

Abstract
The identification of catalytic residues is an essential step in functional characterization of enzymes. We present a purely structural approach to this problem, which is motivated by the difficulty of evolution-based methods to annotate structural genomics targets that have few or no homologs in the databases. Our approach combines a state-of-the-art support vector machine (SVM) classifier with novel structural features that augment structural clues by spatial averaging and Z-scoring. Special attention is paid to the class imbalance problem that stems from the overwhelming number of non-catalytic residues in enzymes compared to catalytic residues. This problem is tackled by: 1) optimizing the classifier to maximize a performance criterion that considers both type I and type II errors in the classification of catalytic and non-catalytic residues; 2) under-sampling non-catalytic residues before SVM training; and 3) during SVM training, penalizing errors in learning catalytic residues more than errors in learning non-catalytic residues. Tested on four enzyme datasets – one specifically designed by us to mimic the structural genomics scenario and three previously-evaluated datasets – our structure-based classifier is never inferior to similar structure-based classifiers and comparable to classifiers that use both structural and evolutionary features. In addition to evaluation of the performance of catalytic residue identification, we also present detailed case studies on three proteins. This analysis suggests that many false positive predictions may correspond to binding sites and other functional residues. A web server that implements the method, our own-designed database, and the source code of the programs are publicly available at http://www.cs.bgu.ac.il/~meshi/functionPrediction.
Keywords: Catalytic Residues, Functional Annotation, Support Vector Machine (SVM), Energy Terms, Spatial Averaging, Feature Selection, Class Imbalance
Introduction
Enzymes mediate virtually all the chemical reactions required for life. They do so by accurately positioning a few catalytic residues within a specific microenvironment known as the active site. Thus, the identification of these residues and ultimately understanding all the electrostatic, entropic and allosteric aspects of the catalytic machinery are among the major goals of enzymology. Such a complete analysis however, requires a considerable scientific effort that includes both experimental and computational studies61. The current study focuses on the first step towards such studies, namely raising initial hypotheses regarding the identity of the catalytic residues.
Computational methods for the prediction of catalytic residues typically rely heavily on evolutionary inference, either direct annotation transfer from characterized proteins to their homologs, or interpretation of conservation patterns.1, 2 Evolutionary inference, however, has three inherent weaknesses. First and foremost, its basic assumptions do not always apply. Proteins may have different functions even if they are evolutionarily related3 and residues may be conserved for reasons other than a catalytic role (e.g., structural stability). Second, evolution-based approaches for the prediction of catalytic residues fail when applied to orphan proteins (ORFans),4 for which we cannot detect homologous proteins in the databases. Third, evolution-based methods are sensitive to errors due to misalignments.
Catalytic residues have characteristic structural features that may augment the evolutionary inference, as they are independent of homologous proteins and alignments.5–7 However, structures are typically determined only after much experimental work has already characterized the protein function. The targets of the structural genomic initiative (SGI)8 constitute a special case. Their structures are known but their function is often uncharacterized. Thus, their computational annotation is essential in order to gain full dividends from the SGI endeavor. The general trend in SGI annotation is that evolution is the major source of knowledge and the structural clues complement it. The current study, following Tong et al.,9 goes one step further and uses only structural clues to predict catalytic residues. The major motivation behind this somewhat puritan approach is the need for methods that can handle ORFans, which are not negligible in number among the structural genomics targets.10 Further, by providing a major challenge to our conceptions, the development of structure-based tools for catalytic residue identification may lead to new insights into the fundamental question of structure-function relationships.
We present here a two-fold strategy for the development of a structure-based predictor of catalytic residues. The first effort focuses on the development and evaluation of a set of structural features that represent the differences between catalytic and non-catalytic residues. The second effort concentrates on a classifier that uses these features to discriminate between the residues.
Our structural features are based on the observations that catalytic residues are typically spatially proximate, deeply occluded,11, 12 and they often destabilize the protein structure.13–24 Further, there is a wide divergence of catalytic propensities among the different residue types.25 These observations have already motivated several purely structural prediction methods for the identification of catalytic residues,12, 26–29 catalytic sites,11, 26 and functional residues.30, 31
Two major problems complicate the use of structural features for catalytic residue prediction. The first is noise. Non-catalytic residues may occasionally have catalytic-like characteristics. Such outliers are relatively rare, but since non-catalytic residues are numerous, compared with the catalytic ones, even a small fraction of them may still result in many false predictions. Previous studies9, 32 coped with this problem by a post-processing step of spatial clustering. Since catalytic residues are clustered within the catalytic site, spatially isolated predictions are most likely false and therefore may be ignored. However, the strength of this solution is limited. Numerous false predictions, due to non-discriminative features, may still be clustered and falsely identified as catalytic. In this study, we present a novel solution that uses the same observation regarding the tendency of catalytic residues to be spatially proximate. However, our approach uses this observation in a pre-processing step. By using spatial averaging over proximate atoms, we smooth out many of these outliers to establish more discriminative features that produce fewer false predictions (Figure 1).
Figure 1. The reasoning behind the use of spatial averaging.
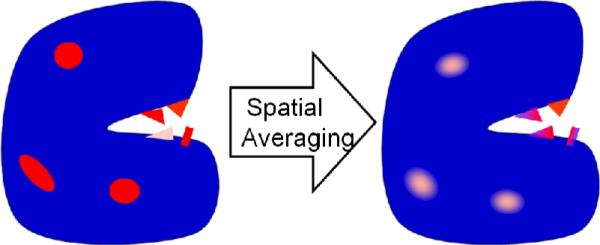
In this schematic illustration of an enzyme (left), blue represents regions with no signal for catalytic residues; pink illustrates regions with a weak signal; red illustrates regions with a strong signal. Random non-catalytic outliers (ellipses) are likely to be distributed all over the structure and their noise is likely to be averaged out (right). Catalytic residues (triangles) are typically clustered and thus their signal is more likely to survive the averaging. Further, the averaging may even propagate some signal from nearby binding residues (rectangle).
The second major problem with structural features is that they are often systematically biased by irrelevant factors such as the protein size or even refinement procedure. We are not aware of any previous explicit treatment for this problem. It turns out, however, that feature normalization, i.e., replacing the feature with its per-protein Z-score (the difference between the feature and its average divided by the feature standard deviation), often results in much less biased features (see Results section).
The second major effort in this study focuses on optimizing a support vector machine (SVM) classifier33, 34 to discriminate residues based on the features selected as part of the first effort. We develop a methodology that tackles the severely low (~1%) ratio of catalytic to non-catalytic residues in enzymes. This so-called “class imbalance problem” has hampered previous studies that used a neural network32 or SVM5, 9, 35, 36 for catalytic residue classification. In a nutshell, machine-learning classifiers that aim to maximize the accuracy of the assignment of residues to their correct classes, tend to over-predict the dominant class under class imbalance. If this problem is not considered, the classifier predicts all residues as non-catalytic and thereby achieves a (misleading) ~99% accuracy. To tackle this problem, previous studies used ad hoc manipulation of the classifier training phase. They either under-sampled (discarded) a random selection of the non-catalytic residues in order to reach better class ratio32, 35 or used different penalties for false predictions of catalytic and non-catalytic residues.9 Ad hoc solutions, however, are prone to less-than-optimal performance and, worse, to overfitting. We present here a new, methodologically solid, implementation of these techniques, as well as a new one described below.
A key element in our methodology is the Matthews correlation coefficient (MCC)37 (see Methods section for definition). MCC was originally used for the assessment of early secondary structure prediction methods, yet another problem that class imbalance complicates.37 Since it is symmetrical with respect to all possible outcomes of a binary classifier, it is more robust with respect to class imbalance than other common measures of classifier performance.38 Thus, MCC is often used as a performance measure in catalytic residue prediction.9, 35 Our new methodology takes a further step and also uses MCC for classifier optimization.
Classifier optimization is performed as part of the SVM training phase by thoroughly sampling the parameter space and selecting the set of parameters that maximize MCC. This selection uses a validation set, which is a subset of the training set not used for the actual training; both are independent of a test set used for performance evaluation.39 In addition to employing MCC for classifier optimization, we systematically under-sample non-catalytic residues and use different penalties for different false predictions. Additionally, our new methodology copes with class imbalance through a “probability threshold method”.40 Usually, the probability estimates (i.e., SVM continuous outputs) are thresholded halfway, at a threshold of 0.5, which is a perfect threshold for maximizing the classification accuracy for a balanced problem. Here, for the imbalanced data, we seek for a probability threshold that maximizes the expected MCC.
For performance evaluation, previous studies used MCC and reported either the results of a single test9 or those achieved using a cross validation experiment,5, 9, 32, 35, 36 which is a more robust approach.41 However, we noticed that even for cross-validation, the average MCC values are somewhat sensitive to the arbitrary partition of the dataset to training and test sets. Thus, we repeat our cross-validation experiment ten times with different partitions of the dataset and report averages and standard deviations. This protocol guarantees a high degree of replicability (i.e., consistency of results derived for random partitions of the data, as, for example, performed by different researchers).42
We evaluate the new prediction method using a benchmark dataset (dataset1 in Table I) that was specifically designed to imitate the most difficult structural genomics scenario. The dataset is non-redundant by both sequence and structure, thus SVM cannot learn any particular active site, only generic characteristics. Furthermore, the dataset does not include ligands bound to the active site, as such ligands might have sharpened structural clues.16 Thus, this dataset is, by design, harder than the ones used in previous studies. Nevertheless, our prediction results are comparable to, or better than, previously published ones, demonstrating the strength of our structural features and SVM methodology. In order to further investigate the strengths and weaknesses of our approach, we also provide three detailed case studies taken from the benchmark dataset.
Table I.
Summary of datasets used in this study.
| dataset1 (benchmark) | dataset2 | dataset3 | dataset4 | |
|---|---|---|---|---|
| Reference | Designed especially for this research | Petrova and Wu35 | Gutteridge et al.32 | Tong et al.9 |
| Annotations | CSA | CATRES | CATRES | CSA |
| # Enzymes | 34 | 79 | 159 | 64 |
| # Catalytic residues | 102 | 252 | 546 | 386 |
| # Non-catalytic residues | 9,546 | 23,548 | 57,614 | 32,977 |
There are minor discrepancies for the non-benchmark datasets between the counts of residues in this table and those reported in the original studies: Petrova and Wu reported 254 catalytic residues and 23,410 non-catalytic residues, Gutteridge et al. reported approximately 55,000 non-catalytic residues and 550 catalytic residues, and Tong et al. reported only 366 catalytic residues. We believe that these discrepancies are due to different preprocessing of the raw PDB files.
A fair and reliable comparison of our approach with other approaches to pure structure-based prediction of catalytic residues requires that we apply our approach to the same datasets on which the other approaches were applied. Thus, in addition to our benchmark results we also report here our prediction performance using three datasets taken from previous studies.9, 32, 35 This direct comparison indicates that for purely structure-based prediction our approach is superior to those of Gutteridge et al.32, and Petrova and Wu35, and comparable to that of Tong et al.9 The relative strengths and weaknesses of our approach and that of Tong et al.9, as well as prospective directions for further advances are discussed below.
Materials and Methods
Datasets
We tested our new methodology on four datasets (Table I). The enzyme structures of all four were extracted from the Protein Data Bank (PDB).43 Annotations of the benchmark dataset (dataset1) and dataset4 were taken from the Catalytic Site Atlas (CSA)44 (version 2.2.2). For the other two datasets, we followed the original studies and used the annotations from the catalytic residue dataset (CATRES).25
Our benchmark dataset was specifically designed to mimic the structural genomics scenario by excluding hetero-oligomers, catalytic site mutants, and structures with hetero-atoms less than 5Å from a catalytic residue. The remaining structures were further filtered for high resolution (up to 2.5Å) and non-redundancy (less than 30% pairwise-sequence identity and no common CATH fold). The final dataset includes 34 enzyme structures with 102 catalytic residues and 9,546 non-catalytic residues. Of these 34 structures, 8 were annotated according to literature references and 26 according to PSI-BLAST alignment with homologous literature entries. Two of these 26 structures (1agi and 1o0x) had E.C. (Enzyme Commission) numbers45 that differed slightly from those of the literature entries they were aligned with, suggesting that the transfer of annotation may be incorrect. However, these structures were included due to their unique CATH topology. Detailed residue information about this dataset (including PDB id, chain, number, type and raw residue feature values, and annotation) is available upon request.
The enzymes of dataset2, dataset3, and dataset4 were taken from the papers of Petrova and Wu,35 Gutteridge et al.,32 and Tong et al.,9 respectively. The datasets are available upon request.
Performance evaluation
Regarding the class of catalytic residues as positive and that of non-catalytic residues as negative, there are four possible outcomes to a binary (two-class) classifier: true positive (TP), false positive (FP) (type I error), true negative (TN), and false negative (FN) (type II error). For example, the outcome is FP when the classifier wrongly predicts a non-catalytic residue as a catalytic residue.
Using these four outcomes, the performance measures considered in this work are38:
Accuracy (ACC), ;
True positive rate (TPR) (sensitivity, recall), ;
False positive rate (FPR) (1-specificity), ;
Precision (PCR), ;
Filtration ratio (FR), ;
Matthews correlation coefficient (MCC); ; and
Area under the curve (AUC).
The range of the first five measures is between 0 and 1. The range of MCC37 values is between −1 and 1, where a value of −1 indicates total disagreement between the predicted and true classes, a value of 1 indicates a perfect prediction, and a value of 0 indicates a completely random prediction. This measure was also used in the studies of Gutteridge et al.,32 Petrova and Wu,35 Pugalenthi et al.,36 and Tong et al.9 for performance evaluation but not for classifier optimization. By employing the same measure for both classifier optimization and performance evaluation – as we do in this study – we guarantee that the classifier will be optimized to the task for which it is being evaluated, i.e., residue classification. AUC is the integral (area) under the receiver operator characteristic (ROC) curve46 that plots TPR as a function of FPR. The highest possible AUC value of 1 is achieved when the classifier obtains a TPR of 1 with FPR of 0, i.e., a perfect classifier. All the performance measures reported in this study are based on a ten-fold cross-validation (CV10) test.41 To increase the test replicability, we repeated it for 10 permutations of the dataset and report the average results over this 10×CV10 procedure.42
The SVM classifier
We developed a novel methodology (Figure 2) for training and optimizing an SVM classifier that aims to alleviate the class-imbalance problem that is detailed in the Introduction. In order to correctly balance the classifier decisions between the majority non-catalytic class and the minority catalytic class, our methodology is based on: 1) optimization of the SVM classifier and its parameters to maximize MCC, and thereby to balance all types of classification errors. This maximization relies on a validation set that is independent of the training set in order to avoid over-fitting; 2) under-sampling non-catalytic residues before SVM training by discarding the non-catalytic residues that are least likely to be misinterpreted as catalytic (those that Kubat and Matwin47 called “redundant”). Under-sampling is performed according to the amount ratio (AR) (Table II) between non-catalytic and catalytic residues providing the greatest value of MCC on a validation set. Values for this parameter are typically between 15 and 93.59 (e.g., see Figure 3A); 3) setting different penalties to erroneous identifications of catalytic residues and non-catalytic residues according to a penalty ratio (PR) (Table II) between these penalties. That is, we balance the enhanced contribution of the majority non-catalytic residue class to learning the SVM classifier by penalizing PR times more false negatives than false positives. Penalizing different SVM misclassifications differently was already applied to residue classification;9 however there, the ratio between the penalties was arbitrarily set. We selected the PR value that maximized SVM MCC measured on a validation set. Values for this parameter are typically between 15 and 20 (e.g., see Figure 3A); and 4) conversion of the probabilistic output of SVM to a binary decision using a probability-threshold (PT) parameter (Table II). SVM outputs that were higher or lower than PT indicated catalytic or non-catalytic residues, respectively. Therefore, PT has implications on the numbers and rates of prediction errors; a too high PT value will increase the rate of catalytic residues that will be missed (i.e., FN error) and a too low value will increase the rate of non-catalytic residues that will wrongly be identified as catalytic (i.e., FP error). Here again, the optimal value of PT was determined as the one maximizing MCC on a validation set. Values for this parameter are typically around 0.05 (e.g., see Figure 3B).
Figure 2. Nested CV10 experiment.

For concurrent parameter optimization and performance evaluation, a nested CV10 experiment is applied to the benchmark dataset. The five training steps (rectangles numbered 1–5) and the final test step (numbered 6) are repeated ten times with alternating test sets. Steps involving inner CV9 experiments are indicated by filled rectangles. Flow of information is indicated by arrows, the colors of which indicate the information type. A summary of the optimized parameters and techniques can be found in Table II.
Step 1. The benchmark dataset is divided into 10 equal-sized and disjoint protein subsets, one of which is set aside as a test set (red). The remaining nine are used for training (blue). Step 2. An exhaustive grid search for the optimal amount and penalty ratios (AR and PR, respectively; see text) is conducted on the training set using a CV9 experiment for every AR-PR grid point. This CV9 experiment uses one subset (orange) for validation and the remaining eight for training (blue). The optimal AR and PR are used in the subsequent steps (plum arrows). Step 3. A CV9 search for the optimal probability threshold (PT) to be used in the test step is performed (cyan arrow). Step 4. The optimal AR is used to balance the training set. Step 5. The resulting balanced training set is used to train SVM with the optimal PR. Step 6. The learned SVM model is evaluated on the test set (red arrow) using the optimal PT (cyan arrow).
Table II.
Techniques employed to cope with the class imbalance problem and the parameters that modulate them.
| Technique | Abbreviation | Description | Associated Parameter |
|---|---|---|---|
| Support vector machine | SVM | The standard binary classifier as described in Burges.33 It uses an unaltered training set, and errors on both classes are penalized the same. | - |
| Under-sampling | us | Reducing class imbalance in the training set by the removal of non-catalytic residues, such that the ratio between non-catalytic and catalytic residues in the training set is AR. | amount ratio (AR) |
| Differential-penalty SVM | dpSVM | The ratio between the penalty on catalytic residue misclassification and the penalty on non-catalytic residue misclassification, which are imposed during SVM-classifier training. | penalty ratio (PR) |
| Probability SVM | probSVM | An SVM classifier that compares the probability of a residue to be catalytic to a probability threshold (PT). | probability threshold (PT) |
| Under-sampling & differential-penalty SVM | us+dpSVM | Consecutive application of us and dpSVM. | AR & PR |
| Under-sampling & differential-penalty & probability SVM | prob+us+dpSVM | A us+dpSVM classifier that compares the probability of a residue to be catalytic to PT. | AR & PR & PT |
Figure 3. Optimization of amount ratio (AR), penalty ratio (PR), and probability threshold (PT) towards high MCC values.

A. MCC values that correspond to a wide range of (AR, PR) pairs (Step 2 in Figure 2). A confined area, 15 ≤ PR ≤ 20 and 15 ≤ AR ≤ 93.59 (dark brown), stands out as optimal with respect to MCC. B. MCC as a function of PT given optimal AR and PR values (Step 3 in Figure 2); the highest MCC value is 0.32 for a PT of 0.05. Error bars represent the standard deviations from the average values on the curve (shown intermittently to maintain clarity). All MCC values were obtained by averaging over the CV experiment (Figure 2).
Our methodology uses an SVM having a polynomial kernel of degree two and an error penalty33 of 55.71 that maximized MCC in a preliminary study. Moreover, before training the SVM classifier, we linearly scaled feature values to the [0,1] range to prevent the dominance of residue features with the largest numeric ranges.
Manipulations of atom properties
The classification problem that we target is defined at the residue level (i.e., catalytic vs. non-catalytic) and thus our classifier uses residue-level features. However, some of these features are based on atomic properties. Below we present our approach to the conversion of eleven atom properties (see Results section) to residue-level features. Two types of manipulations – spatial averaging and Z-score transformation – aid this conversion and considerably increase the power of the atom properties to distinguish between catalytic and non-catalytic residues.
In order to smooth out noise from sparse non-catalytic residues with catalytic-like properties, we apply spatial averaging at the atom level. For an atom property , we generate a family of averaged properties , where a and b are atoms in protein P, and dab is the Euclidian distance between atoms a and b. The parameter α ε {10−3,10−2,10−1,100,101} determines the width of the effective neighborhood of atom a, over which the property is averaged (i.e., the neighborhood expands as α decreases). Each value of α produces a different averaged atomic property, and consequently, different structural features of residues.
In order to minimize systematic biases due to protein size and crystallographic refinement, we linearly transform each atomic property to its Z-score,, where μ is the protein average value of the property and σ is its standard deviation.
Using these manipulations (applying, or not, Z-scoring with either of five neighborhood widths), we convert the basic properties to 110 atomic properties. Finally, in order to transform an atomic property to a residue feature, we assign each residue the greatest value among its atoms.
Residue features
Reasoning about structure-function relationships and the above manipulations suggest a set of 110 residue features that are based on atom properties. In addition, we consider two additional features that are inherently defined at the residue level, namely catalytic propensity25 and hydrophobicity score.25 We estimate the individual predictive power of each feature in two ways. First, we compare the distributions and median values of the feature for catalytic and non-catalytic residues. The distributions of structural features in the residue populations are far from normal. Thus, we use the one-sided Kolmogorov-Smirnov test and two-sided Wilcoxon rank-sum test to validate differences of distributions and medians, respectively. About half of the features indeed show statistically significant differences between their distributions and medians in catalytic and non-catalytic residues (p<0.05). Second, we generate a ROC curve46 for a linear classifier based on each feature and measure its AUC. The AUC values allow us to rank the significant features. The higher the value, the more discriminative is the feature.
The subset of features to be used by SVM is selected by a heuristic two-stage scheme. First, we manually cluster the residue features into thirteen groups, where catalytic propensity and hydrophobicity constitute singleton groups and each of the other eleven groups includes features that result from the same atomic property by different manipulations. Then, the most predictive residue feature (by AUC) from each group is chosen to represent the group. Second, a wrapper approach48 is applied to rank the SVM performance using each of the 213 − 1 = 8,191* feature subsets. For this analysis, we use the benchmark dataset with PR=20, AR=35, and PT=0.05 (Table II), which resulted in good SVM performance in our preliminary studies. A subset of six features (Table III) is selected for catalytic residue prediction although many other subsets of the original 112 features are almost as successful. The selected set is presented in the beginning of the Results section. For the complete list of features, the reasoning behind them, statistical evaluation of their predictive power, and details of the selection process see TableS1 and TableS2 in the Supplementary Material.
Table III.
Performances of the six residue features used by the SVM classifier. These features were selected, first as having the highest linear classifier AUC values within their groups and then as the optimal subset of the latter. The complete set of the features appears in the supplementary material (TableS4).
| Feature | p-Value KS | p-Value WR | Linear Classifier AUC |
|---|---|---|---|
| brl0.001010Z | 3.97E-32 | 0.00E+00 | 0.859 |
| CatalyticP | 3.03E-21 | 0.00E+00 | 0.789 |
| Hydrophobicity | 1.17E-14 | 3.65E-12 | 0.699 |
| Temperature0.01Z | 3.38E-08 | 5.65E-09 | 0.668 |
| Angle0.1Z | 4.66E-07 | 7.89E-08 | 0.654 |
| Solvation0.01Z | 1.83E-05 | 3.33E-06 | 0.633 |
p-values of the Kolmogorov-Smirnov (KS) and Wilcoxon rank-sum (WR) statistical comparison tests, along with values of AUC of linear classifiers discriminating residues, are reported for each of thirteen feature group representatives. The rows are sorted in descending order according to AUC. The KS test statistic can be found in TableS4 of the supplementary material.
Implementation
Protein structure analysis is performed using the MESHI software package.49 The SVM classifier is trained and tested using the MATLAB50 interface of the LIBSVM package.51
All the software developed specifically for this study is publicly available at http://www.cs.bgu.ac.il/~meshi/functionPrediction. This includes a new catalytic residue prediction web server called MESHIfun that is available at http://www.cs.bgu.ac.il/~meshisa/meshifun. The server is trained using our benchmark dataset and it implements the new method. We also provide the raw data that we use for training and testing the SVM classifier to encourage and support testing of other machine learning approaches to the problem.
Results
The structural features used by the SVM classifier
Our SVM uses six residue features that were selected from an initial set of 112 features (see Residue features section). Two of these six features – Hydrophobicity25 and CatalyticP25 – are veteran concepts in the field of catalytic residue prediction and represent some a priori, structure-independent expectation regarding the chemical nature of catalytic residues. The other four features are novel:
Angle0.1Z is a quadratic measure of angle distortion. The subscript indicates that the distortion values are averaged with an α value of 0.1 and Z-scored (see Manipulations of atom properties section). For catalytic residues, distortions from the ideal values are significantly (p<0.02) more common than for non-catalytic residues. However, due to the far larger number of non-catalytic residues, sparse non-catalytic residues with distorted angles dominate. Further, systematic biases occur due to different crystallographic refinement methods. Thus, the feature without averaging or Z-scoring has a low predictive power (AUC=0.57 of the linear classifier). Spatial averaging and Z-score transformation reduce these problems (Figure 4A), resulting in an AUC value of 0.65. Interestingly, the contribution of the two manipulations is not additive; the combined AUC improvement is greater than the sum of the two individual ones (TableS1). The cumulative distributions of the feature after averaging and Z-scoring for catalytic (blue) and non-catalytic (red) residues is presented in Figure 5A. The difference between the distributions is statistically significant (p<0.001 – lower than before the manipulations). We are not aware of any previous work that used a similar feature in the context of structure-based functional prediction.
Figure 4. Effect of spatial averaging and Z-scoring on the predictive power of four of the studied structural features.

Linear classifier ROC curves derived for four pairs of residue features: A. Angle0.1Z vs. Angle10; B. Solvation0.01Z vs. Solvation10; C. Temperature0.01Z vs. Temperature10; D. brl0.001010Z vs. brl0.001010. For all four pairs of features, the ROC curves of the manipulated versions (red) are considerably higher than the curves of their non-manipulated counterparts (blue). The diagonal (dashed line) relates to a random classification (a pure guess). Consequently, manipulation improves AUC values (see text).
Figure 5. Cumulative distributions of four of the studied structural features for catalytic and non-catalytic residues.

Each subplot depicts the difference between the empirical cumulative distribution function (ECDF) of the catalytic residues (blue) and the ECDF of the non-catalytic residues (red) for four studied residue features. The maximal distance between each pair of curves corresponds to the Kolmogorov-Smirnov statistic (see Table III for p-values).
Having a predictive power for this feature is somewhat surprising. The geometry of molecules is determined by the strong interactions between covalently bonded atoms and thus one may expect that all angles would be at their ideal values, up to an experimental error. We see no chemical reason for distortions for either catalytic or non-catalytic residues. Further, the distortions that we observe are smaller than the crystallographic experimental error. We present a speculative explanation of this paradox in the Discussion.
Solvation0.01Z is based on an implicit solvent energy term, which is derived from a non-linear function of the number of carbons in the atom neighborhood (an indicator of burial) and the number of hydrogen bonds that the atom makes. High energies are more common in catalytic residues than in non-catalytic ones, as many of the catalytic residues are polar and pay an energetic price for being occluded in the active site. This difference, however, is not statistically significant. Here again spatial averaging (α = 0.01) and Z-scoring considerably improved the predictive power of this term, resulting in a significant (p<0.02) difference between the ROC curves (Figure 4B) and the distributions (Figure 5B) of catalytic and non-catalytic residues, and an AUC value of 0.63. We are not aware of previous studies that used such a term for functional residue prediction, but it is clearly related to the solvent accessibility feature that is often used.5, 32, 35
Temperature0.01Z is based on the inverse of the atom temperature factor. The significant (p<0.01) difference between its distributions for catalytic and non-catalytic residues (Figure 5C) is consistent with a previous study25 that reported low temperature factors for catalytic residues. The authors of that study suggested that low values of the temperature factor of catalytic residues represent low internal entropy, which is important for the catalytic role. In our study, spatial averaging and Z-score transformation improve performance (Figure 4C), increasing the AUC value from 0.58 to 0.67.
brl0.001010Z is based on atom burial as measured by the number of the atom neighbors, where neighbor contribution is weighted by a Gaussian function of the distance. Catalytic residues are typically buried, but the protein size biases this term; in large proteins the average number of neighbors of all residues is greater than in smaller proteins. This bias renders the number of neighbors non-predictive. Z-score transformation solves this problem (Figure 4D), resulting in a significant (p<10−32) difference in distributions (Figure 5D) and a high AUC value of 0.86. In this case, spatial averaging does not provide any improvement to predictive power, as neighboring atoms always have very similar values. For consistency, though, we do perform spatial averaging but with an α value of 10, which practically amounts to no averaging. This term is apparently related to observations that catalytic residues tend to be close to the protein center of mass. The current formalism however is novel.
SVM performance
We use an SVM classifier to discriminate catalytic from non-catalytic residues using the above six features. Figure 6 assesses the added value of SVM to our methodology by comparing its performance on the benchmark dataset to those of linear classifiers that are based on the individual features. The three plots – ROC (Figure 6A), precision-recall (Figure 6B), and MCC versus probability threshold (Figure 6C) – indicate an advantage of SVM over the linear classifiers with respect to all performance measures. The ROC curve of SVM is higher than those of the linear classifiers (Figure 6A), resulting in an AUC value (0.892) that is above those of the linear classifiers (0.64–0.86). The precision-recall plots (Figure 6B) show a far larger difference in favor of SVM. The precision values of the linear classifiers are relatively low (at best around 0.1) and almost indifferent to the recall value, whereas that of SVM peaks to more than 0.3. Finally and most importantly, since we focus on optimizing MCC, Figure 6C shows that SVM reaches an MCC value above 0.3, where the linear classifier peaks are below 0.2.
Figure 6. Performance of the SVM classifier on the benchmark dataset.

A. ROC curve for the SVM classifier (brown) compared to those of linear classifiers for the six individual features. Each point on the SVM curve is an average of ten CV10 experiments and corresponds to a specific probability threshold (PT). Error bars represent the standard deviations from the averages (not shown for all points in order to maintain clarity). B. Corresponding precision-recall curves are generated in the same way as the ROC curves for the above classifiers. C. MCC for SVM as a function of the probability threshold and for the linear classifiers as a function of feature thresholds. The highest MCC for the SVM is 0.32 for a threshold of 0.05. For both B and C, error bars are as in A.
Table IV presents performance measure values of SVM using the classifier configuration and parameter settings that maximize MCC on the benchmark dataset. The performances of our new catalytic residue predictor, compared to those reported by Petrova and Wu,35 Gutteridge et al.,32 and Tong et al.,9 are shown in Table V, Table VI and Table VII, respectively. In terms of MCC, our predictor is superior to the pure-structure predictors presented by Petrova and Wu and Gutteridge et al., and on a par with the THEMATICS method presented by Tong et al. It should be noted, though, that THEMATICS is inherently restricted to ionizable residues. Thus, while the overall performances of the two methods are similar, they differ remarkably in their actual predictions.
Table IV.
Average performance measures (with standard deviations in parentheses; see Methods for measure definitions) of the SVM classifier on the benchmark dataset.
| Performance Measure | Performance Value |
|---|---|
| MCC | 0.305 (±0.013) |
| TPR | 0.619 (±0.035) |
| PRC | 0.172 (±0.008) |
| ACC (%) | 98.77 (±0.049) |
| AUC | 0.892 (±0.007) |
| FPR | 0.036 (±0.002) |
| FR (%) | 4.3 (±0.2) |
The reported measure values are averages over ten experiments when using the optimal AR, PR, and PT values in each CV fold. The relatively small standard deviations demonstrate the robustness of our method.
Table V.
| Our Method | P&W – S | Our Method | P&W – S+E | |
|---|---|---|---|---|
| The ratio between catalytic and non-catalytic residues in the test set | 1:1 | 1:1 | 1:92 | 1:92 |
| MCC | 0.686 (±0.02) | 0.515 (±0.028) | 0.27 (±0.012) | 0.23 |
| TPR | 0.842 (±0.018) | – | 0.43 (±0.053) | 0.9 |
| PRC | 0.845 (±0.02) | – | 0.198 (±0.026) | – |
| AUC | 0.922 (±0.007) | – | 0.897 (±0.003) | – |
| ACC (%) | 84.43 (±0.96) | 75.55 (±1.382) | 98.9 (±0.018) | 86.96 |
| FPR | 0.164 (±0.027) | – | 0.022 (±0.006) | 0.13 |
| FR (%) | 50.3 (±2) | – | 2.6 (±0.6) | – |
Similarly to Petrova & Wu, we present our performances on both balanced (1:1 ratio of catalytic to non-catalytic residues) and imbalanced (1:92 ratio) test sets. For our method, we report the average performance (with standard deviations in parentheses) of the SVM classifier using a 10×CV10 experiment. The results of Petrova and Wu were taken from their paper. Performance measures that are not explicitly reported by the authors are represented by `–'. S represents the use of only structural features. S+E represents the use of both structural and evolutionary data.
Table VI.
| Our Method | GB–S | GA–S | GB–S+E | GA–S+E | |
|---|---|---|---|---|---|
| MCC | 0.272 (±0.004) | 0.19 | 0.23 | 0.28 | 0.32 |
| TPR | 0.565 (±0.038) | 0.41 | 0.57 | 0.56 | 0.68 |
| PRC | 0.146 (±0.01) | 0.1 | 0.1 | 0.14 | 0.16 |
| AUC | 0.901 (±0.003) | – | – | – | – |
| ACC (%) | 99.04 (±0.008) | – | – | – | – |
| FPR | 0.032 (±0.003) | – | – | – | – |
| FR (%) | 3.7 (±0.375) | – | – | – | – |
For our method, we report the average performance (with standard deviation in parentheses) of the SVM classifier using a 10×CV10 experiment. Performance measures that are not explicitly reported by Gutteridge et al. are represented by `–'. GB and GA represent the results before and after spatial clustering, respectively. We did not perform spatial averaging. S represents the use of only structural features (excluding conservation and DOPS scores). S+E represents the use of both structural and evolutionary data.
Table VII.
| Our Method | Tong et al. | |
|---|---|---|
| MCC | 0.31 (±0.18) | 0.31 |
| TPR | 0.57(±0.35) | 0.61 |
| PRC | 0.22(±0.21) | 0.2 |
| ACC (%) | 96(±2) | – |
| FPR | 0.029(±0.18) | 0.031 |
| FR (%) | 3.4(±1.8) | 3.8 |
Both methods are purely structural. Our method was evaluated by training and testing on the same protein sets used by Tong et al. We report averages and standard deviations over the test set. Performance measures that are not explicitly reported by Tong et al. are represented by `–'. Detailed predictions of dataset4 can be found in TableS10 in the Supplementary Material.
Discussion
The wealth of sequence and structural data poured by the recent genomics and structural genomics initiatives impose an unprecedented challenge to the biological community, to characterize as many newly discovered proteins as possible. High throughput computational methods can provide initial working hypotheses regarding the identity of functional residues. While these hypotheses may include many flaws they can still serve as starting points for systematic studies that combine experiments with time consuming accurate computational methods.61
Not with standing the strength of other current approaches to the problem of catalytic residue identification, purely structural methods are essential for ORFan structural genomics targets,55 for which even conservation patterns cannot be derived. The number of such proteins is not negligible. Within the first 687 Mycoplasma pneumoniae proteins studied by the Berkley Structural Genomics Center, 54 (7.9%) are ORFans.10 It is hard at this stage to estimate how representative this fraction is for other centers, and if and how it will change over time. Still, considering the substantial efforts involved in the structural genomics initiative, we believe that even lower numbers justify the development of methods that would maximize the functional insight gained from this endeavor.
This work presents a purely structural approach to the prediction of catalytic residues. Further, as our approach does not rely on similarity to annotate enzyme structures, it may be able to identify novel types of active sites. The structural features that we use encapsulate much of the common knowledge that has accumulated over the years about catalytic residues. Specifically, we use the observations that catalytic residues tend to have unusual structural characteristics,16, 27–29, 31 to be spatially clustered,32 and to be centrally positioned within the protein structure.11, 12
The current study extends previous ones in several directions. First, it introduces structural clues that were not previously used in this context (e.g., solvation and angle propensity). Second, the study enhances predictive power by spatial averaging and/or Z-score transformation. Third, to the best of our knowledge, our work is the first that attempts to mimic the structural genomics scenario by excluding from the test set any structure in which the catalytic residues are close to a ligand or mutated. Finally, the current study identifies class imbalance as crucial to accurate and practical catalytic-residue prediction. Thus, it proposes a novel methodology based on the MCC performance measure to integratively 1) optimize the SVM classifier, 2) under-sample non-catalytic residues, 3) set different penalties for erroneous identifications of catalytic and non-catalytic residues, and 4) control the threshold used in classification.
By using this methodology and selecting all parameters to maximize MCC instead of classification accuracy, the SVM classifier proves to be highly successful on four distinct datasets of residues. A direct comparison of the performance of our method to three state-of-the-art structure-based prediction methods shows that our method performs, in terms of MCC, better than two of the methods (Table V and Table VI) and on a par with the third one (Table VII). Further, our results are at least comparable to these methods when they are augmented by evolutionary data.
The study of Tong et al.9 is purely structural and based on perturbed theoretical titration curves using a state-of-the-art electrostatic model. This method is very accurate in identifying ionizable catalytic residues. Although the performance of this method (in terms of MCC) is the same as ours, when averaged over all residue types, it clearly highlights a possible route of improvement to our current group of features. In the initial phase of our study, we tested features that are based on a simple Coulomb electrostatic model. These features had low predictive power and were not selected by the SVM wrapper. One may expect that adopting the electrostatic approach of Tong et al. is a promising way to improve our results.
This study, as its predecessors, focuses on the identification of catalytic residues given that the studied protein is an enzyme. This is a somewhat artificial question as often even this bit of information is unknown for structural genomics targets. In a preliminary study, we have tried to perform the same analysis with proteins, such as immunoglobulin, that are not expected to have any enzymatic activity. Qualitatively these proteins were undistinguishable from enzymes in terms of the number of residues predicted as catalytic. Thus, apparently the discrimination between enzymes and non-enzymes cannot be a simple outcome of the current study and requires different classifiers.
Conclusion
The new approach of catalytic residue identification presented here, combines novel residue features with a novel machine-learning methodology. The features manifest new structural clues for a catalytic role of residues. The machine-learning methodology copes with the notorious class imbalance problem. We demonstrated the utility of this approach using a benchmark that was designed specifically to mimic the most difficult structural genomics scenario. In addition, we tested our approach using benchmarks from the literature. The results obtained using these benchmarks suggest that our approach is a promising alternative to the current methods. Detailed case studies we explored suggest that this approach may also prove handy for binding site prediction.
Supplementary Material
This table contains results of the statistical significance tests as well as the AUC and MCC measures for the linear classifiers derived from each residue feature mentioned in the paper. All values were calculated using dataset1. Each row corresponds to a residue feature and the columns are explained below the table in the file itself.
AUC - Area Under the ROC Curve, MCC - Mathews Correlation Coefficient.
This table lists the performance measures achieved in a CV3 experiment by using different feature subsets. Each row corresponds to a feature subset and the columns represent the different performance measures. The rows are sorted according to the value of (TPR + 2*MCC + PRC)/4. Thus, the top row represents the selected feature subset.
CV3 - 3-Fold Cross Validation, TPR - True Positive Rate, MCC - Mathews Correlation Coefficient, PRC - Precision.
Acknowledgements
We would like to thank El-ad David Amir for the helpful discussion and insightful comments. The authors are grateful to Ms. Rise Silverman and Ms. Edna Oxman for carefully editing the manuscript. Funding: Israel Science Foundation (grant no. 289/06); National Institute of Health (award no. 1R01 GM081712-01).
Footnotes
Within a subset each feature may or may not appear. The empty subset was not tested.
References
- 1.Jones S, Thornton JM. Searching for functional sites in protein structures. Curr Opin Chem Biol. 2004;8:3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Kinoshita K, Nakamura H. Protein informatics towards function identification. Curr Opin Struct Biol. 2003;13:396–400. doi: 10.1016/s0959-440x(03)00074-5. [DOI] [PubMed] [Google Scholar]
- 3.Todd AE, Orengo CA, Thornton JM. Evolution of protein function, from a structural perspective. Curr Opin Chem Biol. 1999;3:548–556. doi: 10.1016/s1367-5931(99)00007-1. [DOI] [PubMed] [Google Scholar]
- 4.Yin Y, Fischer D. Identification and investigation of ORFans in the viral world. BMC Genomics. 2008;9:24. doi: 10.1186/1471-2164-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youn E, Peters B, Radivojac P, Mooney SD. Evaluation of features for catalytic residue prediction in novel folds. Protein Sci. 2007;16:216–226. doi: 10.1110/ps.062523907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng G, Qian B, Samudrala R, Baker D. Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design. Nucleic Acids Res. 2005;33:5861–7. doi: 10.1093/nar/gki894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ota M, Kinoshita K, Nishikawa K. Prediction of catalytic residues in enzymes based on known tertiary structure, stability profile, and sequence conservation. J Mol Biol. 2003;327:1053–64. doi: 10.1016/s0022-2836(03)00207-9. [DOI] [PubMed] [Google Scholar]
- 8.Burley SK, Almo SC, Bonanno JB, Capel M, Chance MR, Gaasterland T, Lin D, Sali A, Studier WF, Swaminathan S. Structural genomics: beyond the human genome project. Nat Genet. 1999;23:151–7. doi: 10.1038/13783. [DOI] [PubMed] [Google Scholar]
- 9.Tong W, Williams RJ, Wei Y, Murga LF, Ko J, Ondrechen MJ. Enhanced performance in prediction of protein active sites with THEMATICS and support vector machines. Protein Sci. 2008;17:333–341. doi: 10.1110/ps.073213608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandonia JM, Kim SH, Brenner SE. Target selection and deselection at the Berkeley Structural Genomics Center. Proteins. 2006;62:356–370. doi: 10.1002/prot.20674. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Shimon A, Eisenstein M. Looking at enzymes from the inside out: the proximity of catalytic residues to the molecular centroid can be used for detection of active sites and enzyme-ligand interfaces. J Mol Biol. 2005;351:309–26. doi: 10.1016/j.jmb.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 12.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–46. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 13.Warshel A. Energetics of enzyme catalysis. Proc Natl Acad Sci U S A. 1978;75:5250–4. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warshel A, Sussman F, Hwang JK. Evaluation of catalytic free energies in genetically modified proteins. J Mol Biol. 1988;201:139–59. doi: 10.1016/0022-2836(88)90445-7. [DOI] [PubMed] [Google Scholar]
- 15.Herzberg O, Moult J. Analysis of the steric strain in the polypeptide backbone of protein molecules. Proteins. 1991;11:223–9. doi: 10.1002/prot.340110307. [DOI] [PubMed] [Google Scholar]
- 16.Heringa J, Argos P. Strain in protein structures as viewed through nonrotameric side chains: II. effects upon ligand binding. Proteins. 1999;37:44–55. [PubMed] [Google Scholar]
- 17.Beadle BM, Shoichet BK. Structural bases of stability-function tradeoffs in enzymes. J Mol Biol. 2002;321:285–96. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 18.Gleason FK. Mutation of conserved residues in Escherichia coli thioredoxin: effects on stability and function. Protein Sci. 1992;1:609–16. doi: 10.1002/pro.5560010507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hibler DW, Stolowich NJ, Reynolds MA, Gerlt JA, Wilde JA, Bolton PH. Site-directed mutants of staphylococcal nuclease. Detection and localization by 1H NMR spectroscopy of conformational changes accompanying substitutions for glutamic acid-43. Biochemistry. 1987;26:6278–86. doi: 10.1021/bi00393a048. [DOI] [PubMed] [Google Scholar]
- 20.Meiering EM, Serrano L, Fersht AR. Effect of active site residues in barnase on activity and stability. J Mol Biol. 1992;225:585–9. doi: 10.1016/0022-2836(92)90387-y. [DOI] [PubMed] [Google Scholar]
- 21.Pakula AA, Sauer RT. Amino acid substitutions that increase the thermal stability of the lambda Cro protein. Proteins. 1989;5:202–10. doi: 10.1002/prot.340050303. [DOI] [PubMed] [Google Scholar]
- 22.Schreiber G, Buckle AM, Fersht AR. Stability and function: two constraints in the evolution of barstar and other proteins. Structure. 1994;2:945–51. doi: 10.1016/s0969-2126(94)00096-4. [DOI] [PubMed] [Google Scholar]
- 23.Shoichet BK, Baase WA, Kuroki R, Matthews BW. A relationship between protein stability and protein function. Proc Natl Acad Sci U S A. 1995;92:452–6. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. Localizing frustration in native proteins and protein assemblies. Proc Natl Acad Sci U S A. 2007;104:19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–21. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 26.Wei Y, Ko J, Murga LF, Ondrechen MJ. Selective prediction of interaction sites in protein structures with THEMATICS. BMC Bioinformatics. 2007;8:119. doi: 10.1186/1471-2105-8-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bate P, Warwicker J. Enzyme/non-enzyme discrimination and prediction of enzyme active site location using charge-based methods. J Mol Biol. 2004;340:263–76. doi: 10.1016/j.jmb.2004.04.070. [DOI] [PubMed] [Google Scholar]
- 28.Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol. 2001;312:885–96. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- 29.Ondrechen MJ, Clifton JG, Ringe D. THEMATICS: a simple computational predictor of enzyme function from structure. Proc Natl Acad Sci U S A. 2001;98:12473–8. doi: 10.1073/pnas.211436698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dessailly BH, Lensink MF, Wodak SJ. Relating destabilizing regions to known functional sites in proteins. BMC Bioinformatics. 2007;8:141. doi: 10.1186/1471-2105-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petock JM, Torshin IY, Weber IT, Harrison RW. Analysis of protein structures reveals regions of rare backbone conformation at functional sites. Proteins. 2003;53:872–9. doi: 10.1002/prot.10484. [DOI] [PubMed] [Google Scholar]
- 32.Gutteridge A, Bartlett G, J., Thornton JM. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J Mol Biol. 2003;330:719–34. doi: 10.1016/s0022-2836(03)00515-1. [DOI] [PubMed] [Google Scholar]
- 33.Burges CJC. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min Knowl Discov. 1998;2:121–167. [Google Scholar]
- 34.Vapnik VN. The Nature of Statistical Learning Theory. Springer-Verlag New York, Inc.; New York, NY, USA: 1995. p. 188. [Google Scholar]
- 35.Petrova NV, Wu CH. Prediction of catalytic residues using Support Vector Machine with selected protein sequence and structural properties. BMC Bioinformatics. 2006;7:312. doi: 10.1186/1471-2105-7-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pugalenthi G, Kumar KK, Suganthan PN, Gangal R. Identification of catalytic residues from protein structure using support vector machine with sequence and structural features. Biochem Biophys Res Commun. 2008;367:630–634. doi: 10.1016/j.bbrc.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 37.Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 38.Baldi P, Brunak S, Chauvin Y, Andersen CAF, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 39.Bishop CM. Neural Networks for Pattern Recognition. Oxford University Press; USA: 1995. [Google Scholar]
- 40.David A, Lerner B. Support vector machine-based image classification for genetic syndrome diagnosis. Pattern Recog Lett. 2005;26:1029–1038. [Google Scholar]
- 41.Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. 1995;14:1137–1145. [Google Scholar]
- 42.Bouckaert RR, Frank E. Evaluating the replicability of significance tests for comparing learning algorithms. Advances in Knowledge Discovery and Data Mining. 2004;3:12. [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webb EC. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. Vol. 339. Academic Press Inc; Toronto: 1992. Enzyme Nomenclature 1992; p. 351. [Google Scholar]
- 46.Fawcett T. An introduction to ROC analysis. Pattern Recog Lett. 2006;27:861–874. [Google Scholar]
- 47.Kubat M, Matwin S. Addressing the curse of imbalanced training sets: one-sided selection. Fourteenth International Conference on Machine Learning.1997. p. 186. [Google Scholar]
- 48.Kohavi R, John GH. Wrappers for Feature Subset Selection. Artif Intell. 1997;97:273–324. [Google Scholar]
- 49.Kalisman N, Levi A, Maximova T, Reshef D, Zafriri-Lynn S, Gleyzer Y, Keasar C. MESHI: a new library of Java classes for molecular modeling. Bioinformatics. 2005;21:3931–2. doi: 10.1093/bioinformatics/bti630. [DOI] [PubMed] [Google Scholar]
- 50.The MathWorks, Inc. Matlab. 7.0.0.19901. Natick, MA: 1984–2007. p. R14. [Google Scholar]
- 51.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. 2001 Software available at http://www.csie.ntu.edu.tw/cjlin/libsvm 2001.
- 52.Segelke BW, Nguyen D, Chee R, Xuong NH, Dennis EA. Structures of two novel crystal forms of Naja naja naja phospholipase A2 lacking Ca2+ reveal trimeric packing. J Mol Biol. 1998;279:223–232. doi: 10.1006/jmbi.1998.1759. [DOI] [PubMed] [Google Scholar]
- 53.Sugiura I, Nureki O, Ugaji-Yoshikawa Y, Kuwabara S, Shimada A, Tateno M, Lorber B, Giege R, Moras D, Yokoyama S, Konno M. The 2.0 A crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure. 2000;8:197–208. doi: 10.1016/s0969-2126(00)00095-2. [DOI] [PubMed] [Google Scholar]
- 54.Hofmann B, Tolzer S, Pelletier I, Altenbuchner J, van Pee KH, Hecht HJ. Structural investigation of the cofactor-free chloroperoxidases. J Mol Biol. 1998;279:889–900. doi: 10.1006/jmbi.1998.1802. [DOI] [PubMed] [Google Scholar]
- 55.Fischer D, Eisenberg D. Finding families for genomic ORFans. Bioinformatics. 1999;15:759–62. doi: 10.1093/bioinformatics/15.9.759. [DOI] [PubMed] [Google Scholar]
- 56.Lopez G, Valencia A, Tress M. FireDB--a database of functionally important residues from proteins of known structure. Nucleic Acids Res. 2007;35:D219–23. doi: 10.1093/nar/gkl897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holliday GL, Bartlett GJ, Almonacid DE, O'Boyle NM, Murray-Rust P, Thornton JM, Mitchell JB. MACiE: a database of enzyme reaction mechanisms. Bioinformatics. 2005;21:4315–4316. doi: 10.1093/bioinformatics/bti693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagano N. EzCatDB: the Enzyme Catalytic-mechanism Database. Nucleic Acids Res. 2005;33:D407–D412. doi: 10.1093/nar/gki080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fremont DH, Anderson DH, Wilson IA, Dennis EA, Xuong NH. Crystal structure of phospholipase A2 from Indian cobra reveals a trimeric association. Proc Natl Acad Sci U S A. 1993;90:342–346. doi: 10.1073/pnas.90.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakanishi K, Ogiso Y, Nakama T, Fukai S, Nureki O. Crystal structure of Aquifex aeolicus methionyl-tRNA synthetase complexed with tRNA(Met) and methionyl-adenylate anologue [Google Scholar]
- 61.Olsson MHM, Parson Warshel A. Dynamical Contributions to Enzyme Catalysis: Critical Tests of A Popular Hypothesis. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table contains results of the statistical significance tests as well as the AUC and MCC measures for the linear classifiers derived from each residue feature mentioned in the paper. All values were calculated using dataset1. Each row corresponds to a residue feature and the columns are explained below the table in the file itself.
AUC - Area Under the ROC Curve, MCC - Mathews Correlation Coefficient.
This table lists the performance measures achieved in a CV3 experiment by using different feature subsets. Each row corresponds to a feature subset and the columns represent the different performance measures. The rows are sorted according to the value of (TPR + 2*MCC + PRC)/4. Thus, the top row represents the selected feature subset.
CV3 - 3-Fold Cross Validation, TPR - True Positive Rate, MCC - Mathews Correlation Coefficient, PRC - Precision.