

Abstract
Autosomal dominant facioscapulohumeral muscular dystrophy (FSHD) has an unusual pathogenic mechanism. FSHD is caused by deletion of a subset of D4Z4 macrosatellite repeat units in the subtelomere of chromosome 4q. Recent studies provide compelling evidence that a retrotransposed gene in the D4Z4 repeat, DUX4, is expressed in the human germline and then epigenetically silenced in somatic tissues. In FSHD, the combination of inefficient chromatin silencing of the D4Z4 repeat and polymorphisms on the FSHD-permissive alleles that stabilize the DUX4 mRNAs emanating from the repeat, result in inappropriate DUX4 protein expression in muscle cells. FSHD is thereby the first example of a human disease caused by the inefficient repression of a retrogene in a macrosatellite repeat array.
Facioscapulohumeral muscular dystrophy
Facioscapulohumeral dystrophy (FSHD) was first described by two French neurologists in the late 19th century [1]. The disease was named after its clinical presentation to distinguish it from the well known Duchenne type of muscular dystrophy. Indeed, the core phenotype of FSHD involves progressive weakness and wasting of the facial (facio), shoulder and upper arm (scapulohumeral) muscles (for a detailed clinical description, see Box 1). There is currently no treatment available for FSHD (for outstanding questions, see Box 2) [2].
Box 1. Clinical manifestations of FSHD.
At disease onset, typically in the second decade of life, FSHD is characterized by initially restricted weakness of shoulder and facial muscles [1,48]. With progression, the lower extremities, both distal and proximal, become involved. The spectrum of disease severity is wide, ranging from mildly affected, asymptomatic individuals to severely affected wheelchair bound individuals (about 20%). Non-muscular manifestations include hearing loss and retinal vascular abnormalities that remain largely of no clinical consequence [49–50]. Rarely, however, the retinal vascular disease can result in an exudative retinopathy, Coat’s syndrome, that can result in significant loss of vision. There is no linear and inverse correlation between residual repeat size and disease severity and onset. However, patients having repeat arrays of 1–3 units usually have an infantile onset and rapid progression [51].
Box 2. Outstanding questions.
Which of the D4Z4-derived transcripts confer pathogenicity to the muscle? The D4Z4 repeat has a complex bidirectional expression profile, and it is currently not known how this profile changes in FSHD and which transcripts are causal to pathology.
What is the cause of FSHD2? Patients with FSHD2 are clinically indistinguishable from FSHD1 patients and show comparable chromatin changes in D4Z4, independent of repeat contraction. It is currently not known what causes these changes in D4Z4 chromatin structure.
What creates the bursts of DUX4 expression? Approximately 1:1,000 FSHD muscle nuclei shows robust expression of DUX4. It is currently unknown what causes these bursts of expression.
What is the function of DUX4? DUX4 is highly conserved and robustly expressed in testis, most likely germline. It contains two homeoboxes suggesting that it can bind DNA and may act as a transcription factor. However, its function, and how it confers toxicity to the muscle is as yet poorly understood.
Can we treat FSHD? Assuming that leaky expression of DUX4 is causal to FSHD, it is imperative to pursue strategies to interfere with DUX4 expression in muscle with the objective to ameliorate the disease. These may include RNA interference strategies, ways to silence the DUX4 locus, or molecules that neutralize DUX4 toxicity.
The major form of FSHD (FSHD1: MIM # 158900) is autosomal dominantly transmitted with linkage to the subtelomere of chromosome 4q [3]. The FSHD1 locus was mapped in 1990 [4], the first genetic condition to be mapped with polymorphic microsatellite repeats, and the nature of the genetic defect was resolved in 1993 [5]. Nevertheless, its pathogenic mechanism remains somewhat elusive. This review focuses on recent developments that establish FSHD as the first example of a human disease caused by the inefficient repression of a retrogene array.
FSHD is a repeat disease
FSHD1 is caused by a contraction of the highly polymorphic D4Z4 macrosatellite repeat (MSR) in chromosome 4q (Box 3, Figure 1) [6]. The D4Z4 macrosatellite repeat is located approximately 40–60 kb proximal to the telomere repeat and varies between 11 and >100 copies of D4Z4 units in the unaffected population [6–7]. The D4Z4 repeat unit is defined as a 3.3 kb KpnI fragment, and multiple units are ordered head-to-tail to form the D4Z4 repeat array. Most patients with FSHD1 have a partial and internal deletion of the repeat array leaving only 1–10 units on one of their chromosomes 4. It is believed that at least one unit of D4Z4 is necessary to develop FSHD, as monosomy of 4q does not cause FSHD [8].
Box 3. Repeat sequences in the human genome.
Approximately half of the human genome consists of repetitive DNA, and a significant proportion is organized in tandem arrays. These tandem arrays of DNA embody an extreme example of copy number variation and are classified according to their repeat unit size and their total length. Although different definitions exist, repeat unit sizes 1–4 nucleotides and spanning less than 100 bp are typically defined as microsatellite repeats. Those with repeat unit sizes between 10–40 nucleotides covering several hundreds of base pairs are referred to as minisatellite repeats. The term midisatellite repeat has been proposed for loci contain repeat units of 40–100 nucleotides that may extend over distances of 250 to 500 kb. Macrosatellite repeats, to which D4Z4 belongs, are the largest class of repeat arrays with unit sizes of > 100 nucleotides but which are typically much larger and can span hundreds of kilobases of DNA. While FSHD represents a macrosatellite repeat contraction disease, microsatellite repeat expansions are a frequent cause of neurodegenerative diseases.
Figure 1.
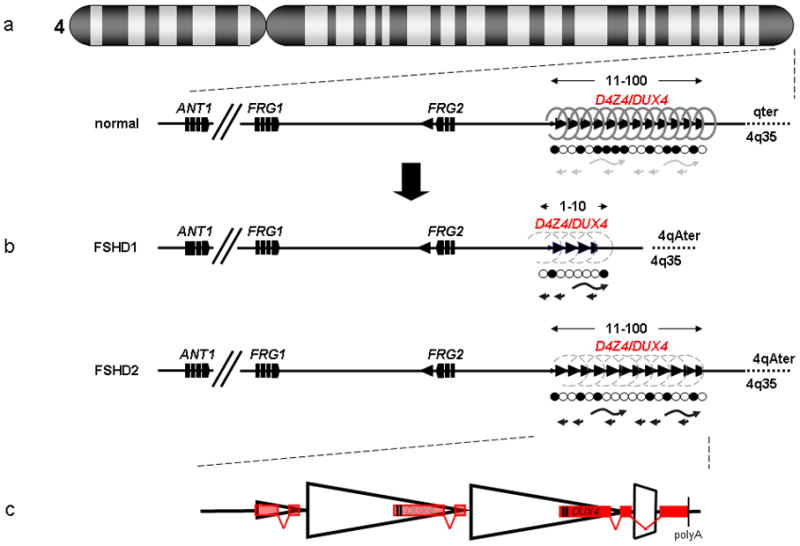
Schematic of the FSHD locus. (a) The D4Z4 repeat (triangles) is located in the subtelomere of chromosome 4q and can vary between 11–100 copies in the unaffected population. This repeat structure has a closed chromatin structure characterized by heterochromatic histone modifications (dense springs), high DNA methylation levels (closed circles) and complex bidirectional transcriptional activity (grey arrows). Candidate genes DUX4, FRG2, FRG1 and ANT1 are indicated. (b) In patients with FSHD, the chromatin structure of D4Z4 adopts a more open configuration (open springs and open circles) leading to inefficient transcriptional repression (black arrows) of the D4Z4 repeat. (c) The DUX4 gene is located within each D4Z4 unit. On permissive chromosomes, the last copy of the DUX4 genes splices to a third exon located in the region immediately flanking the repeat and stabilizing the transcript due to the presence of a polyadenylation (polyA) signal. Figure modified, with permission, from [52].
A contraction of the D4Z4 repeat array only predisposes to the disease as this contraction needs to occur on a specific chromosomal background [9]. Soon after the discovery of the D4Z4 repeat on chromosome 4, it was established that the subtelomere of chromosome 10q is almost identical to that of chromosome 4q and that it also contains a highly homologous and equally polymorphic repeat array [10–11]. A considerable proportion of individuals in the population carry 4q or 10q chromosome ends with repeat arrays that have apparently been entirely or partially transferred between both chromosomes [7,12–13]. However, contracted repeat arrays on chromosome 10 have until recently never been shown to cause FSHD [14–15]. The observation of linkage of the disease with chromosome 4 and the absence of linkage with chromosome 10 led to the hypothesis that interplay of D4Z4 with other, more proximal elements on chromosome 4 could explain the chromosome 4 specificity of the disease. In this scenario, either by spreading [16] or looping [17–18] mechanisms, the D4Z4 repeat contraction would affect the transcriptional regulation of proximal chromosome 4-specific genes. Indeed, closely located genes with high myopathic potential were reported to be transcriptionally upregulated [16] in FSHD, such as FRG1, FRG2 and ANT1. Variability between studies possibly due to biological differences between the tissues sampled and technical differences between the various studies, however, prevented a consensus agreement on any single mechanism [19].
Homologous is not identical
The D4Z4 repeat and its homolog are located in the subtelomere of chromosomes 4 and 10. Subtelomeres are unusual domains, showing a relatively high level of plasticity and resulting in the frequent transfer of sequences between homologous and non homologous chromosome ends [20].
To understand the chromosome 4 linkage with the disease, genetic studies were undertaken leading to the identification of the mechanism of D4Z4 rearrangements and to the identification of large polymorphisms in the subtelomere of chromosome 4q [21–23]. As approximately half of new FSHD cases arise as a consequence of a postzygotic rearrangement of the repeat leading to somatic mosaicism for the D4Z4 repeat contraction, these mosaic cases were instrumental for studying the genetic mechanism [23]. It became apparent that the preferred template for these rearrangements was the sister chromatid, instead of the homologous chromosome, making a frequent occurrence of sequence transfers between homologous chromosomes or between chromosomes 4 and 10 unlikely [23]. In addition, a large polymorphism was identified on chromosome 4 that involved the presence or absence of a β-satellite repeat immediately distal to the D4Z4 repeat array, identifying two major haplotypes of chromosome 4 called 4A (with β-satellite repeat) and 4B (without β-satellite repeat) [21]. On chromosome 10, only the A variant was identified. While 4A and 4B chromosomes are almost equally common in the population, FSHD chromosomes seemed to be exclusively of the 4A type [22], which was later confirmed in independent studies [24–25]. Thus it was concluded that attributes specific to 4A, presumably polymorphisms, confer permissiveness to the D4Z4 repeat.
This led to focused attention on identifying allele-specific polymorphisms associated with FSHD. Initially, a simple sequence length polymorphism (SSLP) was identified immediately proximal to the D4Z4 repeat and was instrumental in our understanding of the genetic basis of FSHD [9,26]. Studies in patients and control individuals from different populations showed that during recent human evolution, there were likely only 4 events in which sequences were transferred between chromosomes 4 and 10. Subsequent detailed genetic studies of the FSHD locus led to the identification of additional polymorphisms subdividing chromosome 4 into at least 17 genetically distinct subtelomeric variants and chromosome 10 into 8 subtelomeric variants. Intriguingly, contractions in only three genetically almost identical chromosome 4 subtelomeres, the common variant 4A161 and the rare variants 4A159 and 4A168, caused FSHD while contractions in other 4q subtelomeres were not associated with disease [9,26–28]. This finding provided strong evidence that genetic factors in the subtelomere of chromosome 4 contribute to FSHD pathology, and indeed detailed sequence analysis of the first and last repeat unit of the array of the most common chromosomal backgrounds identified 4A161-specific sequence variants [26,28]. Thus, not only outside the D4Z4 repeat, but also within the repeat array, it was shown that the permissive chromosomes contained specific sequence variants not shared by other (non-permissive) chromosome ends [26,28]. In summary, these detailed genetic studies revealed that each chromosome maintained specific polymoprhisms, and that specific sequences in the FSHD-permissive variants of 4A confer permissiveness to this repeat.
The D4Z4 unit and its transcriptional landscape
The sequence of the D4Z4 repeat contains the open reading frame (ORF) of a double homeobox transcription factor, DUX4 (Figure 1) [30–31]. The DUX4 ORF is in a single exon, whereas other members of the double-homeobox family have multiple introns, indicating that DUX4 was inserted into the genome as a retrotransposed mRNA from an intron containing the DUX gene, possibly DUXC or less likely Duxbl [32–34]. In contrast to the many pseudogenes retrotransposed to our genome, the DUX4 retrogene maintained a conserved ORF [32]. However, it is unclear whether the conservation of the ORF was a consequence of a conserved functional role propagated to all repeats by concerted evolution.
Although initial attempts to identify DUX4 mRNA expression in normal development or disease were unsuccessful [35], a major advance in understanding FSHD was the identification of poly-adenylated mRNA containing the DUX4 ORF using RT-PCR [36]. Initially identified only in FSHD muscle samples, the polyadenylation site of the DUX4 mRNA was mapped to the region immediately telomeric to the last D4Z4 repeat, a region previously cloned from a phage clone containing the D4Z4 repeat and flanking sequences and called pLAM1 [5]. It was proposed that the contraction of the D4Z4 array results in the transcription of the DUX4 retrogene [36]; however, the abundance of the DUX4 mRNA and protein was extremely low.
In addition, a later study [35] identified D4Z4 and DUX4 transcripts in both FSHD and control muscle. Random priming of RNA identified both sense and anti-sense transcripts throughout the D4Z4 region. Regions of lower transcript abundance correlated with the presence of small si- or mi-RNA-sized fragments, and it was suggested that these bidirectional transcripts and small RNAs contribute to the heterochromatin suppression of this region [35], as discussed in the next section. In addition, several splice forms of a DUX4 polyadenylated mRNA were identified that used the pLAM1 polyadenylation site, but these DUX4 transcripts were also at extremely low abundance [35]. Several groups demonstrated that relatively high levels of DUX4 expression was pathologic to muscle cells and other cell types [37–40]. Therefore, if DUX4 could be shown to be expressed at sufficient levels in FSHD, then it was likely to be a major cause of the muscle pathology.
Chromatin studies
A third important clue that could explain how a repeat contraction can cause disease came from chromatin studies of the D4Z4 array. The D4Z4 repeat is GC rich and contains sequences often residing in heterochromatic domains of the genome [41]. It was therefore postulated that normally the D4Z4 repeat is in a relatively closed chromatin configuration and that, as a consequence of repeat contraction, it would adopt a more open chromatin configuration (Figure 1). DNA methylation studies and studies of histone modifications and other chromatin factors supported this hypothesis [17,42–43]. Normally, the D4Z4 repeat is densely DNA methylated; FSHD chromosomes experience an approximate 30–40% reduction of DNA methylation at specific sites tested in D4Z4. In addition, chromatin immunoprecipitation (ChIP) studies showed that the D4Z4 repeat is normally occupied by both transcriptionally repressive as well as permissive histone modifications, while in FSHD chromosomes, there is a relative loss of repressive histone modifications. These changes in chromatin structure are restricted to the D4Z4 repeat and do not seem to spread proximally. ChIP studies also identified the losses or gains of other chromatin factors such as HP1γ, the cohesin complex, YY1 (losses) and CTCF (gain) at D4Z4 of disease alleles [16,43–44]. Overall, the data support a model in which D4Z4 in FSHD chromosomes adopts a relatively open chromatin structure facilitating the transcriptional activity of the repeat and possibly affecting the processing of the different D4Z4 transcripts.
Interestingly, similar changes in the chromatin structure of D4Z4 were also identified in a small cohort of patients whose disease status could not be confirmed by standard molecular diagnostic tests [43,45–46]. These patients, now classified as FSHD2, have normal, but compared to the general population, smaller-sized D4Z4 repeat arrays; disease alleles of FSHD2 patients do show similar changes in D4Z4 chromatin structure as those of FSHD1 patients [46]. In contrast to FSHD1 where the relative relaxation of the D4Z4 chromatin structure seems restricted to the contracted allele, in patients with FSHD2, the D4Z4 repeats on both chromosomes 4 and 10 seem to be affected. Other repeat structures in the genome of these patients seem to be normally structured [47] and the cause for this change in chromatin structure of D4Z4 in patients with FSHD2 is currently not known. Also in common with FSHD1, patients with FSHD2 have at least one permissive (4A161) chromosome [46]. Thus it seems that patients with FSHD1 and FSHD2 share the commonality of a relative chromatin relaxation of D4Z4 on the genetic 4A161 background.
A new developmental model for FSHD
Very recently, genetic studies directly demonstrated the requirement of the DUX4 polyadenylation site for FSHD [28], and molecular studies have produced a new developmental model for the disease that is consistent with the extremely low abundance of the mRNA and protein [29] (Figure 2). Together, these studies explain the apparent discrepancies in previous models of FSHD and provide compelling support for the expression of DUX4 as a major cause of FSHD.
Figure 2.
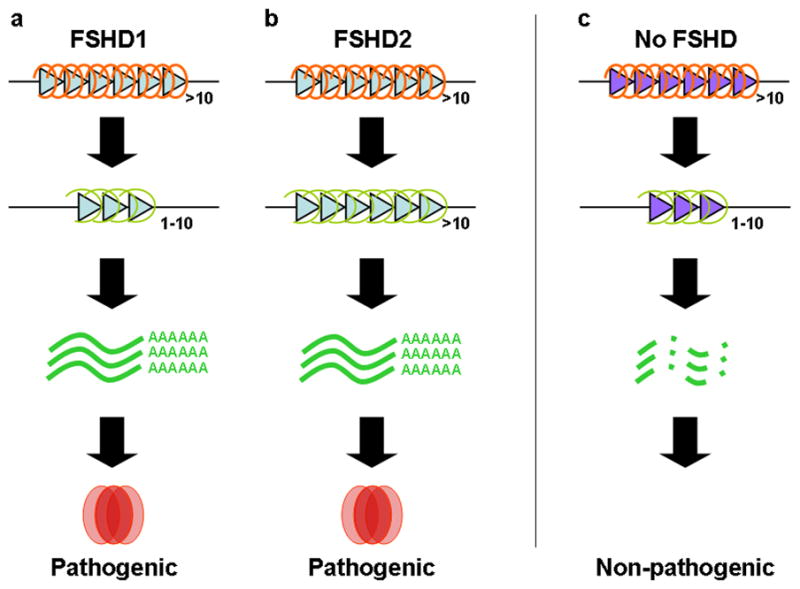
A unifying mechanism for FSHD. Upon (a) contraction of the D4Z4 repeat (FSHD1) or by (b) a yet unknown mechanism (FSHD2), the D4Z4 repeat array (triangles) adopts a more open chromatin configuration (orange > green dots) leading to the leaky expression of DUX4 mRNA. On permissive chromosomes, this mRNA is stabilized due to the presence of a canonical polyadenylation signal immediately distal to the D4Z4 repeat array. (c) Non-permissive chromosomes do not have this polyadenylation signal and therefore DUX4 mRNA gets rapidly degraded. The DUX4 mRNA encodes for a nuclear double homeobox protein that when expressed in muscle induces apoptosis.
Meticulous genetic analysis of patients with unusual hybrid D4Z4 repeat structures containing units with sequence signatures consistent with those originating from chromosomes 4 and 10 revealed a common last portion of the D4Z4 repeat array and flanking pLAM1 sequence [28]. This old-fashioned positional cloning strategy strongly argued that the distal end of the repeat array and flanking pLAM1 sequences are critically important for the development of FSHD [28]. Further corroborating this finding was the identification of an FSHD family in which the disease segregated with a contracted D4Z4 allele of chromosome 10. Importantly, the last part of this disease-associated repeat array was replaced by permissive chromosome 4 sequences. The identification of this family in which FSHD segregates with chromosome 10 essentially confirms the importance of the distal end of the repeat and pLAM1 sequences and precludes a prominent role for other proximal candidate genes on chromosome 4. Further genetic studies of the distal end of the D4Z4 repeat array and flanking sequences allowed an almost perfect separation of permissive and non-permissive chromosome ends based on the identification of consistent polymorphisms in the region sequenced [28]. One noticeable difference between permissive chromosomes 4 and non-permissive chromosomes 10 was the presence of a DUX4 polyadenylation signal on chromosome 4, while on chromosome 10 this polyadenylation signal was lost because of the presence of independent polymorphisms [28]. Indeed, when transfecting the critical region in murine C2C12 muscle cells, stable DUX4 transcripts making efficient use of the DUX4 polyadenylation signal could only be identified when constructs derived from permissive chromosomes with a polyadenylation signal were transfected [28]. Interestingly, a study of the recent hominoid evolution of the 4q subtelomere shows that the permissive chromosome end is ancestral to all 4q chromosome ends, and the data are consistent with an evolutionary pressure to eliminate the third DUX4 exon in pLAM1 [26]. These genetic studies clearly demonstrated the requirement for the polyadenylation site utilized by DUX4 mRNA and, therefore, strongly implicated DUX4 protein as a cause of FSHD. However, as noted above, although DUX4 mRNA was detected in FSHD muscle, it was still at extremely low abundance.
Low abundance mRNA in a population of cells could reflect either a small amount of mRNA in all cells or an abundant amount of mRNA in just a few cells. RT-PCR amplification of DUX4 mRNA in small pools of 100 or 600 differentiated FSHD muscle cells identified relatively abundant transcripts in a subset of the pools [29]. The frequency of positive pools suggested that approximately one-in-1000 FSHD muscle cell nuclei were expressing an abundant amount of DUX4 mRNA. Immuno-detection confirmed that approximately 0.1% of nuclei in cultured FSHD muscle cells expressed an abundant amount of protein. In addition, the DUX4-expressing FSHD muscle nuclei had characteristics consistent with DUX4 induced toxicity, i.e. an aggregation of nuclear DUX4 protein that occurs coincident with DUX4-induced apoptosis. Therefore, the very low abundance of DUX4 mRNA in FSHD muscle represented relatively abundant amounts of DUX4 mRNA and protein in a small subset of the nuclei, likely leading to dysfunction or death of those DUX4-expressing nuclei.
As a retrogene, DUX4 was viewed by some as a dead gene, or pseudogene, brought ‘back to life’ by the contraction of the D4Z4 array, but the conservation of the ORF suggested otherwise [32–33]. Indeed, DUX4 mRNA and protein was shown to be highly expressed in the testes of unaffected individuals, most likely in the germline, both from the permissive 4A161 allele as well as from the non-permissive alleles [29]. It is interesting to note that the germline DUX4 mRNA transcripts from chromosome 10, which lacks the polyadenylation site that is present on the permissive chromosomes, use an alternative polyadenylation site approximately six kilobases telomeric to the end of the D4Z4 array. Use of this alternative polyadenylation was restricted to germline tissues and not identified in somatic tissues [29]. Therefore, the DUX4 retrogene is expressed in early development, i.e., in the human germline, and is epigenetically silenced in somatic tissues. The inefficient chromatin-mediated repression, either related to the contraction of the array in FSHD1 or through unknown mechanisms in FSHD2, results in the occasional escape from repression in muscle cells, and possibly other somatic cells. In this model, the muscle cell nuclei would be lost over time in FSHD because of the inappropriate expression of DUX4 protein (Figure 2).
Summary
These recent studies substantiate a developmental model of FSHD that explains many of the previously unexplained mysteries of this human disease. First, genetic studies demonstrate the requirement for the DUX4 polyadenylation site in the pLAM1 region of the permissive alleles, indicating that DUX4 mRNA is critical for FSHD [28]. Second, molecular studies show decreased density of repressive chromatin modifications in both FSHD1 and FSHD2 [6], indicating that DUX4 mRNA is more likely to be expressed. Third, RNA and protein studies showed an occasional escape from the inefficient chromatin repression leading to high levels of DUX4 expression in a small number of nuclei in FSHD muscle cells [29]. Fourth, abundant expression of DUX4 in testis, most likely the germline cells, indicates that this retrogene might have a normal role in germ cell development [29]. Finally, if a retrogene has subsumed a normal role in germ cell biology, then repression of that gene in somatic cells needs to co-opt regulatory mechanisms distinct from the evolved enhancers and promoters of the parental gene. Therefore, the repression of DUX4 in somatic cells is likely a mechanism adopted from other loci, such as the mechanism of silencing retrotransposons and other repetitive elements, and is not highly evolved for the DUX4 locus. In this case, co-opted mechanisms might not be sufficiently robust to avoid disease, as is evident by the association of FSHD with the contracted D4Z4 array.
This new unifying and substantiated model of FSHD has one additional profound and as yet unexplored implication. DUX4 arose from the retrotransposition of a parental DUX mRNA, possibly either DUXC or less likely Duxbl [32–34]. Both DUXC and Duxbl are expressed in the germline, a requirement for introducing a retrogene into the population, but neither are present in primates [34]. Therefore, primates have retained the retrogene and lost the parental gene, suggesting a selective advantage of the retrogene. Primates have sacrificed upper extremity and facial muscle mass for the advantage of an upright posture and highly expressive facial muscles. Although this remains highly speculative, it is interesting to suggest that the DUX4 retrogene might have been retained in preference to the parental gene because inefficient chromatin repression results in sufficient expression in skeletal muscle to modulate facial and upper extremity muscle mass, even in individuals without the FSHD deletion. If this is correct, then FSHD is a hypermorphic phenotype for traits that are critical for primate evolution, and the mystery of FSHD still has the potential to lead us to new understandings of human biology.
Acknowledgments
Our work is supported by the Fields Center for FSHD and Neuromuscular Research, the National Institutes of Health (NINDS P01NS069539, NIAMS R01AR045203 and NIAMS R21AR059966), the Muscular Dystrophy Association (173202), the Prinses Beatrix Fonds (WAR08-14), the Shaw Family Foundation, the FSH Society, the Dutch FSHD Foundation, and the Pacific Northwest Friends of FSH Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Silvère M van der Maarel, Email: Maarel@lumc.nl.
Rabi Tawil, Email: Rabi_Tawil@URMC.Rochester.edu.
Stephen J. Tapscott, Email: stapscot@fhcrc.org.
References
- 1.Pandya S, et al. Facioscapulohumeral dystrophy. Phys Ther. 2008;88:105–113. doi: 10.2522/ptj.20070104. [DOI] [PubMed] [Google Scholar]
- 2.Tawil R, van der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34:1–15. doi: 10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 3.Wijmenga C, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 4.Wijmenga C, et al. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336:651–653. doi: 10.1016/0140-6736(90)92148-b. [DOI] [PubMed] [Google Scholar]
- 5.van Deutekom JC, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 6.de Greef JC, et al. Epigenetic mechanisms of facioscapulohumeral muscular dystrophy. Mutat Res. 2008;647:94–102. doi: 10.1016/j.mrfmmm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Deutekom JC, et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum Mol Genet. 1996;5:1997–2003. doi: 10.1093/hmg/5.12.1997. [DOI] [PubMed] [Google Scholar]
- 8.Tupler R, et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemmers RJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–894. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakker E, et al. The FSHD-linked locus D4F104S1 (p13E-11) on 4q35 has a homologue on 10qter. Muscle Nerve. 1995;2:39–44. [PubMed] [Google Scholar]
- 11.Deidda G, et al. Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the Facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur J Hum Genet. 1995;3:155–167. doi: 10.1159/000472291. [DOI] [PubMed] [Google Scholar]
- 12.Lemmers RJ, et al. Inter- and intrachromosomal subtelomeric rearrangements on 4q35: implications for facioscapulohumeral muscular dystrophy (FSHD) aetiology and diagnosis. Hum Mol Genet. 1998;7:1207–1214. doi: 10.1093/hmg/7.8.1207. [DOI] [PubMed] [Google Scholar]
- 13.van Overveld PG, et al. Interchromosomal repeat array interactions between chromosomes 4 and 10: a model for subtelomeric plasticity. Hum Mol Genet. 2000;9:2879–2884. doi: 10.1093/hmg/9.19.2879. [DOI] [PubMed] [Google Scholar]
- 14.Lemmers RJ, et al. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann Neurol. 2001;50:816–819. doi: 10.1002/ana.10057. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, et al. Improved characterization of FSHD mutations. Ann Genet. 2001;44:105–110. doi: 10.1016/s0003-3995(01)01075-9. [DOI] [PubMed] [Google Scholar]
- 16.Gabellini D, et al. Inappropriate Gene Activation in FSHD. A Repressor Complex Binds a Chromosomal Repeat Deleted in Dystrophic Muscle. Cell. 2002;110:339–248. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 17.Jiang G, et al. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet. 2003;12:2909–2921. doi: 10.1093/hmg/ddg323. [DOI] [PubMed] [Google Scholar]
- 18.Bodega B, et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41. doi: 10.1186/1741-7007-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dmitriev P, et al. Pearls in the junk: dissecting the molecular pathogenesis of facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2009;19:17–20. doi: 10.1016/j.nmd.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3:91–102. doi: 10.1038/nrg727. [DOI] [PubMed] [Google Scholar]
- 21.van Geel M, et al. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics. 2002;79:210–217. doi: 10.1006/geno.2002.6690. [DOI] [PubMed] [Google Scholar]
- 22.Lemmers RJ, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–236. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 23.Lemmers RJ, et al. Mechanism and timing of mitotic rearrangements in the subtelomeric D4Z4 repeat involved in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2004;75:44–53. doi: 10.1086/422175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas NS, et al. A large patient study confirming that facioscapulohumeral muscular dystrophy (FSHD) disease expression is almost exclusively associated with an FSHD locus located on a 4qA-defined 4qter subtelomere. J Med Genet. 2007;44:215–218. doi: 10.1136/jmg.2006.042804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ZQ, et al. Distinguishing the 4qA and 4qB variants is essential for the diagnosis of facioscapulohumeral muscular dystrophy in the Chinese population. Eur J Hum Genet. 2010 Aug 25; doi: 10.1038/ejhg.2010.143. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemmers RJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete duplication events in human evolution. Am J Hum Genet. 2010;86:364–377. doi: 10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spurlock G, et al. Confirmation that the specific SSLP microsatellite allele 4qA161 segregates with fascioscapulohumeral muscular dystrophy (FSHD) in a cohort of multiplex and simplex FSHD families. Muscle Nerve. 2010;42:820–821. doi: 10.1002/mus.21766. [DOI] [PubMed] [Google Scholar]
- 28.Lemmers RJ, et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snider L, et al. Facioscapulohumeral Dystrophy: Incomplete Suppression of a Retrotransposed Gene. PLoS Genet. 6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewitt JE, et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet. 1994;3:1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 31.Gabriels J, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 32.Clapp J, et al. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:264–279. doi: 10.1086/519311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu SL, et al. Characterization of genomic structures and expression profiles of three tandem repeats of a mouse double homeobox gene: Duxbl. Dev Dyn. 2010;239:927–940. doi: 10.1002/dvdy.22210. [DOI] [PubMed] [Google Scholar]
- 34.Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol. 2010;10:364. doi: 10.1186/1471-2148-10-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snider L, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixit M, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowaljow V, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Bosnakovski D, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosnakovski D, et al. Biphasic myopathic phenotype of mouse DUX, an ORF within conserved FSHD-related repeats. PLoS One. 2009;4:e7003. doi: 10.1371/journal.pone.0007003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wuebbles RD, et al. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 41.Lyle R, et al. The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics. 1995;28:389–397. doi: 10.1006/geno.1995.1166. [DOI] [PubMed] [Google Scholar]
- 42.van Overveld PG, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 43.Zeng W, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ottaviani A, et al. Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J. 2009;28:2428–2436. doi: 10.1038/emboj.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Greef JC, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–1459. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 46.de Greef JC, et al. D4Z4 repeat contraction-independent FSHD2 is clinically identical to FSHD1. Neurology. 75:1548–1554. [Google Scholar]
- 47.de Greef JC, et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology. 2007;69:1018–1026. doi: 10.1212/01.wnl.0000271391.44352.fe. [DOI] [PubMed] [Google Scholar]
- 48.Padberg GW. Thesis. Leiden University; 1982. Facioscapulohumeral disease. [Google Scholar]
- 49.Fitzsimons RB, et al. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain. 1987;110:631–648. doi: 10.1093/brain/110.3.631. [DOI] [PubMed] [Google Scholar]
- 50.Padberg GW, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:S73–S80. [PubMed] [Google Scholar]
- 51.Lunt PW, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35- facioscapulohumeral muscular dystrophy (FSHD) Hum Mol Genet. 1995;4:951–958. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 52.Wilbers J, et al. Facioscapulohumerale spierdystrofie. Ned Tijdschr Tandheelkd. 2010;117:11–14. [PubMed] [Google Scholar]