, because β-VLDL uptake was also reduced in cells expressing this variant. Indeed, the defect in β-VLDL uptake was more severe than the defect in LDL uptake (Fig. 3F vs 4F). The F362A mutation increased the surface expression of the LDLR (Fig. 2), suggesting that this mutation may augment receptor recycling to the cell surface. Augmented recycling may increase the fraction of receptors that recycle before releasing lipoprotein. Normally, this “retro-endocytosis” accounts for approximately 10% of the lipoprotein internalized by the LDLR, but a variety of perturbations can increase the fraction of lipoprotein retro-endocytosed [37, 38]. The F362A mutation may impair β-VLDL uptake more than LDL uptake because β-VLDL binds with higher affinity than LDL (Fig. 6) and is more resistant to release than LDL [17].
One surprise in our findings is that neither the G293S nor the G375S mutations had any detectable effect on lipoprotein uptake. Mutations at both glycines have been implicated in FH and G375S is an FH mutation [23, 29]. We expected a substantial defect with the G375S mutation, because the G375S mutation was identified in an individual with hypercholesterolemia that is typical of individuals heterozygous for a loss-of-function LDLR allele [29]. One possibility is that these mutations impair LDLR function in hepatocytes, but not fibroblasts. Fibroblasts are routinely used to characterize FH mutations [34]; however, a few FH mutations have been identified that impair aspects of LDLR function that are not relevant in fibroblasts. For example, the G823D mutation disrupts basolateral targeting of the LDLR, a function that is required for LDLR function in hepatocytes, which have both apical and basolateral surfaces, but not in fibroblasts, which are not polarized [39]. A second example is the H306Y mutation, which promotes binding of PCSK9 to the LDLR [40]. PCSK9 binding drives degradation of the LDLR in hepatocytes, but not in fibroblasts [41]. Mutations at G293 and G375 may cause FH by impacting unique aspects of LDLR function in the liver, but have little influence on LDLR function in cells such as fibroblasts in the periphery.
The reduced ability of cells expressing the W515A, W541A and H586Y variants to accumulate LDL correlated with reduced ability of these variants to release LDL in response to acidic pH (Fig. 3 and Fig. 4), suggesting that these mutations impair LDL uptake by impairing endosomal release of LDL. Given the severity of the reduction in LDL uptake, it was remarkable how modest a reduction was observed in acid-sensitivity with the W515A, W541A and H586Y variants as compared to ΔBC variant (Fig. 6). This observation suggests either that these three mutations have additional effects not detected in our assays or that LDL uptake is highly sensitive to even small changes in the acid-sensitivity of LDL release. The effect of these three mutations was specific for LDL because no effect was observed with β-VLDL. This specificity is consistent with prior work showing that LDL uptake requires a β-propeller-driven release process, while β-VLDL uptake involves calcium extraction from the LDLR [17].
Cells expressing the W144A and W193A variants failed to support normal uptake of both LDL and β-VLDL (Fig. 3F and 4F) and this failure correlated with a reduced ability of these variants to bind lipoprotein (Fig. 6). The principal defect in binding was reduced binding capacity (Fig. 6). This reduction in lipoprotein binding capacity contrasted with the normal levels of surface receptors expressed on these cells (Fig. 2), indicating that the W144A and W193A variants impair lipoprotein binding by reducing the fraction of surface receptors that are competent to bind lipoprotein. The division of the surface receptors into multiple populations with different ability to bind lipoprotein suggests that W144 and W193 play a role in the proper folding of LA4 and LA5.
The questions of how the LDLR binds to lipoprotein and how acidic pH releases lipoprotein are key to understanding how the LDLR supports lipoprotein uptake. These questions are intertwined because acidic pH drives release by accelerating lipoprotein dissociation [26, 42]. Binding affinity is the ratio of the association (on) rate and the dissociation (off) rate. A change in off rate implies an allosteric mechanism, which in turn implies that protonation of the LDLR or the apolipoproteins to which the LDLR binds results in conformational changes that weaken the interaction between the LDLR and lipoprotein. Acid-dependent release requires the β-propeller module [17, 20, 27], indicating that protonation of the LDLR is the primary driver of acid-dependent release. Consistent with this conclusion, acidic pH causes the LDLR to undergo a conformational change from an extended state to a more compact state in which the β-propeller forms an intramolecular contact with LA 4/5 [19]. Whether this contact is causal in release has not been established.
If the β-propeller contact is causal in release, then LA4/5 should have structural differences in the presence and absence of the β-propeller and acidic pH. To test this possibility, we aligned the structure of LA5 alone at neutral pH (pdb 1AJJ) [43] with LA5 as part of the ectodomain at acidic pH (pdb 1N7D) using the alpha carbons of the two structures. The C-terminal half of LA5 aligned well in both structures; however, the N-terminal half of LA5 did not (Fig. 7). In addition to differences in the main chain, significant side chain differences include a twist in W193, a displacement of H190 toward the LA4/β-propeller interface and a flip in the orientation of the sidechain of E187. These differences suggest that the β-propeller drives structural changes in LA4/5.
A key test of whether the β-propeller contact with LA4/5 is causal in release is to determine where apolipoproteins bind on the LA4/5. If the β-propeller contact is causal then the apolipoproteins should bind to a surface that changes conformation in response to docking of the β-propeller and apolipoproteins must bind to a different surface than the β-propeller in order for the β-propeller to induce the necessary conformational changes in LA4/5. If the β-propeller contact with LA4/5 is not causal in release, but rather acts to prevent rebinding of lipoprotein after release [28], then lipoproteins may bind to the same surface as the β-propeller.
Considerable work has examined the interaction of the LDLR and LDLR family members with ApoE. ApoE can adopt at least two conformations: a poorly lipidated, four-helix bundle state and a highly-lipidated, extended state in which the residues that form the protein core in the four-helix bundle state instead interact with lipids in lipoproteins [44, 45]. The LDLR binds with high affinity to the highly-lipidated state, but not the poorly-lipidated state of ApoE [35, 46]. By contrast, the LDLR family member, LRP1, binds with high affinity to the poorly-lipidated state, but not the highly-lipidated state [46]. Saturation mutagenesis in LA5 of the LDLR suggests that the N-terminal half of LA5 is required for ApoE binding [47]. Consistent with this observation, swapping residues 2809–2816 of LA18 of LRP1 with residues 186–193 of LA5 confers to an LA16-18 recombinant protein the ability to bind to the highly-lipidated form of ApoE [35]. E187 may play a key role in the interaction because the E187K mutation reduces the affinity of the LDLR for β-VLDL 7-fold [26]. Others have suggested that H190 and W193 are also involved in binding to ApoE-lipoproteins [35]; however, while we see reduced β-VLDL binding capacity by LDLRs with mutations at these residues, we do not observe affinity differences (Fig. 6), suggesting that H190 and W193 do not contact ApoE directly. The β-propeller contacts only H190, S191 and W193 within the N-terminal half of LA5, suggesting that the residues involved on LA5 in the apoE interaction are not occluded by the interaction with the β-propeller.
LA5 is not sufficient for apoE binding by the LDLR, but inclusion of LA4 suffices for normal binding [4]. Deletion of LA4 from the LDLR does not impair binding to β-VLDL [2], suggesting that LA3 and LA4 share a determinant that can support the ability of LA5 to bind apoE. The ability of cells expressing W144A to bind β-VLDL with normal affinity suggests that this determinant does not require direct contacts with W144. Consistent with this conclusion, LA3 lacks a tryptophan at the position corresponding to W144 in LA4. Which aspects of LA3 and LA4 promote apoE binding is not clear; however, not all LA repeats can replace LA4 because exchange of LA2 with LA5 does not confer normal ability to bind apoE-lipoproteins [48].
The binding surface on the LDLR for apoB100 is less clear. While the sites for ApoB100 and ApoE overlap because LDL and β-VLDL compete for binding to the LDLR [49, 50], the available data indicates that sites for ApoB100 and ApoE are not identical. Single domain deletions have shown that only deletion of LA5 cripples β-VLDL binding, while deletions of LA3, LA4, LA5, the linker between LA4 and LA5, LA6, LA7 or the EGF-A module all impair LDL binding [2]. Several point mutations in LA4 and LA5 have likewise shown different effects on LDL and β-VLDL binding. For example, the D203K and E208K mutations weaken the binding affinity for β-VLDL, but not for LDL [26], while the W144A and W193A mutations reduce LDL binding affinity, but not β-VLDL binding affinity (Fig. 6). The reduction in LDL binding affinity suggests that W144 and W193 may contact apoB100. These tryptophans are partially occluded by interaction with the β-propeller; however, a recent structure of a tethered ApoE peptide bound to LA17 of LRP provides an example of how an apolipoprotein can interact with tryptophans using a surface different than that occupied by the β-propeller [51]. Thus, apoB100 may interact with W144 and W193, but the surface involved in the interaction may not be occluded by the β-propeller contact surface. It is notable that LDL release is far more sensitive to acidic pH than is β-VLDL release [17]. While the difference in sensitivities is likely due in part to differences in binding affinities of the two lipoproteins, contact of the β-propeller with W144 and W193 may shift these residues away from an optimal conformation for the interaction with apoB100, thereby weakening the ability of the LDLR to bind LDL.
We propose a new model for how the β-propeller drives release. We propose that the N-terminal loop of LA5 provides a critical contact site for both ApoB100 on LDL and ApoE on β-VLDL and that this contact site is what the β-propeller regulates (Fig. 8). During release, acidic pH causes the LDLR to adopt a closed conformation, placing the β-propeller in position to make contact with LA4. The strength of this contact depends upon hydrophobic contacts involving P141, W144, W515 and W541 and ionic contacts involving D149, H562 and H586. The combination of LA4 and the β-propeller generates a surface that then recruits LA5. The interaction of LA5 with this surface strains the structure of the N-terminal loop of LA5. This strain shifts residues of the N-terminal loop that contact apolipoproteins, thereby weakening the ability of the LDLR to bind lipoprotein and increasing the rate of lipoprotein dissociation. E187 may play a key role in binding and release because the E187K mutation weakens the binding affinity of the LDLR for both LDL and β-VLDL and the side chain of E187 points in opposite directions in the structures of LA5 alone and LA5 in the ectodomain structure (Fig. 7D). In this model, the W515A, W541A, H562Y and H586Y mutations impair acid-dependent release by weakening the initial contact with LA4. Consistent with this model is the observation that strengthening the contact by replacing the three histidines with charged residues makes lipoprotein release more sensitive to acidic pH [26]. Future work will test this model by identifying residues necessary for normal lipoprotein binding affinity on LA4 and LA5.
Fig. 8.
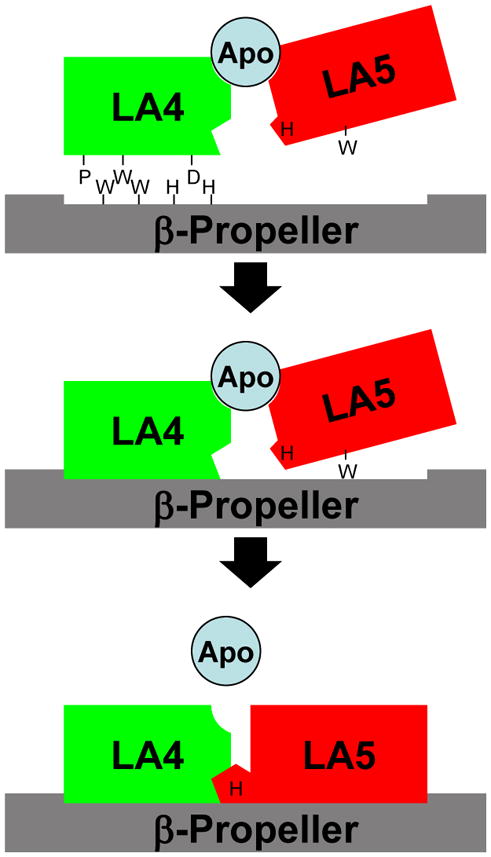
Model for β-propeller driven lipoprotein release - When acidic pH places the β-propeller in position to bind LA4/5, the β-propeller binds first to LA4 through hydrophobic contacts involving P141, W144, W515 and W541 and through ionic contacts involving D149, H562 and H586. The combination of LA4 and the β-propeller then provides a surface that recruits LA5. Binding of LA5 deforms the N-terminal loop of LA5, which interacts with lipoproteins. This deformation weakens the interaction with lipoproteins, accelerating lipoprotein dissociation.
Conclusions
The ability of the LDLR to support lipoprotein uptake is highly sensitive to mutations at residues that form an intramolecular contact between the β-propeller and LA4/5 domains of the LDLR. Mutations at these residues influence the ability of the LDLR to bind lipoprotein, release LDL in response to acidic pH and traffic normally. Comparison of the LA5 structure alone at neutral pH with the structure of LA5 in the ectodomain structure at acidic pH suggests that binding of the β-propeller drives structural changes in a part of LA5 responsible for lipoprotein binding. Collectively, this data supports a new model for how the LDLR binds and releases lipoprotein during the process of lipoprotein uptake.
Supplementary Material
Both the LDLR and GFP are expressed from the same mRNA and thus GFP expression correlates with LDLR expression. GFP expression was determined by flow cytometry. The data shown in the average ± one standard deviation from three independent experiments.
The binding data for LDL binding to cells expressing the indicated LDLR variants is shown in Scatchard format. The χ2 value for goodness of fit for the linear regression was > 0.8 for all assays except W193A, which was 0.25.
The binding data for β-VLDL binding to cells expressing the indicated LDLR variant is shown in Scatchard format. The χ2 value for goodness of fit in the linear regression was >0.8 for all assays.
Acknowledgments
Funding Support:
This work was supported by National Institutes of Health Grant HL085218.
We thank Drs. Michael Brown and Joseph Goldstein for providing human LDL and rabbit β-VLDL. We thank Dr. Joachim Herz for rabbit polyclonal antibody to the LDLR. This study was supported by HL085218 from the National Heart, Lung and Blood Institute.
Abbreviations
The following abbreviations have been used in the text
- LDLR
LDL receptor
- FH
familial hypercholesterolemia
- LA repeat
LDLR type A repeat
- YWTD repeat
repeat having the tyrosine, tryptophan, threonine, and aspartic acid motif
- ApoE
apolipoprotein E
- ApoB100
apolipoprotein B100
- LRP1
LDLR-related protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldstein JL, Hobbs HH, Brown MS. Familial Hypercholesterolemia. In: Scriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2863–2913. [Google Scholar]
- 2.Russell DW, Brown MS, Goldstein JL. Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J Biol Chem. 1989;264:21682–21688. [PubMed] [Google Scholar]
- 3.Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem. 1988;263:13282–13290. [PubMed] [Google Scholar]
- 4.Fisher C, Abdul-Aziz D, Blacklow SC. A two-module region of the low-density lipoprotein receptor sufficient for formation of complexes with apolipoprotein E ligands. Biochemistry. 2004;43:1037–1044. doi: 10.1021/bi035529y. [DOI] [PubMed] [Google Scholar]
- 5.Saha S, Boyd J, Werner JM, Knott V, Handford PA, Campbell ID, Downing AK. Solution structure of the LDL receptor EGF-AB pair: a paradigm for the assembly of tandem calcium binding EGF domains. Structure. 2001;9:451–456. doi: 10.1016/s0969-2126(01)00606-2. [DOI] [PubMed] [Google Scholar]
- 6.Kurniawan ND, Aliabadizadeh K, Brereton IM, Kroon PA, Smith R. NMR structure and backbone dynamics of a concatemer of epidermal growth factor homology modules of the human low-density lipoprotein receptor. J Mol Biol. 2001;311:341–356. doi: 10.1006/jmbi.2001.4867. [DOI] [PubMed] [Google Scholar]
- 7.Blacklow SC, Kim PS. Protein folding and calcium binding defects arising from familial hypercholesterolemia mutations of the LDL receptor. Nat Struct Biol. 1996;3:758–762. doi: 10.1038/nsb0996-758. [DOI] [PubMed] [Google Scholar]
- 8.Basu SK, Goldstein JL, Brown MS. Characterization of the low density lipoprotein receptor in membranes prepared from human fibroblasts. J Biol Chem. 1978;253:3852–3856. [PubMed] [Google Scholar]
- 9.Dirlam-Schatz KA, Attie AD. Calcium induces a conformational change in the ligand binding domain of the low density lipoprotein receptor. J Lipid Res. 1998;39:402–411. [PubMed] [Google Scholar]
- 10.Morris SM, Cooper JA. Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic. 2001;2:111–123. doi: 10.1034/j.1600-0854.2001.020206.x. [DOI] [PubMed] [Google Scholar]
- 11.He G, Gupta S, Yi M, Michaely P, Hobbs HH, Cohen JC. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J Biol Chem. 2002;277:44044–44049. doi: 10.1074/jbc.M208539200. [DOI] [PubMed] [Google Scholar]
- 12.Mishra SK, Watkins SC, Traub LM. The autosomal recessive hypercholesterolemia (ARH) protein interfaces directly with the clathrin-coat machinery. Proc Natl Acad Sci U S A. 2002;99:16099–16104. doi: 10.1073/pnas.252630799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu SK, Goldstein JL, Anderson RG, Brown MS. Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell. 1981;24:493–502. doi: 10.1016/0092-8674(81)90340-8. [DOI] [PubMed] [Google Scholar]
- 14.Gerasimenko JV, Tepikin AV, Petersen OH, Gerasimenko OV. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr Biol. 1998;8:1335–1338. doi: 10.1016/s0960-9822(07)00565-9. [DOI] [PubMed] [Google Scholar]
- 15.Arias-Moreno X, Velazquez-Campoy A, Rodriguez JC, Pocovi M, Sancho J. Mechanism of low density lipoprotein (LDL) release in the endosome: implications of the stability and Ca2+ affinity of the fifth binding module of the LDL receptor. J Biol Chem. 2008;283:22670–22679. doi: 10.1074/jbc.M802153200. [DOI] [PubMed] [Google Scholar]
- 16.Saito M, Hanson PI, Schlesinger P. Luminal chloride-dependent activation of endosome calcium channels: patch clamp study of enlarged endosomes. J Biol Chem. 2007;282:27327–27333. doi: 10.1074/jbc.M702557200. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Z, Michaely P. The role of calcium in lipoprotein release by the low-density lipoprotein receptor. Biochemistry. 2009;48:7313–7324. doi: 10.1021/bi900214u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeon H, Shipley GG. Vesicle-reconstituted low density lipoprotein receptor. Visualization by cryoelectron microscopy. J Biol Chem. 2000;275:30458–30464. doi: 10.1074/jbc.M002583200. [DOI] [PubMed] [Google Scholar]
- 19.Rudenko G, Henry L, Henderson K, Ichtchenko K, Brown MS, Goldstein JL, Deisenhofer J. Structure of the LDL receptor extracellular domain at endosomal pH. Science. 2002;298:2353–2358. doi: 10.1126/science.1078124. [DOI] [PubMed] [Google Scholar]
- 20.Davis CG, Goldstein JL, Sudhof TC, Anderson RG, Russell DW, Brown MS. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature. 1987;326:760–765. doi: 10.1038/326760a0. [DOI] [PubMed] [Google Scholar]
- 21.Amsellem S, Briffaut D, Carrie A, Rabes JP, Girardet JP, Fredenrich A, Moulin P, Krempf M, Reznik Y, Vialettes B, de Gennes JL, Brukert E, Benlian P. Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor gene are a cause of familial hypercholesterolemia. Hum Genet. 2002;111:501–510. doi: 10.1007/s00439-002-0813-4. [DOI] [PubMed] [Google Scholar]
- 22.Hopkins PN, Wu LL, Stephenson SH, Xin Y, Katsumata H, Nobe Y, Nakajima T, Hirayama T, Emi M, Williams RR. A novel LDLR mutation, H190Y, in a Utah kindred with familial hypercholesterolemia. J Hum Genet. 1999;44:364–367. doi: 10.1007/s100380050179. [DOI] [PubMed] [Google Scholar]
- 23.Widhalm K, Dirisamer A, Lindemayr A, Kostner G. Diagnosis of families with familial hypercholesterolaemia and/or Apo B-100 defect by means of DNA analysis of LDL-receptor gene mutations. J Inherit Metab Dis. 2007;30:239–247. doi: 10.1007/s10545-007-0563-5. [DOI] [PubMed] [Google Scholar]
- 24.Sun XM, Patel DD, Webb JC, Knight BL, Fan LM, Cai HJ, Soutar AK. Familial hypercholesterolemia in China. Identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype. Arterioscler Thromb. 1994;14:85–94. doi: 10.1161/01.atv.14.1.85. [DOI] [PubMed] [Google Scholar]
- 25.Punzalan FE, Sy RG, Santos RS, Cutiongco EM, Gosiengfiao S, Fadriguilan E, George P, Laurie A. Low density lipoprotein--receptor (LDL-R) gene mutations among Filipinos with familial hypercholesterolemia. J Atheroscler Thromb. 2005;12:276–283. doi: 10.5551/jat.12.276. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Z, Michaely P. The Epidermal Growth Factor Homology Domain of the LDL Receptor Drives Lipoprotein Release through an Allosteric Mechanism Involving H190, H562, and H586. J Biol Chem. 2008;283:26528–26537. doi: 10.1074/jbc.M804624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beglova N, Jeon H, Fisher C, Blacklow SC. Cooperation between fixed and low pH-inducible interfaces controls lipoprotein release by the LDL receptor. Mol Cell. 2004;16:281–292. doi: 10.1016/j.molcel.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 28.Huang S, Henry L, Ho YK, Pownall HJ, Rudenko G. Mechanism of LDL binding and release probed by structure-based mutagenesis of the LDL receptor. J Lipid Res. 2010;51:297–308. doi: 10.1194/jlr.M000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koivisto UM, Viikari JS, Kontula K. Molecular characterization of minor gene rearrangements in Finnish patients with heterozygous familial hypercholesterolemia: identification of two common missense mutations (Gly823-->Asp and Leu380-->His) and eight rare mutations of the LDL receptor gene. Am J Hum Genet. 1995;57:789–797. [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci U S A. 1997;94:10669–10674. doi: 10.1073/pnas.94.20.10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 32.Michaely P, Zhao Z, Li WP, Garuti R, Huang LJ, Hobbs HH, Cohen JC. Identification of a VLDL-induced, FDNPVY-independent internalization mechanism for the LDLR. EMBO J. 2007;26:3273–3282. doi: 10.1038/sj.emboj.7601769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bolton AE, Hunter WM. The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acylating agent. Biochem J. 1973;133:529–539. doi: 10.1042/bj1330529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 35.Guttman M, Prieto JH, Croy JE, Komives EA. Decoding of lipoprotein-receptor interactions: properties of ligand binding modules governing interactions with apolipoprotein E. Biochemistry. 2010;49:1207–1216. doi: 10.1021/bi9017208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Westhuyzen DR, Stein ML, Henderson HE, Marais AD, Fourie AM, Coetzee GA. Deletion of two growth-factor repeats from the low-density-lipoprotein receptor accelerates its degradation. Biochem J. 1991;277:677–682. doi: 10.1042/bj2770677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aulinskas TH, Oram JF, Bierman EL, Coetzee GA, Gevers W, van der Westhuyzen DR. Retro-endocytosis of low density lipoprotein by cultured human skin fibroblasts. Arteriosclerosis. 1985;5:45–54. doi: 10.1161/01.atv.5.1.45. [DOI] [PubMed] [Google Scholar]
- 38.Greenspan P, St Clair RW. Retroendocytosis of low density lipoprotein. Effect of lysosomal inhibitors on the release of undegraded 125I-low density lipoprotein of altered composition from skin fibroblasts in culture. J Biol Chem. 1984;259:1703–1713. [PubMed] [Google Scholar]
- 39.Koivisto UM, Hubbard AL, Mellman I. A novel cellular phenotype for familial hypercholesterolemia due to a defect in polarized targeting of LDL receptor. Cell. 2001;105:575–585. doi: 10.1016/s0092-8674(01)00371-3. [DOI] [PubMed] [Google Scholar]
- 40.McNutt MC, Kwon HJ, Chen C, Chen JR, Horton JD, Lagace TA. Antagonism of secreted PCSK9 increases low-density lipoprotein receptor expression in HEPG2 cells. J Biol Chem. 2009 doi: 10.1074/jbc.M808802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pitas RE, Innerarity TL, Arnold KS, Mahley RW. Rate and equilibrium constants for binding of apo-E HDLc (a cholesterol-induced lipoprotein) and low density lipoproteins to human fibroblasts: evidence for multiple receptor binding of apo-E HDLc. Proc Natl Acad Sci U S A. 1979;76:2311–2315. doi: 10.1073/pnas.76.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fass D, Blacklow S, Kim PS, Berger JM. Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature. 1997;388:691–693. doi: 10.1038/41798. [DOI] [PubMed] [Google Scholar]
- 44.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 45.Peters-Libeu CA, Newhouse Y, Hatters DM, Weisgraber KH. Model of biologically active apolipoprotein E bound to dipalmitoylphosphatidylcholine. J Biol Chem. 2006;281:1073–1079. doi: 10.1074/jbc.M510851200. [DOI] [PubMed] [Google Scholar]
- 46.Kowal RC, Herz J, Goldstein JL, Esser V, Brown MS. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc Natl Acad Sci U S A. 1989;86:5810–5814. doi: 10.1073/pnas.86.15.5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdul-Aziz D, Fisher C, Beglova N, Blacklow SC. Folding and binding integrity of variants of a prototype ligand-binding module from the LDL receptor possessing multiple alanine substitutions. Biochemistry. 2005;44:5075–5085. doi: 10.1021/bi047575j. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto T, Ryan RO. Domain swapping reveals that low density lipoprotein (LDL) type A repeat order affects ligand binding to the LDL receptor. J Biol Chem. 2009;284:13396–13400. doi: 10.1074/jbc.M900194200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahley RW, Innerarity TL. Interaction of canine and swine lipoproteins with the low density lipoprotein receptor of fibroblasts as correlated with heparin/manganese precipitability. J Biol Chem. 1977;252:3980–3986. [PubMed] [Google Scholar]
- 50.Brown MS, Goldstein JL. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc Natl Acad Sci U S A. 1974;71:788–792. doi: 10.1073/pnas.71.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guttman M, Prieto JH, Handel TM, Domaille PJ, Komives EA. Structure of the minimal interface between ApoE and LRP. J Mol Biol. 2010;398:306–319. doi: 10.1016/j.jmb.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Both the LDLR and GFP are expressed from the same mRNA and thus GFP expression correlates with LDLR expression. GFP expression was determined by flow cytometry. The data shown in the average ± one standard deviation from three independent experiments.
The binding data for LDL binding to cells expressing the indicated LDLR variants is shown in Scatchard format. The χ2 value for goodness of fit for the linear regression was > 0.8 for all assays except W193A, which was 0.25.
The binding data for β-VLDL binding to cells expressing the indicated LDLR variant is shown in Scatchard format. The χ2 value for goodness of fit in the linear regression was >0.8 for all assays.