

Abstract
Chlamydia species are obligate intracellular bacteria that cause sexually transmitted disease, ocular infections and atypical pneumonia. This review highlights recent advances describing the mechanisms by which Chlamydia subvert host cytoskeleton and membrane trafficking pathways to create a replication competent niche.
Keywords: Chlamydia, inclusion, actin, Rab GTPases, sphingomyelin, phosphoinositides, recycling endosomes, mitochondria
1. Introduction – Chlamydia developmental cycle
Chlamydia species are medically important obligate intracellular bacteria that cause sexually transmitted disease, ocular infections and atypical pneumonia [1]. Of the nine species within the family Chlamydiaceae, Chlamydia trachomatis and Chlamydia pneumoniae are responsible for the majority of human Chlamydia infections. Despite differences in clinical manifestations and tissue tropism, all Chlamydia species undergo a unique biphasic developmental cycle that takes place within a non-acidified membrane bound vacuole, termed the inclusion [2]. Infection is initiated by the attachment and subsequent parasite mediated endocytosis of the infectious but metabolically inactive elementary body (EB) into epithelial cells. Once internalized, EBs differentiate into the metabolically active but non-infectious reticulate bodies (RB), which replicate by binary fission. In an asynchronous manner, while some RBs continue to divide, others are triggered by unknown signals to differentiate back into infectious EBs. The developmental cycle ends with release and dissemination of infectious EBs within the host.
Immediately after formation, the properties of the nascent inclusion are modified by processes that are dependent on early Chlamydia gene expression resulting in avoidance of lysosomal fusion and microtubule-dependent trafficking of the inclusion to the microtubule organizing center (MTOC) [2]. Subsequently, the inclusion intercepts vesicular and non-vesicular mediated pathways to obtain host derived lipids such as sphingomyelin [3], cholesterol [4], glycerophospholipids [5] and neutral lipids [6]. Exploitation of host cellular pathways creates an environment not only conducive to differentiation and replication, but also one protected from innate and adaptive host immune responses in part through the inhibition of apoptosis [7] and decreased expression of MHC class I and II molecules [8]. Failure to modify the inclusion results in the eventual targeting of Chlamydiae to and destruction within lysosomes [2]. These interactions between the inclusion and the host are likely mediated by both type three secretion (T3S)-dependent and -independent cytosolic and inclusion membrane localized (Incs) [9] Chlamydia effectors, which are thought to directly target and exploit host cellular pathways [10, 11].
Here we review recent advances in two rapidly developing areas of Chlamydia cell biology, exploitation of the host cytoskeleton and membrane trafficking pathways. Coopting these two basic cellular processes enable Chlamydiae to gain entry into non-phagocytic cells, obtain essential nutrients and precursors, modulate the fusogenicity and direct the intracellular trafficking of the inclusion, and exit the host cell.
2. Exploitation of the host cytoskeleton
Subversion of the host cytoskeleton via translocation of bacterial effectors or activation of signal transduction pathways via receptor binding is a pathogenic trait shared by many microorganisms including Chlamydia species [12]. Chlamydiae target actin, microtubules and intermediate filaments to regulate diverse aspects of their intracellular survival from entry into and exit from the host cell (Fig. 1).
Figure 1. Chlamydia subvert the host cytoskeleton.

A. Chlamydiae enter non-professional phagocytes in a Rac1 and actin-dependent mechanism. In C. trachomatis, the T3SS effector, TARP, is tyrosine phosphorylated by Src family, Abl and Syc kinases resulting in the activation and recruitment of Rac1 GEFs, Sos and Vav2. Rac1 activation leads to Arp2/3-dependent actin polymerization. EB binding and activation of PDGFR is one mechanism that promotes Abl kinase and Rac1 activation. In addition, TARP orthologues from all Chlamydia species nucleate actin via their actin-binding WH2-like domain. TARP mediated actin nucleation is essential for chlamydial invasion. * In C. caviae, Cdc42 and Arf6 also promote actin polymerization. CT166 and CT694, secreted effectors, may also function during entry via inhibition of Rac1 and interaction with AHNAK, respectively. B. EBs are transported via a dynein-dependent, but dynactin-independent mechanism to the MTOC. C. The inclusion is stabilized by the formation of a scaffold consisting of actin and vimentin by a RhoA-dependent mechanism. CPAF-dependent cleavage of vimentin may allow flexibility in the scaffold to accommodate the growth of the inclusion at late stages of development. D. Chlamydiae can exit the host cell via a mechanism that is dependent on actin, myosin II, WASP and Rho GTPases.
2.1 Subversion of the host actin cytoskeleton
In vitro, Chlamydiae enter non-professional phagocytic cells by multiple mechanisms with the choice of strain, method of inoculation, host cell type and culture methods influencing which specific mechanism predominates. Mechanisms dependent and independent of clathrin, microfilaments and lipid rafts have all been documented [13]. Given the essential nature of entry into host cells and the multiple mechanisms that Chlamydiae utilize, it is no surprise that numerous Chlamydia ligands and host cell receptors have been reported [13] [14, 15]. Despite the various entry mechanisms, ligands and receptors that have been proposed, there is a growing consensus that a Rac1-dependent actin remodeling entry mechanism is shared by many Chlamydia species [16–19]. Binding of EBs to the cell surface activates and recruits Rac1 to sites of EB entry of at least two Chlamydia species [18, 19]. During Chlamydia caviae infection, Cdc42 [19] and Arf6 [20] are also activated and are required for entry. Cdc42 is not required for the entry of C. trachomatis [18] and the role of Arf6 during C. trachomatis infection has not been examined. It is not clear why the requirements for small GTPases differ between the Chlamydia species, but highlights the fact that multiple entry mechanisms can be utilized by Chlamydia. During Chlamydia trachomatis invasion, Rac1 is activated by Sos2 and Vav2 [14, 21], Rac1 guanine nucleotide exchange factors (GEFs), and results in the recruitment of the actin regulators WAVE2 and Abi-1 and Arp2/3-dependent actin remodeling and transient pedestal formation [22]. Inhibition of actin polymerization or Rac-dependent signaling pathways by pharmacological inhibition, expression of dominant negative mutants or by siRNA knock down, results in inhibition of Chlamydia invasion demonstrating the importance of these pathways in multiple Chlamydia species [14,16–19, 21]. For C. trachomatis, Rac1 activation may be mediated by tyrosine phosphorylation of the Chlamydia translocated actin-recruiting phosphoprotein (TARP) protein [21] (see below).
Bacterial pathogens subvert actin remodeling through translocation of effector proteins that modulate the activity of Rho family GTPases or directly nucleate actin polymerization. The Chlamydia-specific T3S-dependent effector TARP [23], which is translocated across the host plasma membrane during entry, regulates actin reorganization by both mechanisms. In vitro, TARP nucleates actin polymerization through a direct interaction with actin [24]. Actin polymerization is mediated by a carboxy-terminal WH2-like actin-binding domain common to all Chlamydia TARP orthologues and is essential for invasion [25]. TARP proteins that contain only a single actin-binding domain require oligomerization of TARP via the presence of proline rich repeat domains to nucleate actin [25]. In contrast, some TARP proteins contain multiple actin-binding domains that bypass the need to oligomerize [25]. A possible model that incorporates actin polymerization by both TARP and Arp2/3 proposes that Arp2/3 acts on TARP-nucleated filaments to produce branched chain actin filaments. In addition, at least for C. trachomatis, TARP may also mediate actin polymerization through an alternate Rac-dependent mechanism [14, 21]. TARP orthologues from C. trachomatis, but not other Chlamydia species, are tyrosine phosphorylated by multiple host kinases including Src family [26], Abl [14] and Syc [27] kinases upon translocation into host cells via amino terminal tyrosine rich domain repeats. EB binding and activation of Platelet derived growth factor receptor (PDGFR) or PDGFR-independent activation of Abl kinase activates Rac-dependent signaling pathways via phosphorylation of Tarp, Vav2, and other actin binding proteins [14]. In addition, TARP interacts with Vav2 and Sos in a phosphorylation-dependent manner [21, 28] suggesting that phosphorylation of TARP regulates actin polymerization by Rac-dependent signaling mechanisms via recruitment and activation of the Rac GEFs, Sos1 and Vav2. However, since tyrosine phosphorylation of TARP is not required for entry [29], it may be important for post-entry signaling events [28].
Actin reorganization induced by Chlamydia is transient, and therefore Chlamydiae may also secrete effectors that regulate actin depolymerization. CT166, a C. trachomatis protein that contains regions homologous to clostridial glucosylating toxins (GCTs) is a likely candidate for this process [30, 31]. Chlamydia strains expressing CT166 that contain domains homologous to GCTs induce actin disassembly and are cytotoxic when infected at high multiplicities of infection [30]. Ectopic expression of C. trachomatis CT166 reproduces this cytotoxicity and actin reorganization phenotype via glucosylation and inhibition of Rac1. Ectopic expression also reduces internalization of Chlamydia suggesting that under conditions of a normal infection, CT166 may act to turn off Rac-dependent actin reorganization after entry. Another candidate may be C. trachomatis CT694. Similar to TARP, C. trachomatis CT694 is translocated into the host cell and localizes adjacent to EBs during entry [32]. CT694 interacts and co-localizes with eukaryotic AHNAK, an actin-binding protein, and ectopic expression of CT694 results in loss of actin stress fibers in an AHNAK-dependent manner [32]. The exact function of CT694 during C. trachomatis is unclear, but may play a role in regulating membrane fluidity or actin depolymerization during invasion or early stages of Chlamydia infection.
The inclusion is stabilized by the formation of an actin and intermediate filament scaffold that surrounds the mature inclusion [33]. Scaffold formation is dependent on RhoA, a regulator of actin stress fiber formation, but independent of known RhoA effector mechanisms [33]. Chlamydial proteasome/protease-like activity factor (CPAF)-mediated structural alterations in vimentin are thought to create a dynamic structure that allows for expansion of the inclusion at late stages of Chlamydia development [33]. CPAF is a protease secreted into the host cytosol that targets multiple host proteins with important roles in immune evasion [8]. As mentioned below, sphingomyelin biosynthesis is also required to maintain the integrity of inclusion, but the relationship between sphingomyelin and the actin scaffold is presently unknown [34]. By providing structural integrity to the inclusion, the presence of the cytoskeletal scaffold has been proposed to limit activation of host cytosolic immune surveillance pathways by preventing leakage of luminal contents into the cytosol [33].
Finally, Chlamydiae subvert actin remodeling during exit from the host cell. Live cell imaging studies revealed that Chlamydiae exit by two mutually exclusive mechanisms, cell lysis and inclusion “extrusion” [35]. Cell lysis is dependent on proteases and intracellular calcium signaling and involves sequential lysis of the inclusion, the nucleus and then the plasma membrane. Host cell survival following cell lysis is dependent on lysosomal repair of the plasma membrane, a process requiring actin depolymerization [36]. Inclusion extrusion involves release of an intact inclusion that has been packaged and extruded through the plasma membrane and requires actin polymerization, neuronal Wiskott-Aldrich syndrome protein (WASP), myosin II and Rho GTPase [35].
2.2 Subversion of host microtubules
During the initial stages of infection, the nascent inclusion is trafficked by a dynein and microtubule-dependent mechanism to the peri-Golgi region of the cell where it resides in close contact with the MTOC [37]. Although p150(Glued), a component of dynactin, a multiprotein complex normally required for cargo binding to the dynein motor, localizes to the inclusion, trafficking is independent of dynactin [37]. Interaction with the dynein motor complex is likely mediated through interactions with inclusion membrane proteins, which may mimic the cargo binding activity of dynactin. Consistent with this model, microdomains within the inclusion membrane that contain several Incs, active Src family kinases and cholesterol have been identified that co-localize with centrosomes and dynein [38]. One of these proteins, CT850 co-localizes with centrosomes when ectopically expressed suggesting that these inclusion protein-containing microdomains may play a role in inclusion-centrosome interactions [38]. The importance of Src family kinases in inclusion-centrosome interactions is unclear since their depletion does not affect inclusion development [38]. The inclusion maintains a dynein-dependent association with the centrosome during transit through the host cell cycle resulting in an increase in supranumerary centrosomes, abnormal spindle poles and segregation defects [39]. Chromosomal segregation defects are even observed in cells cured of Chlamydia [39]. Although cytokinesis is inhibited at late stages in Chlamydia infected cells [40], the increase in centrosome numbers does not arise as a result of this defect but instead results from dysregulation of centrosome duplication by a mechanism that requires known regulators of centrosomal duplication and progression through S phase [41]. The relevance for these interactions are not clear since in vivo Chlamydia replicate in terminally differentiated columnar epithelial cells. However, Chlamydia-induced chromosomal segregation defects may explain the observed link between Chlamydia-infections and cervical cancer [42].
3. Subversion of membrane trafficking pathways
3.1 Rab GTPases and phosphoinositides
The biogenesis and maintenance of vacuoles harboring numerous pathogenic organisms involve the selective recruitment or exclusion of Rab GTPases [43]. Rab GTPases are small Ras-like GTPases that function in vesicular trafficking and organelle identity (reviewed in [44]). Localized to distinct organelles or organellar subdomains, Rab GTPases cycle between an inactive cytosolic GDP-bound form and an active membrane associated GTP-bound form. Interaction between the GTP-bound Rab and downstream effectors regulate formation of vesicles at donor membranes, trafficking and fusion of transport vesicles with acceptor membranes.
Chlamydiae target multiple Rab-dependent pathways. Both endocytic (Rab4, 11, 14) [45, 46] and ER-Golgi related (Rab1, 6 and 10) [45] Rab GTPases and their effectors [47, 48] associate with inclusions at different times during the biogenesis of the inclusion by species-dependent and –independent processes. Recruitment of at least several Rab GTPases is mediated through direct interactions with Incs. For example, C. trachomatis CT229 interacts with Rab4 [49], while Cpn0585, interacts with multiple Rab GTPases including Rab1, 10 and 11 [50]. Roles for specific Rab GTPases are only now being elucidated but current data suggests they play multiple essential roles in various aspects of Chlamydia development. Depletion of Rab6, Rab11 [51] or Rab14 [46] and co-expression of dominant negative Rab4 and Rab11 mutants [52], result in varying degrees of impaired Chlamydia growth demonstrating that Rab GTPases play crucial and at times functionally redundant roles during Chlamydia development. As described below, Rab6, Rab11 and Rab14 function in sphingomyelin delivery to the inclusion [46, 51] while Rab4 and Rab11 regulate a slow transferrin recycling pathway intercepted by the inclusion [52]. Genome-wide RNA interference screens confirmed requirements for Rab1 and Rab14 in C. trachomatis development [14] and Rab11 in C. caviae development in Drosophila melanogaster S2 cells [53] as well as identifying additional Rab GTPases (Rab8, 32, 39 and 35) that may play both positive and negative roles during Chlamydia development [14, 53]. The localization of Rab35 to the Chlamydia inclusion has recently been confirmed (M. Scidmore, unpublished data). Further work is needed to clarify roles of specific Rab GTPases, as loss of Rab1 has also been shown to both increase [51] and decrease Chlamydia development [14].
In addition to Rab GTPases, organelle identity is also determined in part by the presence or absence of specific phosphoinositides (PI) and thus many pathogenic bacteria often target PI metabolism [54]. Phosphoinositides are short lived phosphorylated derivatives of inositol that play key roles in intracellular signaling and vesicle transport by controlling the subcellular localization and activity of PI-binding proteins. The intracellular localization of PI is tightly regulated by PI-specific kinases and phosphatases that add or remove specific phosphate groups, respectively, generating seven distinct PI species. Since Rab GTPases function through their interaction with downstream effectors, efforts are currently focused on identifying inclusion localized Rab GTPase effectors [47, 48]. Oculocerebral syndrome of Lowe protein 1 (OCRL1), a Golgi-localized PI-5′ phosphatase that primarily hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 4-phosphate (PI4P) and binds to multiple Rab GTPases localizes to all Chlamydia inclusions [48]. PI4P, the product of OCRL1, is predicted to localize to the inclusion membrane based upon inclusion localization of GFP-tagged PI4P-specific pleckstrin homology (PH) domains of oxysterol binding protein (OSBP) and Good Pasture Binding Protein (GPBP) [48]. A second host enzyme, phosphatidylinositol 4-kinase (PI4KIIα), which produces PI4P from PI, is also present at the inclusion [48]. Although studies with BFA, an inhibitor of anterograde Golgi-to-ER trafficking suggest that Golgi-trafficking is not required for the presence of PI4P at the inclusion, it is currently unknown whether PI4P is produced by OCRL1 or PI4KIIα directly at the inclusion and what specific role it plays. However, the absence of OCRL1 or PI4KIIα reduces the ability of Chlamydia to infect cells suggesting roles in entry or early inclusion modification possibly through recruitment of PI4P-binding proteins or other mechanisms [48].
Requirements for additional PI species during Chlamydia infection have also been documented. Inhibition or depletion of ADP-ribosylating factor 6 (Arf6), the Arf6 effector phosphatidylinositiol 4-phosphate 5-kinase (PIP 5-kinase), or PIP2, the product of PIP 5-kinase, at the plasma membrane alters actin remodeling and reduces the internalization of C. caviae [20]. A role for Arf6 and PIP2 in entry of other Chlamydia species has not been examined. Finally, PI3K-dependent pathways have multiple roles during Chlamydia development including C. pneumoniae [17] and C. caviae entry [19], interaction of the Rac GEF, Vav2 with C. trachomatis TARP [21] and resistance to apoptotic stimuli [7].
3.2 Lipid transport pathways
As obligate intracellular bacteria, Chlamydiae rely on essential nutrients and biosynthetic precursors from its eukaryotic host. It has become increasing evident, that Chlamydiae are extremely proficient at acquiring host-derived lipids. Using the long chain fluorescent derivative of ceramide, N-[7-(4-nitrobenzo-2-oxa-1,3-diazole)]) aminocaproylsphingosine (C6-NBD-Cer), Hackstadt and colleagues demonstrated for the first time, the delivery of a host-derived lipid to the inclusion and uptake of this lipid by EBs [3]. Sphingomyelin is a unique and defining property of all Chlamydia species and is required for Chlamydia development [34, 55]. Furthermore, sphingomyelin trafficking is dependent on a Golgi-dependent vesicular-mediated trafficking pathway [56] that may transport sphingomyelin-containing vesicles to the basolateral surface of polarized cells [57]. These data together, with the close physical proximity to the Golgi apparatus, prompted researchers to propose that the inclusion is trafficked to the peri-Golgi region of the cell where it intercepts a subset of Golgi-derived vesicles normally destined for the plasma membrane as a mechanism to obtain host cell nutrients. This discovery revealed for the first time that inclusion selectively fused with host-derived vesicles and was, in fact, not completely disconnected from host membrane trafficking pathways.
Ten years after the initial discovery, the first clues of how Chlamydiae co-opt sphingomyelin transport pathways were revealed and indicate that Chlamydiae target multiple sources of sphingomyelin. Beatty and colleagues demonstrated that MVBs, components of late endosomes, were trafficked and their contents delivered to the lumen of the inclusion [58]. Disruption of MVB trafficking prevents transport of both cholesterol and sphingomyelin to the inclusion and results in impaired growth of Chlamydiae [58, 59]. Furthermore, inhibition of sphingomyelin synthesis results in loss of inclusion membrane integrity, inhibition of homotypic inclusion fusion and early re-differentiation of RBs into EBs [34]. Inhibition of MVB trafficking, as compared to inhibition of sphingomyelin synthesis, results in a greater reduction of Chlamydia growth suggesting that additional MVB components are essential for Chlamydia development [34].
Subsequently, Myers and co-workers discovered that Chlamydiae induce fragmentation of the Golgi apparatus resulting in the localization of Golgi mini stacks to the inclusion [60]. Golgi fragmentation is mediated by cleavage of Golgin-84, a component of the Golgi matrix, by both inflammatory caspase-and calpain-dependent mechanisms [60]. Inhibition of Golgi-fragmentation reduces transport of sphingomyelin to the inclusion and Chlamydia growth, while the induction of Golgi fragmentation via depletion of the Golgin p115 increases Chlamydia growth [60]. Collectively these data suggest that Chlamydia co-opt an entire organelle to facilitate the essential delivery of Golgi-derived lipids to the inclusion. Rab GTPases also function to maintain Golgi structure [61]. Through siRNA gene induced silencing experiments, it was shown that Rab6 and Rab11, both of which localize to inclusions [45], regulate the ability of Chlamydiae to induce Golgi fragmentation and that Rab6 acts downstream of Golgin 84 cleavage since in the absence of Rab6, Golgin 84 is still cleaved [51]. Rab14, a regulator trans-Golgi to early endosome trafficking, is also implicated in the delivery of sphingomyelin to the inclusion [46]. In contrast to Rab6 and Rab11, there is no evidence that Rab14 destabilizes the Golgi apparatus in infected cells. Further experiments are needed to clarify the roles of Rab GTPases in the destabilization of the Golgi apparatus and in the transport of sphingomyelin and how the formation of mini stacks facilitates sphingomyelin transport to the inclusion. Additionally, the Chlamydia proteins that target Golgin-84 cleavage or regulate sphingomyelin trafficking have not been identified.
The ectopic expression of putative Chlamydia effectors as GFP-tagged proteins in Saccharomyces cerevisiae revealed a tropism for lipid droplets (LD) by four Chlamydia proteins [6, 62] suggesting that in addition to sphingomyelin, Chlamydiae target LDs as a source of lipids. Lipid droplets, ER-derived storage organelles for neutral lipids or long chain fatty acids, were subsequently shown to be recruited to and translocate into the lumen of the inclusion [6, 63]. Lipid homeostasis is altered during Chlamydia infection and pharmacological inhibition of LD formation results in a decrease in inclusion size and a reduction in EB production demonstrating the importance of LDs for Chlamydia development. The presence of neutral lipids in RBs and the absence of a net accumulation of LDs in the lumen suggest that LDs are metabolized possibly by direct scavenging or lipolysis of the associated lipids. Two of the three proteins that display tropism for LDs, Lda1 and Lda3, are translocated into the host cytosol and localize to LDs that are adjacent to the inclusion membrane [6]. Furthermore, ectopically expressed GFP-Lda3 accumulates with LDs that have reduced staining for adipocyte differentiation related protein (ADRP), a surface exposed inhibitor of lipases [63]. Finally, IncA, a protein localized to the inclusion membrane, co-fractionates and partially co-localizes with intralumenal LDs, suggesting that IncA may mark sites of the inclusion that are entry sites for LDs [63]. Based upon the current data, it has been proposed that Chlamydiae translocate Lda3 into the host cytosol, where it acts as bridge to anchor LDs to the inclusion membrane and also may function to remove the protective coat protein ADRP from LDs. Invagination, by as yet some undetermined mechanism, of the inclusion membrane at sites of LD association, traps LDs within the lumen allowing RBs to directly scavenge or metabolize associated lipids, possibly by LD-associated lipases [63]. Scavenging lipids from an organelle from within the lumen of the inclusion may be advantageous as cytosolic surveillance mechanisms and the generation of inflammatory byproducts of fatty acid lipolysis would be avoided.
Eukaryotic glycerophospholipids such as phosphatidyl choline and phosphatidyl inositol are present in purified EBs and delivered to the inclusion by non-vesicular mediated pathways [5]. In contrast to cholesterol and sphingomyelin, glycerophospholipids are selectively modified by Chlamydiae. Chlamydia-induced ERK1/2 dependent activation of cytosolic cPLA2 results in the replacement of eukaryotic-derived straight chain fatty acid at the sn-2 position with a Chlamydia-derived branch chain fatty acid [64]. Inhibition of ERK signaling during Chlamydia infection, results in reduction of glycerophospholipid uptake by Chlamydiae and a decrease in infectivity [64]. Interestingly, although Ras is also activated during Chlamydia infection and usually functions to activate ERK1/2 through Raf, ERK1/2 is activated independently of Ras and Raf1 [65, 66]. In fact, Ras and its down stream target, Raf1, negatively regulate Chlamydia infections [66]. These data highlight the ability of Chlamydiae to activate eukaryotic signaling pathways by non-canonical mechanisms. As we begin to unravel the interactions between Chlamydiae and its eukaryotic host, it is becoming clear that these interactions are quite complex. This is best exemplified by the role of cPLA2, which in addition to being essential in Chlamydia-mediated modification of host derived glycerophospholipids, cPLA2 also regulates the expression of type I interferons and intracellular immunity to C. trachomatis in mouse cells [65].
3.3 SNARE mimicry
The selective targeting of host vesicles suggests that Chlamydiae have evolved mechanisms to regulate the fusogenicity of host transport vesicles and organelles. A clue to unraveling these phenomena was revealed by the discovery that several Incs including IncA, CT223 and CT813, contain eukaryotic soluble NSF (N-ethylmaleimide-sensitive) attachment protein receptors (SNARE) protein domains [67]. SNARE proteins present on opposing membranes drive membrane fusion by bringing vesicle and target membranes close together through the assembly of a SNARE-containing four-helix bundle. Vesicles containing endocytic SNAREs, Vamp3, Vamp7 and Vamp8, accumulate around the inclusion via interactions with IncA that are dependent on IncA’s SNARE motifs [67]. Functional significance for these interactions was revealed during in vitro fusion assays where the presence of IncA inhibited the fusion activity of several endocytic SNAREs [68]. In contrast, IncA has also been shown to be involved in homotypic fusion of Chlamydia-containing vacuoles [67, 69]. Based upon the inhibitory effect of IncA on SNARE-mediated membrane fusion and its role in promoting homotypic vesicle fusion, Subtil and colleagues propose that IncA may act as a switch regulating the maturation of the inclusion. In their model, newly synthesized IncA would first inhibit SNARE-mediated fusion events with the inclusion by binding host SNAREs, and then once IncA-SNARE interactions have been saturated, continued synthesis of IncA would promote IncA-IncA interactions and homotypic vesicle fusion [68]. Since IncA is not expressed until 10 hours post infection and early inclusions resist fusion with lysosomes, additional SNARE-inhibitory Inc activity would be required prior to the expression of IncA.
In addition to SNARE proteins, the Arf family of small Ras-like GTPases, also regulate membrane trafficking. The Golgi-localized Arf1 regulates production of COPI transport vesicles via recruitment of coat proteins and PI homeostasis and membrane trafficking via recruitment of PI4K enzymes and PI4P-binding proteins. Although Arf1 localizes to Chlamydia inclusions in a species-specific manner [48], its role during Chlamydia development is uncertain since loss of Arf1 function has been shown to both inhibit [48] and increase Chlamydia development [66].
3.4 Recycling endosomes and mitochondria
Although non-fusogenic with lysosomes, the inclusion associates with but does not fuse with recycling endosomes containing transferrin [2]. Consistent with these data, the endocytic Rab GTPases, Rab4 and Rab11, also co-localize with the inclusion [45]. Clues to the biological significance of these interactions were uncovered in a screen for low-molecular weight inhibitors of Chlamydia growth [52]. One inhibitor identified as a result of this screen, FR179254, reduces the formation of transferrin-containing Rab4/11 hybrid vesicles and causes a delay in the slow transferrin recycling pathway. Similar phenotypes, reduction in Chlamydia growth and a defect in transferrin recycling, are observed upon co-expression of dominant negative mutants of Rab4 and Rab11 [52]. Surprisingly, growth inhibition caused by FR179254 is reversed by the omission of transferrin-containing serum from the media [52]. Collectively, these data suggest that alteration of the slow recycling pathway and accumulation of iron-laden transferrin-containing endosomes at the inclusion is toxic to the development of Chlamydiae and that Chlamydiae may target Rab4 and Rab11 to ensure proper transferrin recycling as a potential mechanism for iron acquisition.
A genome wide RNA interference screen in D. melanogaster S2 cells revealed a C. caviae-specific requirement for the Tom complex, a multiprotein complex involved in mitochondrial protein export, supporting previous studies that C. caviae, but not C. trachomatis, targets mitochondria [53]. Depletion of two components of the complex, Tom40 and Tom22, reduces C. caviae EB production, but no defects in energy metabolism or susceptibility to apoptotic stimuli are detected and only a small defect in the association of mitochondria with the C. caviae inclusion is observed. Although important for C. caviae development, it is still unclear how mitochondria might specifically benefit C. caviae.
4. Concluding remarks
Once thought to be disconnected from membrane trafficking pathways, we are now discovering that the inclusion intercepts a complex array of host cellular pathways (summarized in Fig. 2) that promote the intracellular survival of Chlamydia. And despite the genetic intractability of Chlamydia, the mechanisms by which these pathways are exploited are rapidly being unraveled. The recent development of potentially powerful genetic tools should hopefully further our understanding of how specific Chlamydia effector proteins target these cellular pathways.
Figure 2. Chlamydia subvert host membrane trafficking pathways.
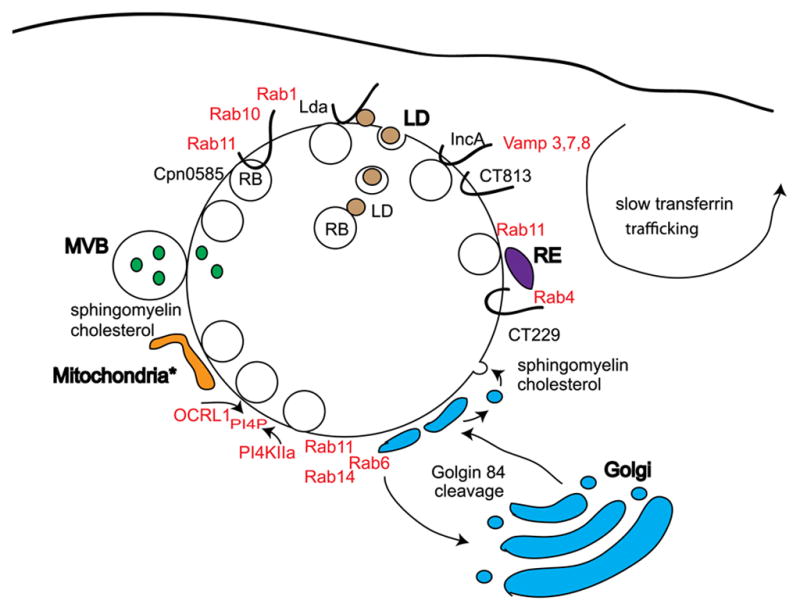
Chlamydiae obtain host derived lipids by intercepting MVB and Golgi-derived vesicles containing sphingomyelin and cholesterol, and lipid droplets (LD) containing neutral lipids via interactions with secreted Lda. Multiple Rab GTPases are associated with Chlamydia inclusions via interaction with Incs and function to mediate Golgi fragmentation, sphingomyelin trafficking and interaction with a slow transferrin recycling pathway. IncA and CT813 interact with and can inhibit host-mediated SNARE trafficking pathways. PI4P is localized to inclusions possibly via recruitment of OCRL1 and PI4KIIα. For C. caviae, the mitochondrial Tom complex is required for infection.
Acknowledgments
I thank members of the Scidmore lab for their comments. Due to space constraints, I apologize to those investigators whose work was not cited. Work in the laboratory is supported by PHS grant RO1A1073831.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schachter J. Overview of human diseases. In: Baron AL, editor. Microbiology of Chlamydia. CRC Press, Inc; Boca Rotan, FL: 1988. pp. 153–165. [Google Scholar]
- 2.Scidmore MA. Chlamydial exploitation of host signaling, cytoskeletal, and membrane trafficking pathways. In: Bavoil PM, Wyrick PB, editors. Chlamydia: Genomics and Pathogenesis. Horizon Bioscience; Norfolk, U. K: 2006. pp. 255–295. [Google Scholar]
- 3.Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells:directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci USA. 1995;92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci USA. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatch GM, McClarty G. Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect Immun. 1998;66:3727–3735. doi: 10.1128/iai.66.8.3727-3735.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16:1646–1651. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- 7.Sharma M, Rudel T. Apotosis resistance in Chlamydia-infected cells: a fate worse than death? FEMS Immunol Med Microbiol. 2009;55:154–161. doi: 10.1111/j.1574-695X.2008.00515.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhong G. Killing me softly: chlamydial use of proteolysis for evading host defenses. Trends Microbiol. 2009;17:467–474. doi: 10.1016/j.tim.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- 10.Valdivia RH. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol. 2008;11:53–59. doi: 10.1016/j.mib.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Betts HJ, Wolf K, Fields KA. Effector protein modulation of host cells: examples in the Chlamydia spp. arsenal. Curr Opin Microbiol. 2009;12:81–87. doi: 10.1016/j.mib.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Dunn JD, Valdivia RH. Uncivil engineers: Chlamydia, Salmonella and Shigella alter host cytoskeleton architecture to invade epithelial cells. Future Microbiol. 2010;5:1219–1232. doi: 10.2217/fmb.10.77. [DOI] [PubMed] [Google Scholar]
- 13.Dautry-Varsat A, Subtil A, Hackstadt T. Recent insights into the mechanisms of Chlamydia entry. Cell Microbiol. 2005;7:1714–1722. doi: 10.1111/j.1462-5822.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- 14.Elwell CA, Caesay A, Kim JH, Kalman D, Engel JN. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008;4:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abromaitis S, Stephens RS. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog. 2009;5:e1000357. doi: 10.1371/journal.ppat.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coombes BK, Mahony JB. Identification of MEK- and phosphoinositide 3-kinase-dependent signaling as essential events during Chlamydia pneumoniae invasion of HEp2 cells. Cell Microbiol. 2002;4:447–460. doi: 10.1046/j.1462-5822.2002.00203.x. [DOI] [PubMed] [Google Scholar]
- 18.Carabeo RA, Grieshaber SS, Hasenkrug A, Dooley C, Hackstadt T. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic. 2004;5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- 19.Subtil A, Wyplosz B, Balana ME, Dautry-Varsat A. Analysis of Chlamydia caviae entry sites and involvement of Cdc42 and Rac activity. J Cell Sci. 2004;117:3923–3933. doi: 10.1242/jcs.01247. [DOI] [PubMed] [Google Scholar]
- 20.Balana ME, Niedergand F, Subtil A, Alcover A, Chavrier P, Dautry-Varsat A. Arf6 GTPase controls bacterial invasion by actin remodeling. J Cell Sci. 2005;118:2201–2210. doi: 10.1242/jcs.02351. [DOI] [PubMed] [Google Scholar]
- 21.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 2008;4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carabeo RA, Dooley CA, Grieshaber SS, Hackstadt T. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell Microbiol. 2007;9:2278–2288. doi: 10.1111/j.1462-5822.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 23.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci USA. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci USA. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jewett TJ, Miller NJ, Dooley CA, Hackstadt T. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog. 2010;6:1–11. doi: 10.1371/journal.ppat.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jewett TJ, Dooley CA, Mead D, Hackstadt T. Chlamydia trachomatis Tarp is phosphorylated by Src family kinases. Biochem Biophys Res Commun. 2008;371:339–344. doi: 10.1016/j.bbrc.2008.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehlitz A, Banhart S, Hess S, Selbach M, Meyer TF. Complex kinase requirements for Chlamydia trachomatis Tarp phosphorylation. FEMS Microbiol Lett. 2008;289:233–240. doi: 10.1111/j.1574-6968.2008.01390.x. [DOI] [PubMed] [Google Scholar]
- 28.Mehlitz A, Banhart S, Mauer AP, Kaushansky A, Gordus AG, Zielecki J, Macbeath G, Meyer TF. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J Cell Biol. 2010;12:143–157. doi: 10.1083/jcb.200909095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect Immun. 2005;73:3860–3868. doi: 10.1128/IAI.73.7.3860-3868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belland RJ, Scidmore MA, Crane DD, Hogan DM, Whitmire W, McClarty G, Caldwell HD. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc Natl Acad Sci USA. 2001;98:13984–13989. doi: 10.1073/pnas.241377698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thalmann J, Janik K, Martin M, Sommer K, Ebeling J, Hofmann F, Genth H, Klos A. Actin re-organization induced by Chlamydia trachomatis serovar D-evidence for a critical role of the effector protein CT166 targeting Rac. PloS One. 2010;5:e9987. doi: 10.1371/journal.pone.0009887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hower S, Wolf K, Fields KA. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol. 2009;72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson DK, Gu L, Rowe RK, Beatty WL. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathog. 2009;5:e1000664. doi: 10.1371/journal.ppat.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci USA. 2007;104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beatty WL. Lysosome repair enables cell survival and bacterial persistence following Chlamydia trachomatis infection. Cell Microbiol. 2007;9:2141–2152. doi: 10.1111/j.1462-5822.2007.00945.x. [DOI] [PubMed] [Google Scholar]
- 37.Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis utilizes host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- 38.Mittal J, Miller NJ, Fischer E, Hackstadt T. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with host microtubule network. Cell Microbiol. 2010;12:1235–1249. doi: 10.1111/j.1462-5822.2010.01465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grieshaber SS, Grieshaber NA, Miller N, Hackstadt T. Chlamydia trachomatis causes centrosomal defects resulting in chromosomal segregation abnormalities. Traffic. 2006;7:940–949. doi: 10.1111/j.1600-0854.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- 40.Greene W, Zhong G. Inhibition of host cell cytokinesis by Chlamydia trachomatis infection. J Infect. 2003;47:45–51. doi: 10.1016/s0163-4453(03)00039-2. [DOI] [PubMed] [Google Scholar]
- 41.Johnson KA, Tan M, Sutterlin C. Centrosome abnormalities during a Chlamydia trachomatis infection are caused by a dysregulation of the normal duplication pathway. Cell Microbiol. 2009;11:1064–1073. doi: 10.1111/j.1462-5822.2009.01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naucler P, Chen HC, Persson K, You SL, Hsieh CY, Sun CA, Dillner J, Chen CJ. Seroprevalence of human papillomaviruses and Chlamydia trachomatis and cervical cancer risk: nested case-control study. J Gen Virol. 2007;88:814–822. doi: 10.1099/vir.0.82503-0. [DOI] [PubMed] [Google Scholar]
- 43.Brumell JH, Scidmore MA. Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol Mol Biol Rev. 2007;71:636–652. doi: 10.1128/MMBR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 45.Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect Immun. 2003;71:5855–5870. doi: 10.1128/IAI.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capmany A, Damiani MT. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through Rab14-mediated transport required for bacterial development and replication. PloS One. 2010;5:e14084. doi: 10.1371/journal.pone.0014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moorhead AR, Rzomp KA, Scidmore MA. The Rab6 effector Bicaudal D1 associates with Chlamydia trachomatis inclusions in a biovar-specific manner. Infect Immun. 2007;74:5632–5673. doi: 10.1128/IAI.01447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moorhead AM, Jung JY, Smirnov A, Kaufer S, Scidmore MA. Multiple host proteins that function in phosphatidylinositol-4 phosphate metabolism are recruited to the chlamydial inclusion. Infect Immun. 2010;78:1990–2007. doi: 10.1128/IAI.01340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rzomp KA, Moorhead AR, Scidmore MA. The GTPase Rab4 interacts with the Chlamydia trachomatis inclusion membrane proteins CT229. Infect Immun. 2006;74:5632–7373. doi: 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect Immun. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipinski AR, Heymann J, Meissner C, Karlas A, Brinkmann V, Meyer TF, Heuer D. Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS Pathog. 2009;5:e1000615. doi: 10.1371/journal.ppat.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ouellette S, Carabeo RA. A functional slow recycling pathway of transferrin is required for growth of Chlamydia. Front Microbio. 2010;1:1–12. doi: 10.3389/fmicb.2010.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Derré I, Pypaert M, Dautry-Varsat A, Agaisse H. RNAi screen in Drosophila cells reveals the involvement of the Tom Complex in Chlamydia infection. PLoS Pathog. 2007;3:1446–1458. doi: 10.1371/journal.ppat.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hilbi H. Modulation of phosphoinositide metabolism by pathogenic bacteria. Cell Microbiol. 2006;8:1697–1706. doi: 10.1111/j.1462-5822.2006.00793.x. [DOI] [PubMed] [Google Scholar]
- 55.van Ooij C, Kalman L, van Ijzendoorn S, Nishijima M, Hanada K, Mostov K, Engel JN. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cell Microbiol. 2000;2:627–637. doi: 10.1046/j.1462-5822.2000.00077.x. [DOI] [PubMed] [Google Scholar]
- 56.Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- 57.Moore ER, Fischer E, Mead DJ, Hackstadt T. The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin-containing exocytic vesicles. Traffic. 2008;9:2130–2140. doi: 10.1111/j.1600-0854.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beatty WL. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci. 2006;119:350–359. doi: 10.1242/jcs.02733. [DOI] [PubMed] [Google Scholar]
- 59.Beatty WL. Late endocytic multivesicular bodies intersect chlamydial inclusion in the absence of CD63. Infect Immun. 2008;76:2872–2881. doi: 10.1128/IAI.00129-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heuer D, Lipinski AR, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- 61.Goud B, Gleeson PA. TGN golgins Rabs and cytoskeleton: regulating the Golgi trafficking highways. Trends Cell Biol. 2010;20:329–336. doi: 10.1016/j.tcb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Sisko JL, Spaeth K, Kumar Y, Valdivia RH. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol Microbiol. 2006;60:51–66. doi: 10.1111/j.1365-2958.2006.05074.x. [DOI] [PubMed] [Google Scholar]
- 63.Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci USA. 2008;105:9379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su H, McClarty G, Dong F, Hatch G, Pan Z, Zhong G. Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J Biol Chem. 2003;279:9409–9416. doi: 10.1074/jbc.M312008200. [DOI] [PubMed] [Google Scholar]
- 65.Vignola MJ, Kashatus DF, Taylor GA, Counter CM, Valdivia RH. cPLA2 regulates expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J Biol Chem. 2010;285:21625–21635. doi: 10.1074/jbc.M110.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gurumurthy RK, Mauer AP, Machuy N, Hess S, Pleissner KP, Schurchhardt J, Rudel T, Meyer TF. A loss-of-function screen reveals Ras- and Raf- independent MEK-ERK signaling during Chlamydia trachomatis infection. Sci Signal. 2010;3:1–11. doi: 10.1126/scisignal.2000651. [DOI] [PubMed] [Google Scholar]
- 67.Delevoye C, Nilges M, Dautry-Varsat A, Subtil A. Conservation of the biochemical properties of IncA from Chlamydia trachomatis and Chlamydia caviae: oligomerization of IncA mediates interaction between facing membranes. J Biol Chem. 2004;279:46896–46906. doi: 10.1074/jbc.M407227200. [DOI] [PubMed] [Google Scholar]
- 68.Paumet F, Wesolowski J, Garcia-Diaz A, Delevoye C, Aulner N, Shuman H, Subtil A, Rothman JE. Intracellular bacteria encode inhibitory SNARE-like proteins. PloS One. 2009;4:e7375. doi: 10.1371/journal.pone.0007375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER. The Chlamydia trachomatis IncA protein is required for homotypic fusion. Cell Microbiol. 1999;1:119–130. doi: 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]