

Abstract
The chemokine CXCL10 is crucial for the control of viral replication through the regulation of mobilization of antigen-specific T cells to sites of infection. CXCL10 is highly expressed both at sites of inflammation as well as constitutively within lymphoid organs by both bone marrow (BM)-derived and non-BM-derived cells. However, the relative immunologic importance of CXCL10 expressed by these divergent sources relative to HSV-1 infection is unknown. Using mouse chimeras reconstituted with either wild type or CXCL10 deficient mouse BM, we show BM-derived, radiation-sensitive cells from wild type mice were solely responsible for resistance to HSV-1 in the trigeminal ganglia and brain stem. The resistance was not reflected by a deficiency in the recruitment of effector cells to sites of inflammation or expression of chemokines or IFN-gamma and likely results from additional, yet-to-be-determined factors emanating from wild type, BM-derived cells.
1. Introduction
Chemokines are a family of small, structurally related proteins which play a central role in the regulation of leukocyte homing. They can be viewed broadly in terms of being either constitutively expressed or inflammatory chemokines. During steady state conditions constitutively expressed chemokines such as the CCR7 ligands CCL19 and CCL21 or the CXCR5 ligand CXCL13, direct the migration of leukocyte subsets to specific spatial compartments within lymphoid organs (Förster, et al., 1999; Stein et al., 2000; Ansel et al., 2002). As a consequence appropriate positioning of individual cell populations for rapid yet controlled immune responses is maintained (Rossi and Zlotnik, 2000; Zlotnik and Yoshie, 2000). In contrast, inflammatory chemokines must regulate the rapid initiation of immune responses at sites of inflammation by co-coordinating the selective recruitment of appropriate leukocytes (Rossi and Zlotnik, 2000). This requires selective up-regulation of chemokine expression at inflammatory sites as well as binding of the chemokine to the extracellular matrix via heparin binding motif/GAG interactions. The localized expression of inflammatory chemokine and chemokine/GAG binding establish a concentration gradient that directs leukocytes towards the chemokine source (Rossi and Zlotnik, 2000; Wuest and Carr, 2008a).
Recent studies have cast doubts as to whether the primary role of inflammatory chemokines is to simply recruit leukocytes towards inflammatory sites where their expression is up-regulated. For example, findings suggest the primary role of the inflammatory chemokine CCL7 towards monocyte mobilization is to drive monocyte egress out of the bone marrow (Tsou et al., 2007). Thus, CCL7 expression at inflammatory sites appears secondary to the CCL7/CCR2-dependent mobilization of monocytes from the bone marrow to the blood stream. In another study, CXCL8 mutants with reduced affinity for extracellular matrix affinity exhibit increased neutrophil chemoattractive properties despite unaltered chemokine receptor affinity (Tanino et al., 2010). This paradox of increased activity despite presumably attenuated ability to establish local concentration gradients correlated with increased serum concentrations of increasingly active CXCL8 mutants suggesting the possibility of a role for CXCL8 in mobilization of PMNs at sites distant from inflammatory loci. However, leukocytes are not simply the recipients of chemokine cues. Rather, many chemokines are highly expressed by cells of the immune system, and leukocyte-derived chemokine expression is a crucial mechanism by which inflammatory first responders, such as PMNs, drive the influx of adaptive effector cells towards inflammatory loci (Tamassia et al., 2007).
Viral and bacterial infections elicit distinct and differing chemoinflammatory signals. The chemokine CXCL10, in particular, plays a major role in the control of viral replication as it is highly up-regulated by type I and II interferon and strongly and specifically promotes the chemotaxis of NK, CD4+, and CD8+ T cells (Trifilo et al., 2004; Klein et al., 2005; Hsieh et al., 2006). CXCL10 is expressed by cells of both hematopoietic and non-hematopoietic origin (Yoneyama et al, 2002; Drennan et al., 2009). Further, CXCL10 is expressed not only at sites of inflammation but also constitutively within lymphoid organs (Drennan et al., 2009). The complex expression pattern of CXCL10 in lymphoid and non-lymphoid tissues, by both hematopoietic and non-hematopoietic-derived cells precludes simple interpretation of the site-and source-specific importance of CXCL10 for control of pathogen replication.
We sought to explore the issue of relative importance of hematopoietic versus non-hematopoietic expression of CXCL10 using herpes simplex virus-1 (HSV-1) infection as a model. HSV-1 infection highly up-regulates CXCL10 expression, and CXCL10 is required for control of HSV-1 replication through the mobilization of NK and HSV-1 specific CD8+ T cells (Wuest and Carr, 2008b). Using bone-marrow (BM) chimeric animals constructed from lethally irradiated animals receiving BM from either wild-type C57BL/6 (WT) or CXCL10 deficient (CXCL10 −/−) mice, we determined genetic deficiency of CXCL10 within the hematopoietic compartment did not impact CXCL10 expression within the brain stems (BS) or trigeminal ganglia (TG) of infected animals. Further, we found BM-derived CXCL10 expression was not required for the recruitment of HSV-1 specific CD8+ T cells. However, we did identify BM-derived CXCL10 expression was necessary to control of HSV-1 replication in the BS and TG. Thus, we conclude that in addition to CXCL10 expression by BM-derived cells, additional factors are expressed that ultimately regulate immune effector cells and anti-viral pathways that control the local spread and replication of HSV-1 in the nervous system of mice.
2. Materials and Methods
2.1 Mice
Animal treatment was consistent with the National Institutes of Health Guidelines on the Care and Use of Laboratory Animals. All procedures were approved by the University of Oklahoma Health Sciences Center and Dean A. McGee Eye Institute Institutional Animal Care and Use Committee. WT C57BL/6 mice were obtained from The Jackson Laboratory. CXCL10−/− mice were generated as previously described (Dufour et al., 2002) and backcrossed to the C57BL/6 background for a minimum of nine generations. Mice were between the ages of 6 weeks and 6 months of age and all mice used in the study were male.
2.2 Bone Marrow Chimeras
Chimeras were generated by lethal irradiation (6.5 Gy/650 RADS/exposure, 2x over 4 hrs) of congenic (CD45.1) C57BL/6 wild type (WT) mice followed by transfer of 1×106 BM cells from CXCL10−/− or WT mice. Mice were allowed a 12 week period to establish the chimera, and chimerism was verified by flow cytometry of peripheral blood leukocytes and after infection with HSV-1 by flow cytometry of cells in the TG and BS of infected animals.
2.3 Virus and Cells
HSV-1 strain McKrae was originally obtained from B. Gebhardt (Louisiana State University Health Sciences Center, New Orleans, LA) and was propagated on Vero cells. Virus stock was maintained at −80°C at a concentration of 1 × 109 PFU/ml, then diluted to 3.33 × 102 PFU/μl immediately before use. Vero cells were maintained in RPMI 1640 medium supplemented with 10% FBS, gentamicin, and antibiotic-antimycotic solution (Invitrogen Life Technologies) at 37°C, 5% CO2, and 95%humidity.
2.4 HSV-1 infection and viral titers
Mice were anaesthetized with ketamine/xylazine followed by corneal scarification with a 25-gaugeneedle. Tear films were removed using a chemwipe, and 1000 PFU of HSV-1 was applied to the cornea. At indicated times post infection (PI), mice were anesthetized and exsanguinated and tissues were harvested. Viral titers within tissues were determined by homogenizing the tissue in RPMI 1640 and clarifying the supernatant (12,000x g, 2 min), then assaying the supernatant by plaque assay on Vero cells.
2.5 Flow cytometry
Antibodies used in this study were sourced as follows; anti-mouse CD3, CD4, CD8, CD45.1, CD45.2, and NK1.1 conjugated with either FITC or PE were purchased from BD Biosciences and CD8+ T cells specific for the HSV-1 glycoprotein B (gB)-derived epitope SSIEFARL(Hanke et al., 1991; Cose et al., 1995) were enumerated using MHC class I tetramer labeled with PE (Baylor University College of Medicine). To analyze leukocytes within trigeminal ganglia (TG) and brain stem (BS), tissues were disrupted with a Wheatley Dounce homogenizer (Fisher Scientific) in RPMI1640 and the suspension was passed through a 40-μm nylon cell strainer (Fisher Scientific). Lymph node cells were extruded by passing cells through a 40-μm cell strainer. Samples were blocked with 2 μl of FcBlock anti-mouse CD16/32 (BD Biosciences) and incubated for 15 minute on ice followed by the addition of 2 μl of rat serum (Jackson ImmunoResearch Laboratories). After another 15 minute incubation on ice, 2 μl of the indicated labeled Abs or 0.5 μl of H-2Kb tetramer reagent, as well as PE-Cy5 rat anti-mouse CD45 (BD Biosciences) was added and incubated. Samples from BS and TG were washed three times with 1.0% BSA in 1x PBS. The single-cell suspensions were fixed overnight in 1.0% paraformaldehyde, then resuspended in 1.0% BSA in PBS for subsequent analysis by flow cytometry. Events were gated by forward and side scatter, as well as by high expression of CD45 Ag to distinguish events from resident CD45low microglia. PanCD45 expression was determined using PE-Cy5-labeled anti-mouseCD45 which we found to be of sufficient intensity and Stokes shift to allow discrimination of labeled cells from auto fluorescent CNS tissue. To determine the absolute counts of leukocytes in the BS and TG, suspensions of these tissues included Countbright fluorescent counting beads (Invitrogen Life Technologies), and the absolute count of leukocytes was extrapolated from the ratio of the count of the gated population to the count of the counting beads present at a known concentration.
2.6 Chemokine Analysis
Tissues were suspended in PBS with 1x protease inhibitor mixture (Calbiochem)and homogenized using a Tissuemiser (Fisher Scientific). Supernatants were clarified by centrifugation (12,000 × g, 2 min). Supernatants were assayed for CXCL9 and CXCL10 concentration by ELISA (R&D Systems). Concentrations of CXCL1, CCL2, CCL5, and IFN-gamma were determined by Bio-Plex suspension array (Bio-Rad). It should be noted as a result of a significant number of deaths (>80%) in the establishment of the CXCL10−/− BM>CXCL10−/− chimera, the results for this group were combined with CXCL10−/− mice for analyte analysis. The CXCL10−/− BM>CXCL10−/− mouse chimeras that did survive (n=2) possessed levels that fell within the range expressed by CXCL10−/− mice.
2.7 Statistics
Analysis of variance followed by the Tukey’s post hoc T-test was used to determine significance (p<.05) comparing groups of mice using the GBSTAT program (Dynamic Microsystems).
3. Results
3.1 Verification of chimerism
We began our study by constructing BM chimeras composed of sub lethally irradiated congenic CD45.1 animals which received BM from either WT or CXCL10 −/− animals (CD45.2). Mice were irradiated twice at 650 rads waiting 4 hr between the first and second dose. Following a 12-week resting period to allow for establishment of chimerism, the success of the mouse chimera was verified by staining of peripheral blood leukocytes for CD45.2 as well as for Pan CD45 expression. Flow cytometry of stained blood, indicated approximately 90% chimerism in animals that received either WT or CXCL10 −/− BM (percentage chimerism calculated as percentage CD45.2+ donor-derived cells divided by percentage of total Pan CD45+ cells) (Fig. 1A and B respectively).
Figure 1.

Verification of chimerism. To verify irradiated congenic CD45.1 animals were chimeric following BM donation, whole blood was analyzed by flow cytometry for expression of CD45.2 and Pan CD45 expression. CD45+ cells from animals which received (A) WT or (B) CXCL10−/− BM were both approximately 90% positive for being CD45.2+ donor-derived cells. (C) Brain leukocytes categorized by low or high expression of CD45 (CD45LO or CD45HI respectively) were analyzed for CD45.2 expression as well with greater than 90% of CD45LO microglia (D) and CD45HI infiltrating leukocytes (E) being donor derived. Data is representative of two experiments with a total of six samples.
The resident leukocyte population of the non-inflamed CNS, microglia, are characterized by low expression of CD45 relative to CD45 high expressing leukocytes entering the CNS during inflammation. Parenchymal microglia are considered radiation resistant whereas perivascular microglia which serve as both APCs and gatekeepers of leukocyte entry into the CNS are a radiation sensitive population (Gehrmann et al., 1995; Heppner et al., 2005). To determine the relative chimerism of CNS microglia, the BS of animals infected with HSV-1 were harvested at day 7 PI and assayed for Pan CD45 expression along with staining for CD45.2. Chimerism of CD45 low expressing microglia was greater than 90% in animals receiving either WT or CXCL10 −/− bone marrow cells (Fig. 1C–D) as was chimerism in high CD45 expressing, infiltrating leukocytes (Fig. 1E).
3.2 Control of viral replication is dependent on CXCL10 expression by BM-derived populations
Upon construction of the appropriate chimeras, we first assayed the impact of BM-derived CXCL10 on the control of viral replication in the BS and TG of HSV-1 infected animals. Following infection of mouse chimeras, tissues were harvested at day 7 PI and assayed for viral burdens via plaque assay in animals which were WT or CXCL10 −/− BM mouse chimeras. Initial experiments also included WT and CXCL10−/− mice. In both the BS and TG, no significant difference in viral burdens was observed between WT and WTBM/WT chimeras or between CXCL10 −/− and CXCL10−/−BM/CXCL10−/− chimeras suggesting that long term effects due to radiation were negligible on resistance to HSV-1 (data not shown). However, a significant elevation in viral burdens in the BS and TG were observed between WT or CXCL10−/− recipients of WT BM compared to CXCL10−/− BM (Fig. 2A and B). Thus, the dominant role of CXCL10 on the control of viral replication appeared to be mediated directly or indirectly by BM-derived cells.
Figure 2.
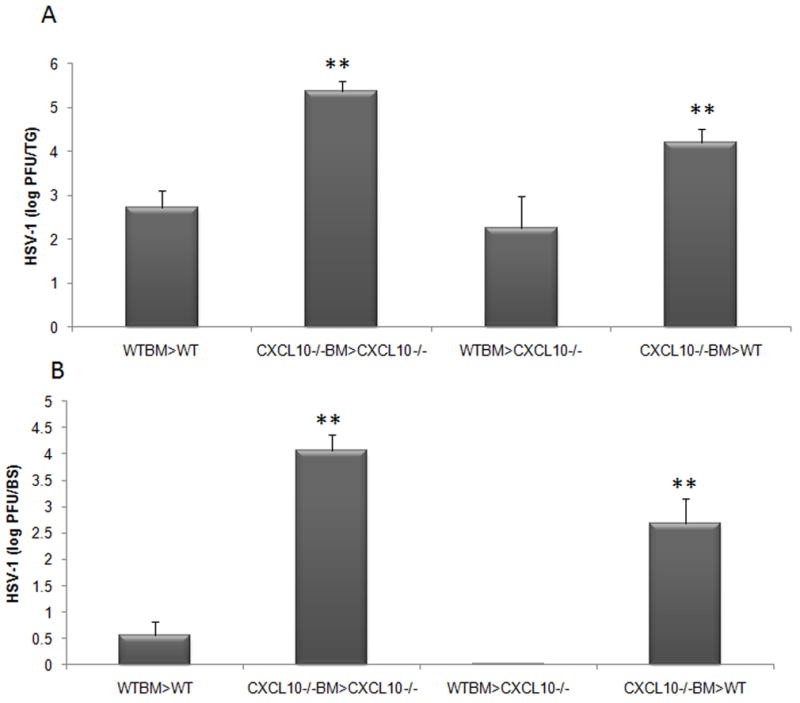
Mouse chimeras in which BM cells are from WT donor mice are resistant to HSV-1 infection compared to CXCL10−/− BM chimeras. The trigeminal ganglia (A) and brain stems (B) of mouse chimeras infected with 1,000 PFU/eye HSV-1 were harvested at day 7 PI, homogenized, and analyzed for viral titer by plaque assay. WT and CXCL10−/− mice receiving congenic WT BM possessed similar levels of virus in the TG and BS and significantly less relative to WT and CXCL10−/− mice receiving congenic CXCL10−/− BM. **p<.01 comparing the indicated groups to WTBM mouse chimeras. The bars represent the mean log PFU HSV-1 ± SEM, n=12/group summarizing four experiments.
3.3 Chemokine expression in the trigeminal ganglia and brain stem
The mouse strain used in this study, C57BL/6, is naturally deficient in the CXCR3 agonist CXCL11 due to a frameshift mutation which results in a premature termination codon (Wuest and Carr, 2008b). This leaves CXCL9 and CXCL10 as the only remaining CXCR3 agonists. To determine if hematopoietic CXCL10 deficiency impacts CXCL9 production in chimeric animals, we assayed CXCL9 levels in the TG and BS along with levels of other chemokines that are up-regulated during HSV-1 infection (Wuest and Carr, 2008b). In the TG, CXCL9 expression was not detectably altered by the presence or absence of CXCL10 expression by BM-derived cells (Fig. 3A). However, CCL2 levels mirrored the results measuring viral titers. Specifically, the mouse chimeras in which BM was donated by CXCL10−/− mice showed an elevation in CCL2 expression compared to mouse chimeras that possessed WT BM (Fig. 3A). These same mice also possessed more virus in the TG relative to the WT BM chimeras (Fig. 2A). However, no other expression patterns comparing levels of cytokines or interferon gamma to viral burden were found in the TG comparing CXCL10−/− BM chimeras to WT BM chimeras (Fig. 3A). Similar to that found in the TG, the absence of CXCL10 production by BM-derived cells had no impact on CXCL9 expression in the BS as well (Fig. 3B). Moreover, there was no appreciable difference in the expression of any chemokine or interferon gamma in the BS of mouse chimeras except as indicated and such isolated changes were not consistent with the viral burden recovered in the BS comparing all groups of mouse chimeras (Fig. 3B).
Figure 3.
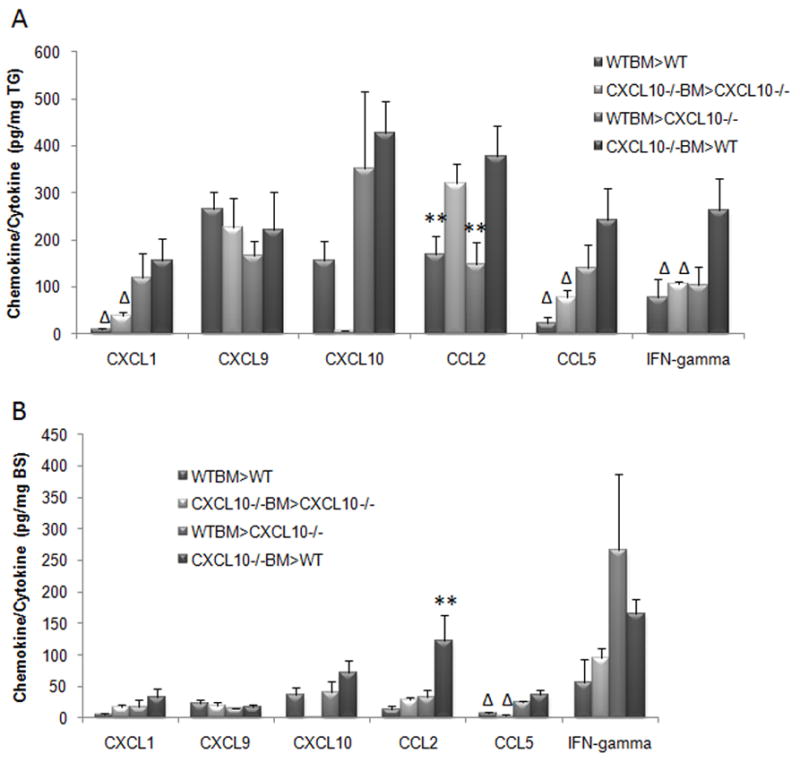
CCL2 levels in the TG of CXCL10−/− BM chimeras correlate to viral burden. Mouse chimeras were infected with 1,000 PFU/eye HSV-1. At day 7 PI, the trigeminal ganglia (TG) (panel A) and brain stems (BS) (panel B) of exsanguinated animals were removed and homogenized. The clarified supernatant was analyzed for cytokine/chemokine content using a suspension array (CXCL1, CCL2, CCL5, and IFN-gamma) or ELISA (CXCL9 and CXCL10) platform. **p<.05 comparing the indicated group(s) to the other groups for the individual analyte. Δp<.05 comparing the indicated group to the WT recipient of CXCL10−/− BM. The bars represent the mean ± SEM pg/mg of tissue wet mass, n = 4–8/group/analyte.
3.4 BM-derived CXCL10 expression does not significantly impact NK or T cell mobilization into the nervous system
To determine the impact of CXCL10 deficiency in BM-derived cells on leukocyte recruitment to the TG and BS, we assayed for the recruitment of CD3+CD4+, CD3+CD8+, HSV-1-sepcific CD8+ T cells (CD8+Tet+), and NK1.1+CD3− populations at day 7 PI. CXCL10 is highly active in the recruitment of activated CD4+ T cells (Dufour et al., 2002). Consistent with this observation, we found a significant reduction in the recruitment of CD4+ T cells to the TG of CXCL10−/−BM>CXCL10−/− mouse chimeras relative to the other mouse chimeras (Fig. 4A). However, other chemokines are apparently able to compensate for the loss of CXCL10 in the recruitment of CD4+ T cells to the BS as neither global deficiency nor BM specific deficiency in CXCL10 expression impacted CD4+T cell numbers at day 7 PI (Figure 4B). Although NK cell infiltration into the HSV-1 infected TG or BS is delayed early during infection in the absence of CXCL10 (Wuest and Carr, 2008b), we did not observe a statistically significant reduction in the numbers of NK1.1+CD3− cells in the TG or BS at day 7 PI in the CXCL10−/− BM>CXCL10−/− mouse chimeras even though there was a trend (Fig. 4). Although not significant, there was also a trend in the reduction of total CD8+ T cells and HSV-specific CD8+ T cells recovered from the BS of CXCL10−/− recipient mouse chimeras in comparison to the WT recipient mouse chimeras (Fig 4B). Consequently, we surmise the absence of CXCL10 has no significant impact on the recruitment of NK cells or T cells to the TG or BS following infection with the exception of total CD4+ T cells in the TG.
Figure 4.
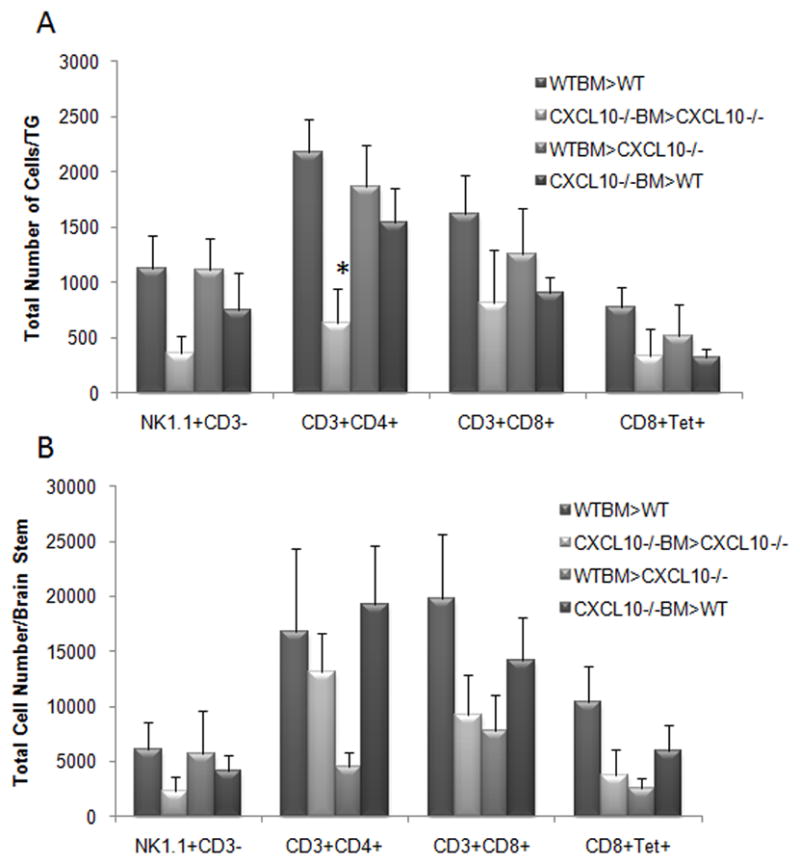
CD4+ T cell recruitment to the TG is diminished in CXCL10−/− mouse recipients of CXCL10−/− BM. Mouse chimeras were infected with HSV-1 (1,000 PFU/eye). At day 7 PI, the trigeminal ganglia (TG) (panel A) and brain stems (BS) (panel B) of exsanguinated mice were removed and processed for flow cytometry analysis. Bars represent the mean ± SEM, n = 4–9/group. *p<.05 comparing the indicated group to all other groups for each phenotype.
3.5 CXCL10−/− recipients possess significantly more T cells in the draining lymph node compared to WT recipients
Previous studies have reported CXCL10 does or does not influence the expansion of effector T cell populations in the lymph nodes (Dufour et al., 2002; Wuest and Carr, 2008b). Since there was a trend in the loss of CD8+ T cells and HSV-specific CD8+ T cells recovered from the brain stem of HSV-1-infected CXCL10−/− recipients, we investigated whether this observation was also reflected in the draining lymph nodes, the mandibular lymph nodes (MLN). Opposite that found in the brain stem, CXCL10−/− recipients possessed significantly more CD4+ and CD8+ T cells as well as HSV-specific CD8+ T cells in the MLN compared to HSV-1-infected WT recipients regardless of the BM source (Fig. 5). The total MLN CD45+ cell population did not differ between the WT and CXCL10−/− recipients suggesting the effect is specific for the T lymphocytes (data not shown). While there was a trend for a reduction in CXCL10 levels in the MLN of the CXCL10−/− recipients, the levels did not reach significance compared to the WT recipients (data not shown). Likewise, the MLN level of the other functional CXCR3 ligand, CXCL9, did not show significant changes comparing the WT to CXCL10−/− chimeras (data not shown).
Figure 5.
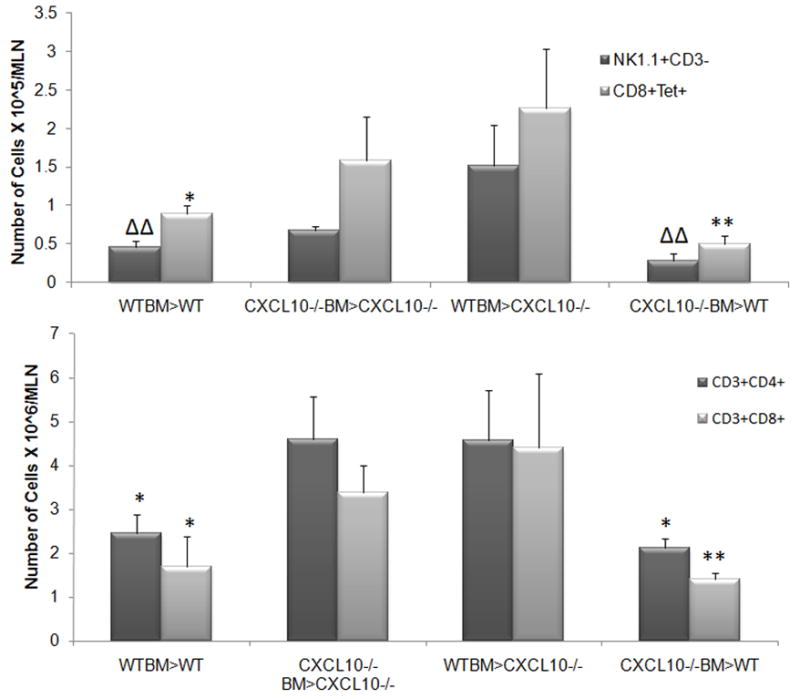
T cell populations are elevated in CXCL10−/− recipient chimeras. Mouse chimeras were infected with HSV-1 (1,000 PFU/eye). At day 7 PI, the mandibular lymph nodes (MLN) of exsanguinated mice were removed and processed for flow cytometry analysis. Bars represent the mean ± SEM, n = 5–9/group. **p<.01, *p<.05 comparing the indicated group to the CXCL10−/− recipient mouse chimeras. ΔΔp<.01 comparing the indicated group to the WTBM>CXCL10−/− mouse chimera.
4. Discussion
Chemokines regulate the coordinated mobilization of leukocytes between lymphoid organs, blood, and inflamed tissues (Robertson, 2002; Luster et al., 2005). The dominant function of inflammatory chemokines is to drive leukocyte recruitment along concentration gradients to the chemokine source (Baggiolini, 1998; Zlotnik and Yoshie, 2000). Some inflammatory chemokines also act to drive leukocyte mobilization from lymphoid stores (Tsou et al., 2007) and many stimulate cellular activities extending beyond influencing motility (Ward et al., 1998; Kinashi, 2005; Allen et al., 2007) including CXCL10 which has been demonstrated to alter signaling through the TCR (Kumar, et al., 2006; Dar and Knechtle, 2007). As the expression pattern of CXCL10 is unique in that it is both highly and rapidly up-regulated during inflammation and constitutively expressed in lymphoid organs (Trifilo et al., 2004; Klein et al., 2005; Hsieh et al., 2006; Zhang et al., 2008), the nature of its protective role during infection is unclear.
Our study on the role of CXCL10 expression by BM-derived cells began with the hypothesis that BM-derived cells may make a detectable contribution to overall CXCL10 expression in the BS and TG of HSV-1 infected animals and act through the recruitment of effector leukocytes. The anti-viral effect of CXCL10 was strictly dependent on CXCL10 expression by BM-derived cells. We observed no difference in viral burdens between WT BM>WT and WT BM> CXCL10−/− chimeras. However, WT BM mouse chimeras were found to harbor significantly less HSV-1 in the TG and BS compared to CXCL10−/− BM chimeras whether the recipient was WT or CXCL10−/−. Analysis of expression of CXCL10 in the chimeras did not parallel susceptibility to infection but rather, both BM-derived and resident cell populations contributed to the level of CXCL10 in the nervous system in response to HSV-1. Thus, control of viral replication depended on expression of CXCL10 by BM-derived cells while CXCL10 levels in inflamed tissues did not. Our results appear to be in contradiction to the presiding view that the function of CXCL10 during viral infection is to promote local leukocyte recruitment along the concentration gradient of CXCL10 (Baggiolini, 1998; Zlotnik and Yoshie, 2000). Of the chemokines investigated, only CCL2 levels were consistent with viral titers in the TG; the more infectious virus, the more CCL2 protein. We have previously found such levels of this chemokine correspond to an increase in macrophage and activated monocyte infiltration into the nervous system of HSV-infected mice (Conrady et al., 2009; Thapa and Carr, 2009). However, we have also noted the expression is not tissue specific (as in this case) but rather mirrors antigenic stimulus. Therefore, additional work is required to determine the source of CCL2 in comparison to HSV-1 spread and monocyte/macrophage recruitment in a longitudinal study to ascertain the relevance of CCL2 expression in this model.
Although not significant, there was a noticeable decline in the recruitment of total CD8+ and HSV-specific CD8+ T cells to the BS of CXCL10−/− recipient chimeras. The deficiency in recruitment was not due to a loss of CXCL10 levels or other T cell recruiting chemokines investigated as there was no pattern of chemokine expression that would explain the loss of CD8+ T cell infiltration. Whether this phenotype is reflected by a loss of another chemokine not included in the current study or possibly a change in the integrity of the endothelium or resident population in the CNS is currently unknown. However, perivascular microglia play a crucial role in the presentation of antigen to CNS infiltrating T cells, chemokine expression, and are notably radiation sensitive in contrast to parenchymal microglia (Hickey and Kimura, 1988; Gehrmann et al., 1995; Heppner et al., 2005) and similar cell populations exist in the TG (Mori et al, 2003). The majority of microglial cells in both groups of chimeric animals were donor-derived by day 7 PI (Figure 1D). Thus, donor-derived perivascular microglial cells could play a crucial role in the recruitment of HSV-1 specific CD8+ T cells including the production of soluble factors that modify endothelial addressins and thus, facilitate entry into the tissue. Unlike the TG, the BS is a segregated tissue via the blood-brain-barrier. The exact mechanism by which BM-derived CXCL10 expression is required for HSV-1 specific CD8+ T cell recruitment is unclear. However, our study provides evidence of the requirement for BM-derived CXCL10 expression for control of viral replication.
Acknowledgments
The authors would like to acknowledge Christopher D. Conrady for thoughtful discussion regarding the manuscript as well as Dr. Andrew Luster for graciously providing the original CXCL10−/− breeder mice for our re-derived colony. This work was supported by NIH R01 AI067309 (to DJJC). Additional support includes P20 RR017703 and an unrestricted grant from Research to Prevent Blindness. DJJC is a recipient of a PHF presidential professorship.
References
- Allen S, Crown S, Handel T. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Ansel KM, Harris RBS, Cyster JG. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity. 2002;16:67–76. doi: 10.1016/s1074-7613(01)00257-6. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- Conrady CD, Thapa M, Wuest T, Carr DJJ. Loss of mandibular lymph node integrity is associated with an increase in sensitivity to HSV-1 infection in CD118-deficient mice. J Immunol. 2009;182:3678–3687. doi: 10.4049/jimmunol.0803878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cose SC, Kelly JM, Carbone FR. Characterization of diverse primary herpes simplex virus type 1 gB-specific cytotoxic T-cell response showing a preferential Vβ bias. J Virol. 1995;69:5849–5852. doi: 10.1128/jvi.69.9.5849-5852.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar WA, Knechtle SJ. CXCR3-mediated T-cell chemotaxis involves ZAP-70 and is regulated by signalling through the T-cell receptor. Immunology. 2007;120:467–485. doi: 10.1111/j.1365-2567.2006.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennan MB, Franki AS, Dewint P, Van Beneden K, Seeuws S, van de Pavert SA, Reilly EC, Verbruggen G, Lane TE, Mebius RE, Deforce D, Elewaut D. Cutting edge: the chemokine receptor CXCR3 retains invariant NK T cells in the thymus. J Immunol. 2009;183:2213–2216. doi: 10.4049/jimmunol.0901213. [DOI] [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN- -inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Hanke T, Graham FL, Rosenthal KL, Johnson DC. Identification of an immunodominant cytotoxic T-lymphocyte recognition site in glycoprotein B of herpes simplex virus by using recombinant adenovirus vectors and synthetic peptides. J Virol. 1991;65:1177–11786. doi: 10.1128/jvi.65.3.1177-1186.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nature Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hsieh MF, Lai SL, Chen JP, Sung JM, Lin YL, Wu-Hsieh BA, Gerard C, Luster AD, Liao F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J Immunol. 2006;77:1855–1863. doi: 10.4049/jimmunol.177.3.1855. [DOI] [PubMed] [Google Scholar]
- Kinashi Intracellular signalling controlling integrin activation in lymphocytes. Nature Rev Immunol. 2005;5:546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, Hedin KE. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity. 2006;25:213–224. doi: 10.1016/j.immuni.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Luster A, Alon R, von Andrian U. Immune cell migration in inflammation: present and future therapeutic targets. Nature Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Mori I, Goshima F, Koshizuka T, Imai Y, Kohsaka S, Koide N, Sugiyama T, Yoshida T, Yokochi T, Kimura Y, Nishiyama Y. Iba1-expressing microglia respond to herpes simplex virus infection in the mouse trigeminal ganglion. Mol Brain Res. 2003;120:52–6. doi: 10.1016/j.molbrainres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71:173–183. [PubMed] [Google Scholar]
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Stein JV, Rot A, Luo Y, Narasimhaswamy M, Nakano H, Gunn MD, Matsuzawa A, Quackenbush EJ, Dorf ME, von Andrian UH. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, Exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J Exp Med. 2000;191:61–76. doi: 10.1084/jem.191.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamassia N, Calzetti F, Ear T, Cloutier A, Gasperini S, Bazzoni F, McDonald PP, Cassatella MA. Molecular mechanisms underlying the synergistic induction of CXCL10 by LPS and IFN-gamma in human neutrophils. Eur J Immunol. 2007;37:2627–2634. doi: 10.1002/eji.200737340. [DOI] [PubMed] [Google Scholar]
- Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AE, Wells TN, Parks WC, Wight TN, Martin TR, Frevert CW. Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J Immunol. 2010;184:2677–2685. doi: 10.4049/jimmunol.0903274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa M, Carr DJJ. CXCR3 deficiency increases susceptibility to genital herpes simplex virus type 2 infection: Uncoupling of CD8+ T-cell effector function by not migration. J Virol. 2009;83:9486–9501. doi: 10.1128/JVI.00854-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilo MJ, Montalto-Morrison C, Stiles LN, Hurst KR, Hardison JL, Manning JE, Masters PS, Lane TE. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J Virol. 2004;78:585–594. doi: 10.1128/JVI.78.2.585-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–9. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SG, Bacon K, Westwick J. Chemokines and T lymphocytes: more than an attraction. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]
- Wuest T, Carr DJJ. The role of chemokines during herpes simplex virus-1 infection. Front Biosci. 2008a;13:4862–4872. doi: 10.2741/3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Carr DJJ. Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol. 2008;181:7985–93. doi: 10.4049/jimmunol.181.11.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama H, Narumi S, Zhang Y, Murai M, Baggiolini M, Lanzavecchia A, Ichida T, Asakura H, Matsushima K. Pivotal role of dendritic cell- derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med. 2002;195:1257–1266. doi: 10.1084/jem.20011983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180:2641–2649. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O. Chemokines: A New Classification System and Their Role in Immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]