

SUMMARY
The speed of circadian clocks in animals is tightly linked to complex phosphorylation programs that drive daily cycles in the levels of PERIOD (PER) proteins. Using Drosophila we identify a time-delay circuit based on hierarchical phosphorylation that controls the daily downswing in PER abundance. Phosphorylation by the NEMO/NLK kinase at the per-short domain on PER stimulates phosphorylation by DOUBLETIME (DBT/CK1δ/ε) at several nearby sites. This multi-site phosphorylation operates in a spatially-oriented and graded manner to delay progressive phosphorylation by DBT at other more distal sites on PER, including those required for recognition by the F-box protein SLIMB/β-TrCP and proteasomal degradation. Highly phosphorylated PER has a more open structure, suggesting that progressive increases in global phosphorylation contribute to the timing mechanism by slowly increasing PER susceptibility to degradation. Our findings identify NEMO as a clock kinase and demonstrate that long-range interactions between functionally distinct phospho-clusters collaborate to set clock speed.
Keywords: circadian rhythms, Drosophila, PER, NEMO/NLK, CK1/DBT, SLIMB/β-TrCP
INTRODUCTION
Circadian (≅24 hr) rhythms are a widespread feature of the temporal organization exhibited by life forms and are driven by cellular pacemakers or clocks that are based on species or tissue specific sets of clock genes (Schibler, 2006). A hallmark feature of these clocks is that even in the absence of external time cues they oscillate with free-running periods of ~24 hr. Based on many lines of evidence obtained from a range of model systems it is clear that dynamic changes in the phosphorylation state of one or more clock proteins is a key state-variable in rhythm generating mechanisms driving the pace of these clocks (Bae and Edery, 2006; Gallego and Virshup, 2007; Markson and O'Shea, 2009; Mehra et al., 2009). In animals, PERIOD (PER) proteins are the clock components behaving as the primary phospho-timer (Bae and Edery, 2006; Gallego and Virshup, 2007), whereas in Neurospora it is FREQUENCY (FRQ) (Brunner and Schafmeier, 2006; Heintzen and Liu, 2007; Mehra et al., 2009). Another shared feature of these phospho-timing clock proteins is that they connect to gene expression by functioning in a phase-specific manner to inhibit the activities of positively acting core clock transcription factors, leading to daily cycles in gene expression which ultimately underlie many of the observed circadian rhythms in cellular, physiological and behavioral phenomena.
How phosphorylation regulates clock speed in eukaryotes is not clear. Recent findings indicate that there are 25–30 phosphorylation sites on PER proteins in animals (Chiu et al., 2008; Vanselow et al., 2006) and 80–100 on FRQ (Baker et al., 2009; Tang et al., 2009), many of which undergo daily changes in phospho-occupancy. The dynamic regulation of PER and FRQ phosphorylation involves multiple kinases and phosphatases (Bae and Edery, 2006; Gallego and Virshup, 2007; Mehra et al., 2009). In these eukaryotic systems a major effect of phosphorylation on regulating clock pace is via controlling the stabilities of PER and FRQ proteins, which yields daily cycles in their levels that drive clock progression. Studies in Drosophila melanogaster have been instrumental in our understanding of clock mechanisms in general and mammalian ones in particular (Allada and Chung, 2010). Indeed, that time-dependent changes in the phosphorylated state of a key clock protein might be a critical aspect of circadian timing mechanisms was initially suggested based on the Drosophila PER (dPER) protein, the first clock protein to be biochemically characterized (Edery et al., 1994).
DOUBLETIME (DBT) (homolog of mammalian CKIδ/ε) is the major kinase controlling the temporal program underlying dPER phosphorylation and stability (Kloss et al., 1998; Price et al., 1998). Newly synthesized dPER is initially present as a hypophosphorylated variant(s) in the late day/early night, progressively increasing in extent of phosphorylation such that by the late night/early day only hyperphosphorylated species are detected (Edery et al., 1994). During this temporal phosphorylation program, dPER enters the nucleus around mid-night, where it acts to repress the central clock transcription factors, dCLOCK (dCLK) and CYCLE (CYC). The F-box protein SLIMB (Drosophila homolog of β-TrCP) (Grima et al., 2002; Ko et al., 2002) preferentially recognizes highly phosphorylated isoforms of dPER, targeting them for proteasomal degradation, which relieves repression of dCLK-CYC enabling the next round of circadian transcription. A strikingly similar scenario also occurs in mammals, whereby CKIδ/ε plays a major role in regulating daily cycles in the abundance of mammalian PERs (mPER1-3) by targeting them to the proteasome via β-TrCP (Gallego and Virshup, 2007). The importance of PER phosphorylation to human health is highlighted by studies showing that mutations in either a phosphorylation site on human PER2 or CKIδ underlie several familial advanced sleep phase syndromes (FASPS) (Toh et al., 2001; Vanselow et al., 2006; Xu et al., 2005; Xu et al., 2007).
Because the daily downswing in the levels of PER proteins coincides with when they attain highly phosphorylated states, it was thought that the targeting of these proteins to the proteasome is based on the recognition of many phosphorylation signals. However, several years ago we showed that although the first 100 aa of dPER contain only a fraction of the total phosphorylated residues, the phospho-signals in this region are necessary and sufficient for SLIMB binding (Chiu et al., 2008). Most notably, the phospho-occupancy of Ser47 is the key signal regulating the efficiency of SLIMB binding, a critical event in setting the pace of the clock (Chiu et al., 2008). However, if recognition of clock proteins such as PER to their respective F-box proteins only requires a few phosphorylated residues, how do other phosphorylation events contribute to setting the clock speed?
As an entrée into this problem we sought to understand the role of the classic perShort (perS) allele, which exhibits behavioral rhythms of ~19 hr (Konopka and Benzer, 1971) and accelerates the biochemical cycles in dPER phosphorylation and abundance (Edery et al., 1994; Marrus et al., 1996). The perS mutation changes a Ser residue at amino acid (aa) position 589 to Asn (S589N) (Baylies et al., 1987; Yu et al., 1987), and recent findings from the Young lab and our group raised the possibility that Ser589 is phosphorylated in vivo (Chiu et al., 2008; Kivimae et al., 2008). Herein we show that not only is Ser589 phosphorylated in flies but that it also functions as part of a hierarchical phospho-cluster. This phospho-cluster is gated by phosphorylation at Ser596, which stimulates the phosphorylation by DBT at the perS site and several nearby Ser/Thr residues. The per-short phospho-cluster functions as a major control center of the dPER phosphorylation program by delaying the timing of DBT-dependent phosphorylation at other more distal regions on dPER, including Ser47. We also show that Ser596 is phosphorylated by the proline-directed kinase, NEMO/NLK, identifying a new kinase in the central clockworks. We propose that time-delay phospho-circuits that slow down phosphorylation at other phospho-clusters are likely to be key aspects of the timer in eukaryotic circadian clocks and also provide a working model for understanding how mutations in clock protein phosphorylation sites and/or the kinases that phosphorylate them can yield both fast and slow clocks.
RESULTS
The per-short phospho-cluster is a major determinant regulating dPER stability by controlling the rate of DBT-mediated binding of dPER to SLIMB
Our prior mass spectrometry data indicated that the perS site (Ser589) and several nearby sites (i.e., Ser596, Ser585 and Thr583) are phosphorylated (Chiu et al., 2008), suggesting that this region functions as a discrete phospho-cluster. Indeed, earlier work showed that a variety of missense mutations in the region encompassing aa585-601 of dPER (Fig. 1A) lead to short period phenotypes (termed the “per-short-domain”) (Baylies et al., 1992; Rothenfluh et al., 2000). As an initial attempt to probe the effects of phosphorylation in the per-short domain, we used a previously described assay based on cultured Drosophila Schneider (S2) cells (Ko and Edery, 2005; Ko et al., 2002). In this system, the DBT-dependent progressive phosphorylation and subsequent SLIMB-mediated degradation of dPER can be recapitulated by transfecting S2 cells with recombinant dper and dbt, whereby expression of dbt is controlled by the copper-inducible metallothionein promoter (pMT) and that of dper by the constitutive actin5C promoter (pAc).
Figure 1. Differential effects of phospho-site mutations in the perS phospho-cluster on DBT-dependent phosphorylation kinetics and degradation rate of dPER in cultured S2 cells.

(A) Sequence of dPER from amino acid 579 to 601, encompassing the original “per-short” region (aa 585 to 601), highlighting the phosphorylation site mutations generated in this study (labeled in red) as well as locations of the perS and perT mutations. (B, C) S2 cells were co-transfected with wild type (WT) or mutant variants of pAc-3XFLAG-His-dper-6Xcmyc (simplified as dper) and pMT-dbt(WT)-V5-His (simplified as dbt, left of panels), and collected at the indicated times (hr) post-dbt induction. The dPER variants (top of panels) were detected by immunoblotting in the presence of α-c-myc antibodies. (D) dPER staining intensity values normalized to Hsp70 loading control. At least two independent experiments were used to generate the average for each time point. Error bars represent SEM. See Fig. S1 for supporting data.
Replacing each of the Ser/Thr phosphorylated residues we identified by mass spectrometry in the per-short domain to an Ala [dPER(TS583-596A)] greatly accelerated the timing of DBT-mediated degradation of dPER (Figs. 1B, D, and S1A). In addition, the transition from hypo- to hyper-phosphorylated dPER isoforms occurs earlier with slowly migrating species of dPER(TS583-596A) readily visible 6 hr after dbt induction (Fig. 1B, compare lanes 2 and 6). The kinetics of DBT-mediated phosphorylation and degradation were still accelerated when the Ser/Thr phospho-acceptor sites in the per-short domain were replaced with Asp [dPER(TS583-596D)], suggesting that in this case the negatively charged Asp residues do not function as phospho-mimetics (Fig. 1B, lanes 13-16). Our results are consistent with recent findings showing that deletion of the per-short domain renders dPER less stable when expressed in S2 cells (Kivimae et al., 2008).
We next replaced individual phospho-acceptor sites in the per-short domain with an Ala residue. In addition, we evaluated another per-short domain mutant called perT (G593D), which yields a severe short phenotype with 16 hr behavioral rhythms (Hamblen et al., 1998; Konopka et al., 1994) (Fig. 1A). All the different versions of dPER were degraded faster, except for dPER(T583A), which behaved similar to that of wild type dPER (Figs. 1C, D, and S1B). Intriguingly, there is a clear trend towards faster DBT-mediated degradation for mutations closer to the carboxy (C)-terminal end of the per-short domain, with dPER(S596A) showing the greatest instability amongst the single-site mutants (Fig. 1D; e.g., compare values at 6 hr post-dbt induction). A C-terminal bias in the rate of DBT-induced increases in dPER phosphorylation was also observed in the presence of MG132 to inhibit proteasome-mediated degradation (Figs. 2A, S2A and B; see Fig. S2B for measurement of a hyperphosphorylation index). Because all the different dPER versions are eventually converted to hyper-phosphorylated isoforms when degradation is inhibited (Figs. 2A and S2A), these results indicate that the per-short domain functions in a spatially oriented manner to regulate the rate by which DBT progressively increases the global phosphorylation of dPER, whereby eliminating phosphorylation at sites closer to the C-terminal of this cluster lead to faster kinetics.
Figure 2. Differential effects of phospho-site mutants in the perS phospho-cluster on dPER/SLIMB interactions and S47 phosphorylation.

(A) S2 cells were co-transfected with dbt and dper variants and collected at the indicated times (hr) post-dbt induction (top of panels). The culture media contained MG132 to inhibit degradation of dPER. (B) S2 cells were co-transfected with WT or mutant variants of pAc-3XFLAG-His-dper/Tev100-6Xc-myc and pMT-dbt-V5-His, treated with MG132, and collected at the indicated times (hr) post-dbt induction (top of panels). Extracts were subjected to TEV protease cleavage. Equal amounts of cleaved protein were incubated with glutathione resins bound with GST-SLIMB. The relative amounts of the dPER(aa1–100) fragment in the starting material (lysate; top panels) and that bound to the resins (GST-SLIMB; bottom panels) were visualized by immunoblotting in the presence of αFLAG antibodies. Non-specific bands in the lysate (top panels) are marked by an asterisk (*, left of panel). (C) S2 cells co-expressing pAc-dper-V5-His (WT and mutants) and pMT-dbt-V5-His were incubated with MG132 and harvested at 6 hours post-dbt induction. Extracts were immunoprecipitated with α-V5 antibodies and dPER isoforms phosphorylated at Ser47 detected using α-pS47 antibodies (top), while total dPER protein was visualized using α-V5 antibodies (bottom). (D) dPER total protein and pS47 signal intensity in panel (C) were quantified, and the ratio of dPER pS47 over total dPER proteins were presented in histogram format. See Fig. S2 for supporting data.
Although DBT phosphorylates dPER at numerous regions, phosphorylation events in the first 100aa of dPER are sufficient and necessary for SLIMB binding, with phosphorylation at Ser47 a critical signal generating the SLIMB phospho-degron (Chiu et al., 2008). Identification of the SLIMB binding site on dPER was partly accomplished by generating a “TEV/TAG” version of dPER, which contains a FLAG epitope at the amino-terminus and a TEV protease site inserted at aa100 (dPER/TEV100) (Chiu et al., 2008). This strategy enables the normal phosphorylation of full-length dPER in S2 cells followed by extract preparation and TEV cleavage to determine the biochemical properties of the aa1-100 SLIMB binding fragment in isolation. We applied this approach to probe whether the per-short phospho-cluster affects phosphorylation within the first 100aa by generating TEV/TAG versions of different per-short domain mutants under conditions where S2 cells were incubated with MG132 to block degradation. A trend towards more C-terminal phospho-site mutants exhibiting faster DBT-mediated phosphorylation kinetics was also observed for the first 100aa of dPER (Fig. 2B, top panels; Fig. S2C). For example, at 6 hr post-dbt induction there is clearly a higher fraction of hypo-phosphorylated to hyper-phosphorylated isoforms in the first 100aa of wild type dPER compared to the S596A and S589A versions (Fig. 2B, top panels, compare lane 2, 6, 10 and 14; Fig. S2C). To further link phosphorylation at the per-short domain to SLIMB-mediated events, we evaluated a double mutant whereby phosphorylation at S47 and S596 are both abolished [dPER(S47/596A)] (Fig. S3). Prior work showed that highly phosphorylated species of dPER are stabilized and accumulate if phosphorylation at S47 is blocked [dPER(S47A)] (Chiu et al., 2008). This was also the case for dPER(S47/596A) (Fig. S3 compare lanes 4, 8, 12 and 16), indicating that the enhanced degradation of dPER by blocking phosphorylation at S596 is linked to the phosphorylated status at S47.
To evaluate the effects of the per-short phospho-cluster within a more functional context, we measured binding of the dPER(aa1-100) fragment to SLIMB fused to glutathione-S-transferase (GST) (Chiu et al., 2008). For the wild type dPER(aa1-100) fragment, peak binding to SLIMB occurs at 20 hr post-dbt induction, a pattern also observed for the T583A mutant (Fig. 2B, lower panel, compare lanes 1-4 with 17-20; see Fig. S2D for quantitation of results). In sharp contrast, binding of the 1-100aa fragment from the S596A and S589A dPER mutants to SLIMB peaks much earlier by 6-12 hr post-dbt, whereas maximal binding is attained by 12 hr post-dbt induction for the S585A mutant (Fig. S2D). Strikingly, although little binding occurred in the absence of exogenous dbt (naïve S2 cells have some levels of endogenous dbt), relative differences were still observed in the binding affinities of the different 1-100aa dPER fragments to SLIMB, with the S585A mutation yielding less binding compared to the S596A and S589A versions but more than wild type and the T583A mutation (Fig. 2B, lower panels, compare lanes 1, 5, 9, 13 and 17). Finally, we also compared the intensity of Ser47 phosphorylation at 6 hr post-dbt induction using our previously generated phospho-specific antibodies (Chiu et al., 2008). Even though it was difficult to obtain a clear order between the mutants [e.g., dPER(S596A) isoforms phosphorylated at Ser47 are relatively more unstable compared to the other dPER variants evaluated even in the presence of proteasome inhibitors; data not shown], the results consistently indicated higher phospho-occupancy of Ser47 for the S596A, S589A and S585A mutants compared to the wild type control or dPER(T583A) (Fig. 2C, upper panel and Fig. 2D).
In summary, while there is some variation in the relative ordering of mutants depending on the experimental platform used, the cumulative biochemical data indicate that the per-short phospho-cluster is a major determinant of dPER stability by functioning in a spatially-oriented and graded manner to regulate the rate of DBT-mediated phosphorylation events underlying the binding of SLIMB to dPER, with phospho-sites closer to the C-terminal side of the cluster having more potent effects.
The per-short domain is a time-delay phospho-cluster that slows down the pace of the clock in a spatially oriented and graded manner
To investigate the physiological significance of our findings in whole animal systems, we generated transgenic flies that produce tagged versions of the dPER(TS583-596A), dPER(S596A), dPER(S589A) and dPER(S585A) proteins. We did not generate a dPER(T583A) version because the biochemical results indicated no observable differences with control dPER (Figs. 1 and 2). The effects of the different transgenes were examined in the per-null wper0 (Konopka and Benzer, 1971), whereby the only functional copy of dper is provided by the transgene.
As previously shown, wper0 flies expressing the wild type dper transgene exhibit strong rhythms with ~24 hr periods (Figure 3 and Table S1) (Kim et al., 2007; Ko et al., 2007). Also as expected, the p{dper(S589A)} transgenic flies, which replaces the Ser at the perS site with Ala, manifest ~19 hr rhythms that are similar in length to the original perS mutant (Konopka and Benzer, 1971; Rutila et al., 1992). Flies bearing dper transgenes that either abolish phosphorylation at all the Ser/Thr residues within the per-short phospho-cluster identified by mass spectrometry [p{dper(TS583-596A)}] or just Ser596 [p{dper(S596A)}] manifested ultra-short behavioral rhythms of ~16 hr. Intriguingly, p{dper(S585A)} flies had less severe period-shortening effects with rhythms of ~21 hr. We also evaluated the p{dper(TS583-596A)} transgene in a wild type per+ background and obtained a period of ~21 hr (Table S1), consistent with the semi-dominant nature of dper alleles (Konopka and Benzer, 1971). The ability of the different dper versions to shorten behavioral rhythms is ordered as follows; p{dper(TS583-596A)} = p{dper(S596A)} > p{dper(S589A)} > p{dper(S585A)} > p{dper(WT)}.
Figure 3. Spatially oriented and graded responses of per-short phospho-cluster site mutants on clock speed.

(A to E, left panels) Flies bearing different dper transgenes (indicated at top of panels) in a wper0 genetic background were collected at the indicated times (top of panels) and head extracts analyzed for dPER by immunoblotting in the presence of αHA. (A to E, right panels) The corresponding locomotor activity profiles of the transgenic flies in LD are shown. The second and third days worth of activity data during LD were averaged, generating the 24-hr profiles shown in the panels. The free-running periods (τ) are also shown. Arrow represents the timing of the clock-controlled evening peak. Vertical bars represent the activity recorded in 15-minute bins during times when the lights were on (grey bars) or off (black bars). ZT0 is lights-on, whereas ZT12 is lights-off. Horizontal bars at bottom; open, lights-on; closed, lights-off. (F) dPER signal intensity (Y-axis) was normalized by setting the peak intensity value for each dPER variant to a value of 1. Two independent experiments were used to generate the average and error bars represent SEM. See Table S1 for supporting data.
These findings reveal a remarkable congruence between the period-altering effects of mutations in the per-short phospho-cluster on behavioral rhythms and the biochemical results obtained when analyzing the same dPER versions in cultured S2 cells. Thus, the effects of the per-short phospho-cluster on the speed of circadian pacemakers in flies is closely linked to attenuating the efficiency of DBT-dependent binding of SLIMB to dPER. In addition, blocking phosphorylation at Ser596 eliminates the function of the entire cluster, suggesting that the 16 hr rhythms observed in the perT mutant (G593D) are due to a similar biochemical defect.
Under standard entraining conditions of 12hr light: 12 hr dark cycles (LD) at 25oC, D. melanogaster manifest a bimodal distribution of locomotor activity with a morning peak centered around the dark-to-light transition and an evening peak centered around the light-to-dark transition (Rosato and Kyriacou, 2006) (Fig. 3A-E, right panels). As expected, the shorter the free-running period of the dPER mutant, the earlier the timing of evening activity in LD cycles. Daily cycles in dPER abundance and phosphorylation were also advanced in LD, consistent with the behavioral results and prior findings analyzing the perS and perT mutants (Fig. 3A-E, left panels and Fig. 4C-E, top panels; see Fig. 3F for quantitation of dPER abundance rhythms) (Edery et al., 1994; Hamblen et al., 1998; Marrus et al., 1996). In this analysis we collected flies every 4 hr during a daily cycle, precluding a more reliable comparison of the different per-short phospho-cluster mutants.
Figure 4. Phospho-specific antibodies reveal that DBT phosphorylates S589 but not S596, and that phosphorylation at S596 stimulates phosphorylation at S589 and S585, which delays phosphorylation at S47.

(A) Extracts were prepared from S2 cells co-transfected with a wild type (WT) version of dper (pAc-dper-V5-His) and pMT-dbt-V5-His (+) or an empty pMT plasmid, pMT-V5-His (−). Cells were treated with MG132 and cycloheximide 16 hours post-dbt induction and harvested 4 hrs later. dPER was immunoprecipitated (IP) with α-V5 beads and split into equal parts that were either treated in the absence (-λPP) or presence (+λPP) of λ-phosphatase. Recovered immune complexes were probed by western blotting (WB) in the presence of the indicated antibody (left of panel). (B) Head extracts were prepared from wper0;p{dper(WT)} or wper0;p{der(Δaa755-809)} flies collected at the indicated times (ZT; top of panels). dPER-HAHis-containing immune complexes were recovered using α-HA beads and split into two equal parts that were treated in the absence (-λPP) or presence (+λPP) of λ-phosphatase. Recovered immune complexes were probed by western blotting (WB) in the presence of the indicated antibody (left of panel). (C, D, E) Head extracts were prepared from wper0;p{dper(WT)}, wper0;p{dper(S589A)}, and wper0;p{dper(S596A)} flies collected at the indicated times (ZT). dPER-HAHis-containing immune complexes were recovered using α-HA beads, followed by western blotting (WB) in the presence of the indicated antibody (left of panel). Positions of non-specific signals from α-pS589 and α-pS47 antibodies are indicated by asterisks (*; right of panels). See Figs. S4 and S5 for supporting data.
Phosphorylation at Ser596 enhances subsequent phosphorylation of Ser589 by DBT
To confirm and better analyze individual phosphorylation events within the per-short phospho-cluster we sought to generate phospho-specific polyclonal antibodies, and were successful in generating good quality antibodies that recognize phosphorylated S589 (α-pS589) and α-pS596). Several lines of evidence demonstrate the high specificity of our α-pS589 and α-pS596 antibodies for phosphorylated Ser589 and Ser596, respectively (Figs. 4 and S4; and data not shown). For example, phosphatase treatment of wild type dPER expressed in either S2 cells or flies either eliminated or strongly diminished detection by these antibodies (Fig. 4A, compare lanes 3 and 4 to 1 and 2, top two panels; Fig. 4B, compare lanes 3 and 4 to 1 and 2, top two panels). In addition, the staining intensities of dPER(S589A) and dPER(S596A) mutant proteins produced in flies and S2 cells were severely diminished when probed with their respective phospho-specific antibodies (Figs. 4C-E and S4).
Results obtained using mass spectrometry of dPER produced in either S2 cells or phosphorylated in vitro in the presence of DBT strongly suggest that the perS site is phosphorylated by DBT (Chiu et al., 2008; Kivimae et al., 2008), consistent with earlier work demonstrating allele-specific genetic interactions between a presumed hypomorphic allele of dbt, termed dbtAR and the perS mutation (Rothenfluh et al., 2000). Indeed, induction of dbt in S2 cells greatly enhanced phosphorylation of Ser589 (Fig. 4A top panel, compare lanes 1 and 2). We also generated an α-pS585 antibody that unfortunately only resulted in a detectable signal when probing dPER produced in S2 cells. Nonetheless, like pS589, the intensity of the signal was strongly increased following induction of dbt (Fig. S5A, compare lanes 1 and 2, top panel). In contrast, the Ser residue at position 596 is followed by a proline residue, indicating that S596 is phosphorylated by a “pro-directed” kinase, making it highly unlikely that DBT directly phosphorylates S596 (discussed below). Furthermore, mass spectrometry analysis shows that unlike S589, S585 and T583, induction of exogenous dbt is not required to detect phosphorylated S596 (Chiu et al., 2008), indicating the presence of one or more endogenous kinase(s) in S2 cells that can modify S596 (see below). In agreement with this notion, phosphorylated Ser596 is readily detected when S2 cells are singly transfected with only dper-containing plasmids (Fig. 4A, middle panel, compare lanes 1 and 2).
To investigate whether Ser589 is phosphorylated in a DBT-dependent manner in vivo, we used transgenic flies bearing the dPER(ΔdPDBD) internal deletion (Fig. 4B), which removes aa755-809 of dPER and abrogates the ability of DBT to stably bind and phosphorylate dPER (Kim et al., 2007; Nawathean et al., 2007). Indeed, whereas the intensity of phosphorylated Ser589 in the delta mutant is reduced to phosphatase treated non-specific background levels (Fig. 4B, top panel, compare lanes 5 and 6 to 7 and 8), phosphorylation at Ser596 remains strong (Fig. 4B, middle panel, compare lanes 5 and 6 to 7 and 8).
Remarkably, blocking phosphorylation at S596 abolished phosphorylation at S589 in flies (Fig. 4D) and strongly delayed the kinetics of DBT-mediated phosphorylation at S589 in S2 cells (Fig. S4). In sharp contrast, the phosphorylated status of S589 has no bearing on the efficiency of phosphorylation at S596 (Fig. 4E; see also Figs. 6B and S4). A similar relationship was also observed for S585, whereby phosphorylation of S596 in flies was normal in the dPER(S585A) mutant (Fig. S5B, middle panels), and in cultured S2 cells S596 strongly enhances phosphorylation at S585 (Fig. S5A, top panels, compare lanes 2 and 14). Although blocking phosphorylation at S589 does not impede phosphorylation at S585, and vice-versa, it is possible that blocking one site reduces phosphorylation at the other site (Fig. S5A, compare lanes 2 and 10, top panel; Fig. S5B, lower panels). Immunoblotting of extracts prepared from wild type flies collected at different times in a daily cycle shows that phosphorylation at S596 precedes that at S589, and that phosphorylation at S589 peaks prior to that of S47 (Fig. 4C). Moreover, dPER isoforms containing phosphorylated S47 are readily observed by ZT12-16 for the S596A and S589A dPER mutants, whereas detection of pS47 for wild type dPER begins around ZT20 and peaks in the early day (Fig. 4C-E). This striking temporal order is further evidence for a model whereby phosphorylation of S596 enhances phosphorylation by DBT of S589 and presumably the other phospho-acceptor sites on the per-short cluster domain (i.e., S585 and possibly T583), which collectively act to delay DBT-mediated phosphorylation at the SLIMB recognition site on dPER centered on the phospho-occupancy of S47.
Figure 6. Nmo is a clock kinase that phosphorylates S596 on dPER.

(A) Targeted expression of nmo RNAi in tim-expressing clock neurons was achieved by crossing w; UAS-DICER2; tim-UAS-GAL4 (TUG) males with females from two independent responder RNAi lines [v101545 (line 1) and v104885 (line 2)] that are in a w1118 genetic background. Flies were collected at ZT 2 or 18, and head extracts prepared. dPER-containing immune complexes were recovered using α-dPER antibodies (GP339), followed by western blotting (WB) with α-pS596 (top). The relative input of dPER for the IP was assayed by western blotting (WB) with α-dPER (GP73; bottom). (B) S2 cells were transfected with pAc-dper-V5-His (WT, S589A, or S596A mutant) in the presence of an empty plasmid (pAc-3XFLAG-His) or nmo-expressing plasmid [pAc-3XFLAG-His-nmo (WT or K69M kinase-dead mutant)]. dPER-V5-containing immune complexes were recovered using α-V5 beads, followed by western blotting (WB) in the presence of the indicated antibody (left of panel) to detect dPER isoforms phosphorylated at S596 (top panel), total dPER proteins (middle) and NMO (bottom). (C) Model showing how the per-short phospho-cluster regulates the pace of the clock. NMO phosphorylates S596 on dPER, which stimulates DBT-dependent phosphorylation of S589, S585 and (perhaps) T583. Phosphorylation in the per-short phospho-cluster somehow delays phosphorylation at other DBT-dependent sites on dPER, including S47, thus delaying the dPER/SLIMB interaction and the daily downswing in dPER levels. See Figs S4 to S6 for supporting data.
NEMO/NLK kinase phosphorylates Ser596
As noted above, the Ser residue at position 596 of dPER is followed by a proline residue, indicating that Ser596 is phosphorylated by a “pro-directed” kinase. These kinases are part of the CMGC family of kinases that include several subfamilies, such as mitogen-activated protein kinases (MAPK), cyclin-dependent kinases (Cdks) and dual-specificity tyrosine-regulated kinases (DYRKs) (Kannan and Neuwald, 2004). Based on sequence motifs there are ~34 known/predicted CMGC kinases in Drosophila melanogaster (www.kinase.com). It is unlikely that the kinase phosphorylating S596 would be from outside the CMGC family as the flanking Pro residue on the substrate greatly constrains the kinds of kinases that can recognize and phosphorylate the amino-proximal Ser/Thr acceptor site (Kannan and Neuwald, 2004).
To try and identify physiologically relevant kinases that might phosphorylate Ser596 we used the Gal4/UAS system (Brand and Perrimon, 1993) to screen available transgenic RNAi lines from the Vienna-based Drosophila stock center (VDRC; (Dietzl et al., 2007) that were directed against known/predicted Drosophila CMGC kinases. We used the well-characterized tim-Gal4 driver (TUG) (Blau and Young, 1999) to drive RNAi expression in clock cells (TUG>UAS-RNAi). We reasoned that inhibiting expression of the Ser596 kinase in clock cells should, at least partially, phenocopy the S596A mutation and yield behavior rhythms with short free-running periods. Of all the RNAi lines we tested only those targeting the kinase NEMO (NMO) manifested significantly short periods compared to parental controls (Fig. 5 and data not shown). Three independent TUG>UAS-RNAnmoi lines yielded free-running periods of ~21–22 hr (Fig. 5). Although the 21–22 hr rhythms observed in nmo RNAi flies are not as short as the ~16 hr periods observed in p{dper(S596A)} flies, severe nmo hypomorphs also manifest 21–22 hr rhythms (Yu et al., 2011). Thus, it is highly unlikely that the 21–22 hr rhythms observed using three independent nmo RNAi lines are because of incomplete silencing of nmo. Also, NMO might phosphorylate other sites on dPER and/or other clock proteins which could explain why reducing its levels/activity does not have equivalent effects on clock speed as eliminating phosphorylation at S596 (Fig. 6B; data not shown). Genetic manipulations using the UAS/GAL4 system to overexpress nmo in clock cells did not lead to significant changes in behavioral periods, suggesting its levels are not rate-limiting for clock speed (data not shown).
Figure 5. RNAi knockdown of nmo in pacemaker cells speeds up the pace of the clock.
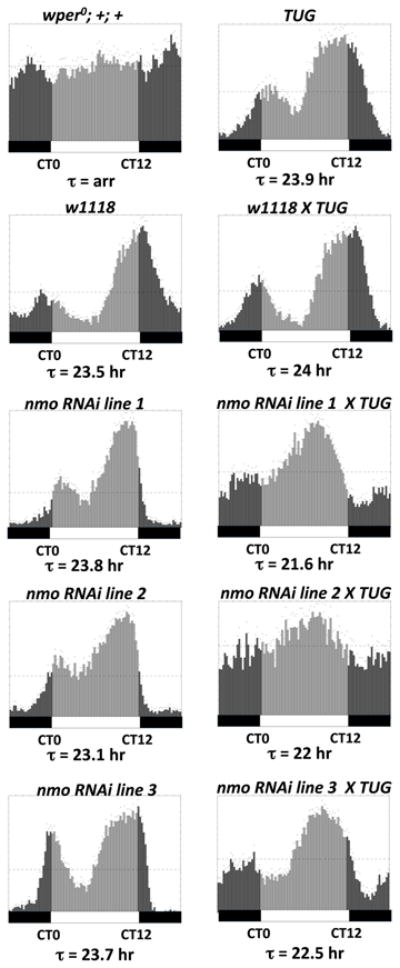
Shown are group averages of the 24-hr activity profiles (average of first three days during constant dark conditions) for males of the different genotypes. The free-running periods (τ) are also shown. Vertical bars represent the activity recorded in 15-minute bins during the subjective day (light grey bars; CT0-12) or subjective night (dark grey bars; CT12-24). Horizontal bars at bottom; open, subjective day; closed, subjective night. Targeted expression of nmo RNAi in tim-expressing clock neurons was achieved using the UAS/GAL4 system. The GAL4 driver line used here is w; UAS-DICER2; tim- -UAS-GAL4 (TUG). Three independent responder RNAi lines (line 1= v101545, line 2 = v104885, line 3 = v3002) were used and they were all in w1118 genetic background. Appropriate parental and genetic background (w1118) controls were included in the experiment. wper0; +; + represents the control for arrhythmicity (arr).
To verify that NMO phosphorylates S596 in flies we generated head extracts from TUG>UAS-nmoRNAi flies and parental controls collected at ZT18 and ZT2 and probed by immunoblotting in the presence of phospho-specific α-pS596 antibodies. Two different nmoRNAi lines were evaluated to ensure reproducibility. Expressing RNAi directed against nmo in clock cells led to striking decreases in the staining intensity of phosphorylated S596 (Fig. 6A, top panel and Fig. S6E, middle panel) that are not due to variations in the total abundance of dPER (Figs. 6A and S6E, bottom panels). We also evaluated the status of phosphorylation at S589, which is readily observed at ZT2 but not ZT18 in control flies (Fig. S6E, top panel). Importantly, the phospho-occupancy of S589 is also diminished in the nmo RNAi lines (Fig. S6E, compare lane 1–3), further establishing that in flies phosphorylation of S596 by NMO stimulates downstream phosphorylation of S589.
Additional evidence for NMO as a dPER kinase was obtained in cultured S2 cells and in vitro (Figs. 6 and S6). For example, expression of wild type NMO in S2 cells but not a negative dominant version (K69M), increased phosphorylation of S596 in wild type dPER (Fig. 6B, lanes 1–3, and Fig. S6B), whereas no phospho-specific signal was observed with the S596A mutant (Fig. 6B, compare lanes 2 and 6; Fig. S6B, compare lanes 2 and 5). It is noteworthy that NMO still evokes a mobility shift in the dPER(S596A) mutant (Fig. 6B, middle panel, lanes 4–6), suggesting additional phosphorylation sites on dPER (data not shown). Blocking phosphorylation at S589 does not affect phosphorylation at S596 by NMO (Fig. 6B, lanes 4 and 5). We also expressed all the known/suspected pro-directed kinases and only induction of nmo led to significant increases in the phospho-occupancy of S596 (Fig. S6A and data not shown). In agreement with this observation, dPER specifically interacts with NMO but not other kinases of the CMGC family (Fig. S6C). To obtain more direct evidence that NMO phosphorylates S596, dPER was purified and phosphatase treated to remove any phosphorylation that occurred in S2 cells and subsequently incubated with a range of commercially available kinases, including the highly structurally related MAP kinase. Of all the kinases we tested in vitro, only NMO led to dramatic increases in the phospho-occupancy of S596 (Fig. S6D).
Phosphorylation-induced conformational changes
While the phospho-occupancy of Ser47 plays a critical role in the direct binding of SLIMB to dPER, a general role of global phosphorylation might be to evoke conformational changes that slowly make dPER a more efficient substrate for degradation. As an initial attempt to address this issue we purified hyper-phosphorylated dPER and treated part of the sample with phosphatase to reduce/eliminate phosphorylation, while the other part was mock-treated. Finally, both samples were subjected to limited proteolysis using a variety of proteases. Intriguingly, hyper-phosphorylated dPER is more susceptible to degradation, implying a more open structure (Fig. 7A and B). This was also the case when using the TEV protease with our dPER/T100 version of dPER. For example, some full-length dPER/T100 is still observed after 2 hr incubation with TEV for the hypo-phosphorylated version, whereas little to no full-length product is detected in the hyper-phosphorylated version (Fig. 7C, compare lanes 2 and 5), suggesting that the region encompassing the SLIMB binding region in the first 100aa is also more accessible in highly phosphorylated dPER. Similar results were also obtained when we compared hyper- and hypo-phosphorylated isoforms of the dPER(S596A) mutant (Fig. 7). Using this assay we were not able to observe a reproducible difference in the susceptibility to limited proteolysis between wild type dPER and dPER(S596A). Although this experimental strategy does not provide high resolution structural information, the results are consistent with the notion that the more rapid DBT-mediated degradation kinetics for dPER(S596A) (Fig. 1) is not due to a grossly different conformation compared to wild type dPER but rather due to changes in the speed with which the mutant version attains isoforms more prone to degradation.
Figure 7. Limited proteolysis suggests highly phosphorylated dPER has a more open conformation compared to hypo-phosphorylated isoforms.

(A, B) Extracts were prepared from S2 cells co-transfected with WT of S596A variants of pAc-dper-V5-His and pMT-dbt that were harvested 20 hr after dbt induction. dPER protein was immunoprecipitated (IP) with α-V5 beads and subsequently treated in the absence (-λPP) or presence (+λPP) of λ-phosphatase. Immunoblotting with α-dPER antibodies was used to detect dPER proteins after limited proteolysis using (A) Trypsin (T1 and T2) or (B) Factor Xa (Fx). (C) Extracts were prepared from S2 cells transfected with WT or S596A variants of pAc-dper/Tev100-V5-His. Hyperphosphorylated dPER was generated by addition of 30nM of Calyculin A (Calbiochem) 2 hours before cell harvest. 50uM MG132 (Sigma) and 10ug/ml cycloheximide (Sigma) were also added to maximize the amount of hyperphosphorylated dPER and to prevent degradation. Cells were harvested 48 hrs after transfection. Immunoblotting with α-c-myc antibodies was used to detect dPER (full length and aa101-1224) proteins after limited proteolysis using AcTEV (Invitrogen).
Discussion
Herein, we show that the per-short domain functions as a discrete hierarchical phospho-cluster that delays DBT-mediated phosphorylation at the SLIMB recognition site on dPER, providing new insights into how clock protein phosphorylation contributes to circadian timing mechanisms (Fig. 6C). The cumulative effect of this delay circuit is to slow down the pace of the clock by ~8 hr. We propose that DBT functions in a step-wise manner to phosphorylate clusters on dPER that have distinct biochemical functions and effects on the rate of dPER degradation; e.g., elements such as the per-short phospho-cluster that delays dPER degradation and those such as the SLIMB binding site and global phosphorylation that enhance instability. NMO plays a major role in the relative timing of DBT activity at these different elements because it stimulates multi-site phosphorylation at the per-short delay cluster by DBT, which slows down the ability of DBT to phosphorylate instability elements. Thus, a large portion of the phosphorylation events dictating when in a daily cycle dPER is targeted for rapid degradation is not directly linked to binding of SLIMB per se. Our findings demonstrate that presumptive long-range interactions between distinct positively and negatively acting phospho-clusters collaborate to set clock speed and helps explain why mutations in clock protein phosphorylation sites and/or the kinases that phosphorylate them can yield both fast and slow clocks.
Our proposed mechanism for the function of the per-short domain (Fig. 6C) is supported by the congruence between in vitro biochemical studies based on purified recombinant dPER protein from cultured S2 cells and in vivo changes in the pace of behavioral rhythms using transgenic models. This suggests that a primary biochemical effect of the per-short domain on clock speed in the fly is via modulating the rate of DBT-mediated phosphorylation at the SLIMB phospho-degron on dPER. The physiological role of T583 phosphorylation is not clear as mutating this site does not have detectable effects on the binding of dPER to SLIMB in S2 cells (Figs 1-2 and data not shown). In this regard it is interesting to note that the original per-short domain was identified as encompassing aa585-601 of dPER (Baylies et al., 1992). Thus, it is likely that the 8hr per-short delay circuit is governed by the dynamics underlying the phosphorylated status of 3 sites (i.e., S596, S589 and S585).
At present it is not clear how phosphorylation in the per-short cluster slows down subsequent phosphorylation by DBT at Ser47 and other sites. Inactivating the per-short cluster leads to increases in the rate of DBT-mediated phosphorylation at not only the N-terminus (Fig. 2) but also the C-terminus of dPER (data not shown), suggesting it is a major control center for regulating the relative efficiency of DBT phosphorylation at many sites on dPER. We suggest that the per-short phospho-cluster acts as a transient temporal trap for DBT. Once the sites in the per-short domain are phosphorylated by DBT this somehow allows it to continue its normal rate of phosphorylation at other phospho-clusters. Although speculative, progressive increases in phosphoryation at some of these other phospho-clusters might generate time-dependent local/overall conformational changes in dPER (Fig. 7), possibly via electrostatic repulsion, eventually leading to a more open dPER structure that is either more accessible to phosphorylation by DBT at the SLIMB binding site and/or a more efficient substrate for degradation. Thus, the rapid degradation of dPER during the early day is likely due to a combination of synchronous increases in the phospho-occupancy of Ser47 and overall phosphorylation of dPER. Other factors such as protein phosphatases and the action of TIMELESS also play major roles in regulating the speed of the dPER phosphorylation program (Bae and Edery, 2006).
How might phosphorylation at S596 enhance phosphorylation at S589 and S585 by DBT? Phosphorylation by the CK1 kinase family is generally enhanced by priming (Flotow et al., 1990). However, phosphorylation at the per-short domain by DBT does not follow the consensus priming-dependent recognition motif for the CK1 family of kinases (i.e., S/Tp-X-XS/T, where S/Tp refers to the primed site, X is any amino acid, and the underlined residues the CK1 target site; (Flotow et al., 1990), as the S596 priming site is located C-terminal to the DBT sites. Thus, it is likely that phosphorylation of S596 by NMO stimulates DBT phosphorylation at the per-short region in a non-priming dependent manner. Ongoing studies are aimed at understanding the biochemical events underlying the ability of phosphorylation at S596 to enhance phosphorylation by DBT in the per-short region.
The discovery of a delay phospho-circuit also sheds light on why mutations in different phosphorylation sites on PER or FRQ proteins, although affecting stability, can speed up or slow down the clock (e.g., (Baker et al., 2009; Chiu et al., 2008; Tang et al., 2009; Toh et al., 2001; Vanselow et al., 2006). Our findings also offer a logical explanation for why mutations that lower the kinase activity of CKI, which overall is expected to slow down the rate of PER degradation, can yield fast clocks. For example, although other mechanisms have been offered to explain the short period phenotypes observed for the CKIεtau mutation in hamsters (Gallego et al., 2006; Meng et al., 2008) and a CKIδ mutation associated with familial advanced sleep phase syndrome (FASPS) in humans (Xu et al., 2005), it is possible that phosphorylation at a per-short type delay cluster is preferentially compromised by the mutant kinase, which could appear as a substrate-specific gain-of-function mutation.
Negatively acting phospho-clusters are likely to be a general feature of the timing mechanisms regulating the daily abundance cycles of clock proteins such as PERs in animals and FRQ in Neurospora. However, other regulatory modules that operate in a phase-specific manner must participate to generate an ~24 hr oscillator. Most conspicuously, clock speed is intimately linked to the PER and FRQ abundance cycles necessitating daily phases of de novo synthesis to replenish the pools of previously degraded proteins. As recently shown the transcriptional negative feedback aspect of dPER regulating dCLK-CYC-mediated transcription is also a component of the period setting mechanism (Kadener et al., 2008). Therefore, the ~24 hr dPER abundance cycle is based on a combination of time constraints that are generated using different regulatory modules. We propose that the per-short based timer mainly functions once dPER has accumulated and begins participating in transcriptional repression, controlling dPER abundance once it is disengaged from the dynamics of its cognate mRNA by setting in motion a series of sequential phosphorylation events that are calibrated to stimulate dPER degradation in the nucleus at the appropriate time in a daily cycle, enabling the next round of circadian gene expression. In this context, it is interesting to note that a prior study analyzing the per-short domain suggested that it functions with a nearby perSD domain to increase the transcriptional repressor function of dPER (Kivimae et al., 2008). It is possible that the same phosphorylation events leading to dPER degradation also function to increase its potency within the repressor complex.
Our studies also identify NEMO as a clock kinase. NEMO is the founding member of the evolutionarily conserved Nemo-like kinase (Nlk) family of proline-directed serine/threonine kinases closely related to mitogen-activated protein kinases (MAPK) (Brott et al., 1998). It was originally characterized in Drosophila as required for planar cell polarity during eye development (Choi and Benzer, 1994) and is now known to function in many pathways. Nmo/Nlk is localized in the nucleus (Brott et al., 1998) and is another factor in the circadian clock that also functions in the Wnt/Wg signaling pathway (Thorpe and Moon, 2004; Zeng and Verheyen, 2004), such as CKIε/DBT, β-TrCP/SLIMB, and GSK-3β/SGG. It will be of interest to determine if Nlk functions in the mammalian clock. Intriguingly, the phosphorylation sites on dPER are largely clustered and several of them have the same spatial arrangement as the per-short cluster with a predicted pro-directed kinase site at the C-terminal end of the phospho-cluster (Chiu et al., 2008). This suggests that Nmo and/or other pro-directed kinases serve as control points to activate spatially and perhaps functionally distinct phospho-clusters (Fig. 6; data not shown). Indeed, we recently showed that phosphorylation at Ser661 of dPER by an as yet unidentified pro-directed kinase primes further phosphorylation by SGG at Ser657 to regulate the timing of dPER nuclear entry in key pacemaker neurons (Ko et al., 2010).
In summary, a central aspect of circadian clocks is the presence of one or more clock proteins that provide a dual function by behaving as phospho-based timers and linking its timer role to gene expression by operating in a phase-specific manner to recruit repressor complexes that inhibit central clock transcription factors. Our studies suggest that a major part of the timing mechanism underlying these phospho-clock proteins is based on spatially and functionally discrete phospho-clusters that interact to impose calibrated and sequentially ordered biochemical time constraints. In the case of dPER, the per-short phospho-cluster functions as a central timing module by slowing down the ability of DBT to phosphorylate instability elements regulating dPER degradation and hence when dPER repressor activity is terminated and the next round of circadian gene expression begins.
Experimental Procedures
S2 cell culture and transfection
S2 cells and DES expression medium were obtained from Invitrogen and transient transfections were performed using Effectene (Qiagen) according to manufacturers instructions and as previously described (Chiu et al., 2008; Ko et al., 2002). For each transient transfection, 0.8 μg of different dper containing plasmids and 0.2 μg of pMT-dbt-V5-His or empty control pMT-V5-His plasmids were used. For expression of proline-directed kinases, 0.4μg of plasmids were used. Expression of dbt and other kinases under pMT promoter was induced by adding 500 μM CuSO4 to the culture media 36 hrs after transfection. Stable cell lines expressing pMT-gst-slimb were generated using calcium phosphate transfection kit (Invitrogen), and induction of pMT-driven expression was achieved using 500 μM CuSO4. For experiments in which the proteasome inhibitor MG132 (50 μM; Sigma) and cycloheximide (10 μg/ml; Sigma) were used (Fig. 2, 4A, S2A, S4, S5A), they were added 4 hrs prior to cell harvest.
Transgenic flies and locomotor activity assays
Transgenic flies carrying dper mutations were generated as described in greater detail in the supplemental material. Fly locomotor activity rhythms were measured using the Drosophila Activity Monitoring (DAM) System from Trikinetics (Chiu et al., 2010).
Generation of phosphospecific antibodies and immunoblotting
Phospho-specific antibodies were generated by Proteintech Group, Inc. (U.S.A.). Rabbits were immunized with three different peptides to generate three different phosphospecific antibodies: pS585 = N -C-PHENELTVpSERDSVM; pS589 = N -C-ELTVSERDpSVMLGEI; pS596 = N -C-VMLGEIpSPHHDYYDS (where p=phosphate). Immunoblotting analysis was performed as previously described (Lee et al., 1998) with modifications detailed in supplemental materials. The guinea pig α-dPER antibodies (GP339 and GP73) were previously described (Ko et al., 2010; Sidote and Edery, 1999).
Immunoprecipitation (IP) and phosphatase (λ-PP) treatment
IP and λ-PP treatment were performed as described (Lee et al., 1998) with modifications detailed in supplemental materials.
TEV enzyme cleavage and GST pull-down assay
TEV cleavage and GST-SLIMB binding assays were performed as previously described (Chiu et al., 2008).
In vitro kinase assay
S2 cells were transiently transfected with pAc-dper-V5-His. S2 cells were harvested 48 hrs after transfection and dPER proteins were extracted using EB2 (see supplemental materials). After IP with α-V5 agarose (Sigma) (see supplemental materials) to obtain dPER proteins, samples were subjected to λ-PP (NEB) or mock treatment at 30 C for 30 min. Samples were subsequently washed two times with appropriate reaction buffers (NLK from Millipore; CDC2, MAPK, and GSK3β from NEB). Kinase assays were performed at 30 C for 30 min following manufacturer s suggested protocol.
Supplementary Material
Acknowledgments
We thank Paul Hardin (Texas A&M, USA) for communicating unpublished results (Yu et al., in press), Thomas Kusch (Rutgers University, USA) for Sfx cells, and the VDRC (Vienna, Austria) for RNAi lines. We thank Axel Diernfellner and Michael Brunner (Heidelberg University, Germany) for conditions for limited proteolysis. This work was supported by NIH grant (R01NS03498) to I.E and NIH grant (K99/R00NS061952) to J.C.
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allada R, Chung BY. Circadian organization of behavior and physiology in Drosophila. Annu Rev Physiol. 2010;72:605–624. doi: 10.1146/annurev-physiol-021909-135815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae K, Edery I. Regulating a circadian clock's period, phase and amplitude by phosphorylation: insights from Drosophila. J Biochem. 2006;140:609–617. doi: 10.1093/jb/mvj198. [DOI] [PubMed] [Google Scholar]
- Baker CL, Kettenbach AN, Loros JJ, Gerber SA, Dunlap JC. Quantitative proteomics reveals a dynamic interactome and phase-specific phosphorylation in the Neurospora circadian clock. Mol Cell. 2009;34:354–363. doi: 10.1016/j.molcel.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylies MK, Bargiello TA, Jackson FR, Young MW. Changes in abundance or structure of the per gene product can alter periodicity of the Drosophila clock. Nature. 1987;326:390–392. doi: 10.1038/326390a0. [DOI] [PubMed] [Google Scholar]
- Baylies MK, Vosshall LB, Sehgal A, Young MW. New short period mutations of the Drosophila clock gene per. Neuron. 1992;9:575–581. doi: 10.1016/0896-6273(92)90194-i. [DOI] [PubMed] [Google Scholar]
- Blau J, Young MW. Cycling vrille expression is required for a functional Drosophila clock. Cell. 1999;99:661–671. doi: 10.1016/s0092-8674(00)81554-8. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brott BK, Pinsky BA, Erikson RL. Nlk is a murine protein kinase related to Erk/MAP kinases and localized in the nucleus. Proc Natl Acad Sci U S A. 1998;95:963–968. doi: 10.1073/pnas.95.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, Schafmeier T. Transcriptional and post-transcriptional regulation of the circadian clock of cyanobacteria and Neurospora. Genes Dev. 2006;20:1061–1074. doi: 10.1101/gad.1410406. [DOI] [PubMed] [Google Scholar]
- Chiu JC, Low KH, Pike DH, Yildirim E, Edery I. Assaying locomotor activity to study circadian rhythms and sleep parameters in Drosophila. J Vis Exp. 2010 doi: 10.3791/2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu JC, Vanselow JT, Kramer A, Edery I. The phospho-occupancy of an atypical SLIMB-binding site on PERIOD that is phosphorylated by DOUBLETIME controls the pace of the clock. Genes Dev. 2008;22:1758–1772. doi: 10.1101/gad.1682708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KW, Benzer S. Rotation of photoreceptor clusters in the developing Drosophila eye requires the nemo gene. Cell. 1994;78:125–136. doi: 10.1016/0092-8674(94)90579-7. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Edery I, Zwiebel LJ, Dembinska ME, Rosbash M. Temporal phosphorylation of the Drosophila period protein. Proc Natl Acad Sci U S A. 1994;91:2260–2264. doi: 10.1073/pnas.91.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem. 1990;265:14264–14269. [PubMed] [Google Scholar]
- Gallego M, Eide EJ, Woolf MF, Virshup DM, Forger DB. An opposite role for tau in circadian rhythms revealed by mathematical modeling. Proc Natl Acad Sci U S A. 2006;103:10618–10623. doi: 10.1073/pnas.0604511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8:139–148. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- Grima B, Lamouroux A, Chelot E, Papin C, Limbourg-Bouchon B, Rouyer F. The F-box protein slimb controls the levels of clock proteins period and timeless. Nature. 2002;420:178–182. doi: 10.1038/nature01122. [DOI] [PubMed] [Google Scholar]
- Hamblen MJ, White NE, Emery PT, Kaiser K, Hall JC. Molecular and behavioral analysis of four period mutants in Drosophila melanogaster encompassing extreme short, novel long, and unorthodox arrhythmic types. Genetics. 1998;149:165–178. doi: 10.1093/genetics/149.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzen C, Liu Y. The Neurospora crassa circadian clock. Adv Genet. 2007;58:25–66. doi: 10.1016/S0065-2660(06)58002-2. [DOI] [PubMed] [Google Scholar]
- Kadener S, Menet JS, Schoer R, Rosbash M. Circadian transcription contributes to core period determination in Drosophila. PLoS Biol. 2008;6:e119. doi: 10.1371/journal.pbio.0060119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Neuwald AF. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci. 2004;13:2059–2077. doi: 10.1110/ps.04637904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Ko HW, Yu W, Hardin PE, Edery I. A DOUBLETIME kinase binding domain on the Drosophila PERIOD protein is essential for its hyperphosphorylation, transcriptional repression, and circadian clock function. Mol Cell Biol. 2007;27:5014–5028. doi: 10.1128/MCB.02339-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivimae S, Saez L, Young MW. Activating PER repressor through a DBT-directed phosphorylation switch. PLoS Biol. 2008;6:e183. doi: 10.1371/journal.pbio.0060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, Young MW. The Drosophila clock gene double-time encodes a protein closely related to human casein kinase Iepsilon. Cell. 1998;94:97–107. doi: 10.1016/s0092-8674(00)81225-8. [DOI] [PubMed] [Google Scholar]
- Ko HW, DiMassa S, Kim EY, Bae K, Edery I. Cis-combination of the classic per(S) and per(L) mutations results in arrhythmic Drosophila with ectopic accumulation of hyperphosphorylated PERIOD protein. J Biol Rhythms. 2007;22:488–501. doi: 10.1177/0748730407306929. [DOI] [PubMed] [Google Scholar]
- Ko HW, Edery I. Analyzing the Degradation of PERIOD Protein by the Ubiquitin-Proteasome Pathway in Cultured Drosophila Cells. Methods Enzymol. 2005;393:394–408. doi: 10.1016/S0076-6879(05)93018-8. [DOI] [PubMed] [Google Scholar]
- Ko HW, Jiang J, Edery I. Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature. 2002;420:673–678. doi: 10.1038/nature01272. [DOI] [PubMed] [Google Scholar]
- Ko HW, Kim EY, Chiu J, Vanselow JT, Kramer A, Edery I. A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3beta/SGG in circadian clocks. J Neurosci. 2010;30:12664–12675. doi: 10.1523/JNEUROSCI.1586-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka RJ, Benzer S. Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1971;68:2112–2116. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka RJ, Hamblen-Coyle MJ, Jamison CF, Hall JC. An ultrashort clock mutation at the period locus of Drosophila melanogaster that reveals some new features of the fly's circadian system. J Biol Rhythms. 1994;9:189–216. doi: 10.1177/074873049400900303. [DOI] [PubMed] [Google Scholar]
- Lee C, Bae K, Edery I. The Drosophila CLOCK protein undergoes daily rhythms in abundance, phosphorylation, and interactions with the PER-TIM complex. Neuron. 1998;21:857–867. doi: 10.1016/s0896-6273(00)80601-7. [DOI] [PubMed] [Google Scholar]
- Markson JS, O'Shea EK. The molecular clockwork of a protein-based circadian oscillator. FEBS Lett. 2009;583:3938–3947. doi: 10.1016/j.febslet.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrus SB, Zeng H, Rosbash M. Effect of constant light and circadian entrainment of perS flies: evidence for light-mediated delay of the negative feedback loop in Drosophila. Embo J. 1996;15:6877–6886. [PMC free article] [PubMed] [Google Scholar]
- Mehra A, Baker CL, Loros JJ, Dunlap JC. Post-translational modifications in circadian rhythms. Trends Biochem Sci. 2009;34:483–490. doi: 10.1016/j.tibs.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng QJ, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sladek M, Semikhodskii AS, Glossop NR, Piggins HD, et al. Setting clock speed in mammals: the CK1epsilontau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron. 2008;58:78–88. doi: 10.1016/j.neuron.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawathean P, Stoleru D, Rosbash M. A small conserved domain of Drosophila PERIOD is important for circadian phosphorylation, nuclear localization, and transcriptional repressor activity. Mol Cell Biol. 2007;27:5002–5013. doi: 10.1128/MCB.02338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, Young MW. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell. 1998;94:83–95. doi: 10.1016/s0092-8674(00)81224-6. [DOI] [PubMed] [Google Scholar]
- Rosato E, Kyriacou CP. Analysis of locomotor activity rhythms in Drosophila. Nat Protoc. 2006;1:559–568. doi: 10.1038/nprot.2006.79. [DOI] [PubMed] [Google Scholar]
- Rothenfluh A, Abodeely M, Young MW. Short-period mutations of per affect a double-time-dependent step in the Drosophila circadian clock. Curr Biol. 2000;10:1399–1402. doi: 10.1016/s0960-9822(00)00786-7. [DOI] [PubMed] [Google Scholar]
- Rutila JE, Edery I, Hall JC, Rosbash M. The analysis of new short-period circadian rhythm mutants suggests features of D. melanogaster period gene function. J Neurogenet. 1992;8:101–113. doi: 10.3109/01677069209084155. [DOI] [PubMed] [Google Scholar]
- Schibler U. Circadian time keeping: the daily ups and downs of genes, cells, and organisms. Prog Brain Res. 2006;153:271–282. doi: 10.1016/S0079-6123(06)53016-X. [DOI] [PubMed] [Google Scholar]
- Sidote D, Edery I. Heat-induced degradation of PER and TIM in Drosophila bearing a conditional allele of the heat shock transcription factor gene. Chronobiol Int. 1999;16:519–525. doi: 10.3109/07420529908998725. [DOI] [PubMed] [Google Scholar]
- Tang CT, Li S, Long C, Cha J, Huang G, Li L, Chen S, Liu Y. Setting the pace of the Neurospora circadian clock by multiple independent FRQ phosphorylation events. Proc Natl Acad Sci U S A. 2009;106:10722–10727. doi: 10.1073/pnas.0904898106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe CJ, Moon RT. nemo-like kinase is an essential co-activator of Wnt signaling during early zebrafish development. Development. 2004;131:2899–2909. doi: 10.1242/dev.01171. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Vanselow K, Vanselow JT, Westermark PO, Reischl S, Maier B, Korte T, Herrmann A, Herzel H, Schlosser A, Kramer A. Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS) Genes Dev. 2006;20:2660–2672. doi: 10.1101/gad.397006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptacek LJ. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell. 2007;128:59–70. doi: 10.1016/j.cell.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Jacquier AC, Citri Y, Hamblen M, Hall JC, Rosbash M. Molecular mapping of point mutations in the period gene that stop or speed up biological clocks in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1987;84:784–788. doi: 10.1073/pnas.84.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Houl JH, Hardin PE. NEMO kinase contributes to core period determination by slowing the pace of the Drosophila circadian oscillator. Current Biology. 2011 doi: 10.1016/j.cub.2011.02.037. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng YA, Verheyen EM. Nemo is an inducible antagonist of Wingless signaling during Drosophila wing development. Development. 2004;131:2911–2920. doi: 10.1242/dev.01177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.