

Abstract
We evaluated the genetic contribution of the leukotriene (LT) pathway to risk of coronary artery disease (CAD) in 4,512 Caucasian and African American subjects ascertained through elective cardiac evaluation. Of the three previously associated variants, the shorter “3” and “4” alleles of a promoter repeat polymorphism in ALOX5 increased risk of CAD in African Americans (OR = 1.4, 95% CI 1.0–1.9; p = 0.04), whereas a haplotype of LTA4H (HapK) was associated with CAD in Caucasians (OR = 1.2, 95% CI 1.01–1.4; p = 0.03). In Caucasians, first-stage analysis of 254 haplotype-tagging SNPs in 15 LT pathway genes with follow-up of 19 variants in stage 2 revealed an LTA4H SNP (rs2540477) that increased risk of CAD (OR = 1.2, 95% CI 1.1–1.5; p = 0.003) and a PLA2G4A SNP (rs12746200) that decreased risk of CAD (OR = 0.7, 95% CI 0.6–0.9; p = 0.0007). The PLA2G4A rs12746200 variant also decreased risk of experiencing a major adverse cardiac event (MACE = myocardial infarction, stroke, or death) over 3 years of follow-up (HR = 0.7, 95% CI 0.5–0.9; p = 0.01), consistent with its cardioprotective effect. Functional experiments demonstrated that stimulated monocytes from carriers of LTA4H variants HapK or rs2540477 had 50% (p = 0.002) and 33% (p = 0.03) higher LTB4 production, respectively, compared to non-carriers. These ex vivo results are consistent with LTB4 being the direct product of the reaction catalyzed by LTA4H and its role in promoting monocyte chemotaxis to sites of inflammation, including the artery wall of atherosclerotic lesions. Taken together, this study provides additional evidence that functional genetic variation of the LT pathway can mediate atherogenic processes and the risk of CAD in humans.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-011-0963-3) contains supplementary material, which is available to authorized users.
Introduction
Class four leukotrienes (LTs) are potent pro-inflammatory mediators synthesized from arachidonic acid, an omega-6 polyunsaturated fatty acids (PUFAs) (Peters-Golden and Henderson 2007). The rate-limiting step in this pathway is catalyzed by the enzyme arachidonic acid 5-lipoxygenase (ALOX5). The biologically active LTs are synthesized by subsequent conversion to LTB4 and the cysteinyl LTs (LTC4, LTD4, and LTE4) via enzymatic reactions by LTA4 hydrolase (LTA4H) and LTC4 synthase (LTC4S), respectively (Peters-Golden and Henderson 2007). LTs then affect the function of target cells, including monocytes and other pro-inflammatory leukocytes, through receptor-mediated signal transduction.
While LTs have long been known to be involved in chronic allergic inflammatory conditions, such as asthma, the LT pathway has also recently garnered attention for its potential role in coronary artery disease (CAD)-related traits. This stems from a series of biochemical, genetic, and pharmacological studies over the last few years that have provided evidence for the pro-atherogenic role of LTs (Back and Hansson 2006; Mehrabian and Allayee 2003; Tymchuk et al. 2006). For example, genetic deficiency for ALOX5 in mice protects against aortic lesion formation and leads to other metabolic disturbances (Mehrabian et al. 2002, 2005, 2008). Other mouse studies have reported the involvement of LT pathway genes in atherosclerosis-related traits as well, including the LT receptors and ALOX5 activating protein (ALOX5AP) (Ahluwalia et al. 2007; Aiello et al. 2002; Heller et al. 2005; Jawien et al. 2006, 2008; Subbarao et al. 2004).
Studies in humans have also provided evidence supporting the notion that LTs participate in atherosclerotic processes. Immunohistochemical studies have shown that ALOX5, ALOX5AP, and LTA4H are abundantly expressed in arterial walls of CAD patients, with ALOX5 having markedly increased expression in advanced lesions and localizing to macrophages, dendritic cells, and neutrophilic granulocytes (Qiu et al. 2006; Spanbroek et al. 2003). In addition, individuals carrying the shorter alleles of a functional ALOX5 promoter polymorphism, consisting of tandem Sp1 binding sites, have significantly increased carotid atherosclerosis and risk of myocardial infarction (MI), particularly in the context of high dietary arachidonic acid levels (Allayee et al. 2008; Dwyer et al. 2004). This is supported by studies that have reported associations between other ALOX5 and ALOX5AP variants with CAD-related phenotypes (Burdon et al. 2010; Carlson et al. 2007; Crosslin et al. 2009; Helgadottir et al. 2004). More recently, a 10-SNP haplotype of LTA4H, designated HapK, has been associated with MI in Caucasians and African Americans, with a more pronounced effect in the latter group (Helgadottir et al. 2006), and LTC4S variants have been associated with surrogate measures of CAD, including coronary artery calcification and carotid atherosclerosis (Iovannisci et al. 2007). Importantly, these genetic studies are bolstered by functional data showing that the associated variants/haplotypes lead to increased gene expression or LT production (Allayee et al. 2008; Helgadottir et al. 2004, 2006; Sanak et al. 2000; Vikman et al. 2009). Despite these reports, evidence for association of LT pathway genes with CAD traits has not been consistently observed across all studies (Assimes et al. 2008; Koch et al. 2007; Zee et al. 2006; Zintzaras et al. 2009). Thus, the aim of the present study was to comprehensively evaluate the genetic contribution of the LT pathway to CAD in a large cohort of subjects undergoing elective cardiac evaluation.
Materials and methods
Study subjects
GeneBank is a single site (Cleveland Clinic) sample repository generated from patients undergoing elective diagnostic coronary angiography or elective cardiac computed tomographic angiography with extensive clinical and laboratory characterization and longitudinal observation (Bhattacharyya et al. 2008; Nicholls et al. 2010). Ethnicity was self-reported and information regarding demographics, medical history, and medication use was obtained by patient interviews and confirmed by chart reviews. All clinical outcome data were verified by source documentation. CAD was defined as adjudicated diagnoses of stable or unstable angina, MI (adjudicated definition based on defined electrocardiographic changes or elevated cardiac enzymes), angiographic evidence of ≥50% stenosis of one or more major epicardial vessel, and/or a history of known CAD (documented MI, CAD, or history of revascularization). Prospective cardiovascular risk was assessed by the incidence of major adverse cardiac events (MACE) during 3 years of follow-up from the time of enrollment, which included nonfatal MI, nonfatal stroke, and all-cause mortality. Nonfatal events were defined as MI or stroke in patients who survived at least 48 h following the onset of symptoms. Adjudicated outcomes ascertained over the ensuing 3 years for all subjects following enrollment were confirmed using source documentation. All patients provided written informed consent prior to being enrolled in GeneBank and the study was approved by the Institutional Review Board of the Cleveland Clinic. The present genetics study was approved by the Institutional Review Boards of the Cleveland Clinic and USC Keck School of Medicine.
Clinical laboratory measurements
Samples were collected from overnight fasted subjects on the day of elective cardiac catheterization. Plasma aliquots were isolated from whole blood collected into EDTA tubes, maintained at 0–4°C immediately following phlebotomy, processed within 4 h of blood draw, and stored at −80°C until analysis. Plasma levels of total cholesterol, low-density lipoproteins (LDL), high-density lipoproteins (HDL), triglycerides, and high sensitivity C-reactive protein (CRP) were measured on the Abbott ARCHITECT platform (Abbott Diagnostics, Abbott Park IL).
Genotyping
Genomic DNA was extracted from isolated buffy coats using DNeasy isolation kits (Qiagen, Valencia, CA). Genotyping of the 274 haplotype-tagging SNPs was carried out using the Illumina GoldenGate System, which involves using allele-specific primer extension in combination with multiplex PCR with universal primers. Technical details regarding this methodology are available from Illumina, Inc. (http://www.illumina.com). Haplotype-tagging SNPs were selected using the HapMap data for Caucasians and the Tagger program (de Bakker et al. 2005). Genotyping of previously associated SNPs/variants and those selected for replication in stage 2 was performed using either fragment analysis, as described elsewhere (Dwyer et al. 2004), or the TaqMan Allelic Discrimination system from Applied Biosystems, Inc. (Foster City, CA) (Livak 1999, 2003). For determination of HapA and HapK, we genotyped the same SNPs as reported by deCode Genetics (Helgadottir et al. 2004, 2006), which were rs17222814, rs10507391, rs4769874, and rs9551963 for HapA and rs61937881 (SG12S16), rs2660880, rs6538697, rs1978331, rs17677715, rs2247570, rs2660898, rs2540482, rs2660845, and rs2540475 for HapK (Supplemental Table 1).
Statistical analyses
Prior to analysis, all variants were tested for Hardy–Weinberg equilibrium in subjects without CAD using a χ 2 test. SNPs deviating from HWE (p < 0.05) were excluded from further analysis. Haplotypes of ALOX5AP (HapA) and LTA4H (HapK) were estimated using an expectation-maximization (EM) algorithm to generate maximum likelihood estimates of haplotype frequencies, which assigns the probability that each individual possesses a particular haplotype pair. Unconditional multiple logistic regression was used to independently test for association with CAD with adjustment for age and sex under dominant genetic models (as a means to increase sample size and power). The fully adjusted regression models included age, sex, medication use, plasma CRP levels, alcohol consumption, and Framingham ATP-III risk score (which includes smoking and diabetes status) and the results are reported as odds ratios (OR) with 95% confidence intervals (CI). Since we were testing specific hypotheses with respect to the HapA and HapK haplotypes of ALOX5AP and LTA4H, respectively, these analyses were performed as tests of a single haplotype compared to all other haplotypes (i.e., carriers of HapA or HapK vs. non-carriers). For analyses of HapK, we also performed a haplotype score test with all haplotypes having frequencies greater than 1%, as implemented in the Haplo.Stats package. All analyses were performed using SAS version 9.2 (SAS Institute Inc, Cary, NC) or R 2.10.1 (http://www.R-project.org) and carried out separately in Caucasian and African American subjects.
Kaplan–Meier failure estimates were plotted to illustrate differences in rates for developing a MACE over 3 years of follow-up as a function of genotype. Subjects experiencing a MACE within 14 days of enrollment were excluded. Due to sample size limitations, we assumed dominant genetic models and significance was assessed using a log-rank test. Relative risk for experiencing a MACE was assessed using Cox proportional hazard models with adjustment for age, sex, medication use, plasma CRP levels, alcohol consumption, and Framingham ATP-III risk score (which includes diabetes status). Other variables tested, but not included in the final model, were BMI and smoking. Adjusted hazard ratio (HR) and 95% CI are reported with 2-sided p values that were considered significant when <0.05. Time-to-event analyses were carried out using Stata 8.2 (StataCorp LP, College Station, TX).
Monocyte samples for functional studies
Monocyte samples for the functional analysis of genetic variants came from the baseline visit of a randomized, double blind, placebo-controlled intervention trial to examine the effect of omega-3 fatty acid supplementation in subjects with different ALOX5 promoter variants. The registry number for this study is NCT00536185 and details of the study design can be found at the ClinicalTrials.gov website. Healthy adults between 20 and 59 years of age who had not been diagnosed with any major disorder and who self-identified as African American, Black, or a person of African descent, were recruited into the study from three study sites in Davis, Sacramento, and Oakland, California. Potential study participants received a brief interview to characterize general health, previous diagnosis of major diseases, smoking and alcohol usage, medication, nutritional supplements, and other factors. Eligible subjects that had one of the six 5-LO promoter repeat genotypes of interest (33, 34, 44, 35, 45, 55) were subsequently invited to participate in the study. A fasting blood sample was used for a complete blood count, lipid and chemistry panel analysis to identify any undiagnosed medical problems. Study participants that had a physician-diagnosed chronic inflammatory disease (arthritis, autoimmune disease, or asthma), CVD, hypertension, diabetes, or a lipid disorder that required regular use of anti-inflammatory or lipid-lowering medication were excluded. Subjects with abnormal results on standard chemistry and lipid panels or a complete blood count that suggested underlying undiagnosed disease were also excluded and referred to their physician for further evaluation. The institutional review boards of The University of California, Davis, Alta Bates Summit Medical Center, the USC Keck School of Medicine reviewed and approved ethical permission for all procedures involving human volunteers and the protocols. Written informed consent was obtained from all study participants.
Cell isolations
At the baseline visit subsequent to enrollment, a fasting blood sample (80 ml) from each participant was collected into heparinized tubes and processed within 4 h. Plasma was separated after low-speed centrifugation and the buffy coat containing mononuclear cells (lymphocytes and monocytes) and granulocytes was then removed and diluted with approximately one volume of Hank’s Balanced Salt Solution (HBSS) into a total volume of approximately 32 ml. Monocytes were purified using two-step gradients and washed once with HBSS, followed by positive selection using CD14-labeled magnetic microbeads with an LS magnetic column (Miltenyi Biotec, Auburn, CA), as described previously (Vikman et al. 2009). Purity of monocytes was assessed by FACS analysis (BD FACSCalibur) using PE-labeled anti-CD14 antibody (Miltenyi Biotec) and an isotype control reagent (IgG2a; BD Biosciences, San Jose, CA). The median percent purity (25th/75th percentiles) of monocytes was 93% (89%/96%). SNP genotyping was carried out as described above.
Real-time RNA quantitation
Total RNA was isolated using RNeasy kits from Qiagen, Inc. (Valencia, CA) of which 1 μg was reverse transcribed using cDNA Archive kits from ABI (Foster City, CA). Real-time mRNA levels for LTA4H and beta-glucuronidase (GUSB) as an endogenous control were determined in triplicate using pre-developed assays (LTA4H, Hs00168505_m1; GUSB, Hs99999908_m1) from Applied Biosystems Inc. (Foster City, CA). LTA4H mRNA levels were normalized to those of GUSB and the replicates were averaged to determine transcript abundance in each sample relative to a reference sample. The mRNA levels of GUSB, as determined by the average Ct values for the triplicate reactions, did not differ across LTA4H genotypes in these monocyte samples.
Ex vivo LTB4 production analyses
Within 1 h after isolation from blood, monocytes (1 × 106/ml) were cultured at 37°C in 5% CO2 using RPM1 1640 complete media supplemented with 10% heat-inactivated autologous plasma. The cells were then stimulated with either the calcium ionophore A23187 (Sigma, St. Louis, MO) at a final concentration of 10 μM or with DMSO as a control culture. After a 60-min incubation, the supernatants and cell pellets were collected and the supernatants were extracted using 60 mg HLB solid phase extraction cartridges (Waters, Inc; Milford, MA). Oxylipid analytes, including LTB4, were chromatographically separated on an ultra-performance liquid chromatography system equipped with a 2.1 × 150 mm Acquity BEHC18 reversed phase column and quantified by negative mode electrospray ionization on a Quattro Micro tandem mass spectrometer (Waters, Inc.). One in 20 samples was analyzed in replicate to assess analytical precision. Differences in mRNA and LTB4 levels were determined using t tests (Statview version. 5.0; SAS Institute Inc., Cary, NC) with p values <0.05 considered as statistically significant.
Results
Clinical characteristics of GeneBank subjects
The general characteristics of the Caucasian and African American GeneBank subjects used in the present study are presented in Table 1. For both ethnicities, subjects with CAD at baseline exhibited the known traditional risk factors for cardiovascular disease, including being older, more likely to be male, and having higher plasma levels of triglycerides and CRP and lower HDL-cholesterol levels (Table 1). However, plasma total and LDL-cholesterol levels were lower in subjects with CAD, which is likely due to these patients having a higher prevalence of co-morbidities and taking lipid-lowering medications.
Table 1.
General characteristics of the GeneBank participants
| Caucasian | African American | |||||
|---|---|---|---|---|---|---|
| Without CAD (n = 913) | With CAD (n = 2,834) | p value | Without CAD (n = 263) | With CAD (n = 502) | p value | |
| Age (years) | 59 ± 12 | 66 ± 11 | <0.0001 | 57 ± 11 | 61 ± 11 | <0.0001 |
| Male/female (n) | 478/435 | 2,140/694 | <0.0001 | 99/164 | 264/238 | <0.0001 |
| BMI (kg/m2) | 29.7 ± 6.8 | 29.4 ± 5.6 | 0.92 | 33.6 ± 8.1 | 31.2 ± 7.1 | <0.0001 |
| Total cholesterol (mg/dl) | 177 ± 37 | 168 ± 40 | <0.0001 | 182 ± 38 | 176 ± 50 | 0.01 |
| HDL-cholesterol (mg/dl) | 42 ± 13 | 37 ± 11 | <0.0001 | 51 ± 15 | 46 ± 15 | <0.0001 |
| LDL-cholesterol (mg/dl) | 108 ± 31 | 100 ± 33 | <0.0001 | 111 ± 72 | 106 ± 43 | 0.13 |
| Triglycerides (mg/dl) | 129 ± 81 | 155 ± 105 | <0.0001 | 123 ± 81 | 128 ± 83 | 0.32 |
| CRP (mg/l) | 2.2 (3.4) | 3.1 (5.7) | <0.0001 | 2.2 (3.7) | 3.0 (5.6) | 0.008 |
| DBP (mmHg) | 75 ± 12 | 74 ± 12 | 0.004 | 81 ± 13 | 77 ± 13 | 0.0002 |
| SBP (mmHg) | 133 ± 20 | 135 ± 21 | 0.003 | 139 ± 22 | 140 ± 23 | 0.53 |
Data are shown as mean ± SD with the exception of CRP levels which are shown as median (IQR). Two-sided p values are reported between subjects with and without CAD at baseline
Evaluation of previously reported LT pathway variants
Table 2 lists the LT pathway genes selected for the present study and their biological functions. We first evaluated several variants of ALOX5, ALOX5AP, and LTA4H that were previously associated with CAD-related phenotypes (Dwyer et al. 2004; Helgadottir et al. 2004, 2006) in 3,747 Caucasian and 765 African American subjects from the GeneBank cohort. These included a variable number of tandem Sp1 repeats in the ALOX5 promoter as well as haplotypes of ALOX5AP (HapA) and LTA4H (HapK) (Supplemental Table 1). As in our prior analyses of the ALOX5 repeats (Allayee et al. 2008), carriers of the functional “short” promoter alleles with “3” and “4” Sp1 repeats (i.e. genotypes of 3/3,3/4,4/4,3/5,4/5,3/6,3/7,4/6,4/8) were compared with subjects carrying alleles of ≥“5” repeats (i.e. genotypes of 5/5,5/6,5/7,5/8,5/9,6/6,6/7). In the overall GeneBank cohort, the frequencies of the “3” and “4” repeats were ~1 and 14%, respectively, in Caucasians and 30 and 14%, respectively, in African Americans, which are consistent with previous reports for these ethnicities (Assimes et al. 2008; Dwyer et al. 2004; Lima et al. 2006). In Caucasians, there was no association of the “3” and “4” alleles with CAD (Table 3), whereas African American carriers of these shorter repeats had increased risk compared to non-carriers (OR = 1.4; 95% CI 1.01–1.9; p = 0.04). Analyses of the ALOX5AP and LTA4H haplotypes, adjusted for age and sex, yielded significant evidence of association with HapK and CAD in Caucasians (OR = 1.2; 95% CI 1.01–1.4; p = 0.03) but not with HapA (Table 3). Further adjustment for CRP levels, medication use, alcohol consumption, and Framingham risk score, which includes diabetes and smoking status slightly attenuated these results but still yielded a significant p value of 0.05 for HapK. An analysis using a haplotype score test that included all possible LTA4H haplotypes having frequencies greater than 1% (n = 13) also yielded consistent evidence for association with only HapK (p = 0.07). By comparison, neither haplotype was associated with CAD in African Americans.
Table 2.
The major genes of LT biosynthetic pathway and their function
| Gene (symbol) | Function |
|---|---|
| Cytosolic phospholipase A2 group 4A (PLA2G4A) | Releases arachidonic acid from cell membranes |
| Cytosolic phospholipase A2 group 5 (PLA2G5) | Releases arachidonic acid from cell membranes |
| Arachidonic acid 5-lipoxygenase (ALOX5) | Incorporates oxygen into arachidonic acid and forms LTA4 |
| ALOX5 activating protein (ALOX5AP) | Presents arachidonic acid to and activates ALOX5 |
| LTA4 epoxide hydrolase (LTA4H) | Converts LTA4 to LTB4 |
| LTC4 synthase (LTC4S) | Converts LTA4 to the first cysteinyl LT, LTC4 |
| LTB4 receptor 1 (LTB4R) | Receptor for LTB4 |
| LTB4 receptor 2 (LTB4R2) | A second receptor for LTB4 |
| Cysteinyl leukotriene receptor 1 (CysLTR1) | Receptor for cysteinyl LTs (LTC4, LTD4, and LTE4) |
| Cysteinyl leukotriene receptor 2 (CysLTR2) | A second receptor for the cysteinyl LTs |
| Microsomal glutathione S-transferase 1 (MGST1) | Cysteinyl LT metabolism |
| Microsomal glutathione S-transferase 2 (MGST2) | Cysteinyl LT metabolism |
| Microsomal glutathione S-transferase 3 (MGST3) | Cysteinyl LT metabolism |
| Prostaglandin reductase 1 isoform 1 (PTGR1) | Catalyzes degradation of LTB4 |
| Cytochrome P450 4F3 (CYP4F3) | Catalyzes degradation of LTB4 |
Table 3.
Association of previously reported LT pathway variants in Caucasian and African American GeneBank subjects
| SNP/variant (gene) | Risk allele/haplotype frequency | OR (95% CI) | |||
|---|---|---|---|---|---|
| Without CAD (n) | With CAD (n) | Non-carriers | Carriers | p value* | |
| Caucasian | |||||
| Promoter repeats (ALOX5)a | 0.15 (886) | 0.17 (2,736) | 1.0 | 1.1 (1.0–1.4) | 0.2 |
| HapA (ALOX5AP)b | 0.15 (913) | 0.15 (2,834) | 1.0 | 1.1 (0.9–1.3) | 0.3 |
| HapK (LTA4H)c | 0.14 (912) | 0.16 (2,830) | 1.0 | 1.2 (1.01–1.4) | 0.03 |
| African American | |||||
| Promoter repeats (ALOX5)a | 0.43 (261) | 0.48 (495) | 1.0 | 1.4 (1.01–1.9) | 0.04 |
| HapA (ALOX5AP)b | 0.06 (263) | 0.06 (502) | 1.0 | 1.1 (0.7–1.6) | 0.8 |
| HapK (LTA4H)c | 0.05 (263) | 0.06 (502) | 1.0 | 0.9 (0.5–1.4) | 0.5 |
Sample sizes are shown based on complete genotype and phenotype data for each variant
OR (95% CI) odds ratio (95% confidence intervals)
* p values were obtained from a logistic regression with adjustment for age and gender
aCombined frequencies of the “3” and “4” repeat alleles are given; non-carriers = genotypes of 5/5, 5/6, 5/7, 5/8, 5/9, 6/6, 6/7; carriers = genotypes of 3/3, 3/4, 4/4, 3/5, 4/5, 3/6, 3/7, 4/6, 4/8
bHapA = GTGA derived from SNPs rs17222814 (G), rs10507391 (T), rs4769874 (G), rs9551963 (A)
cHapK = CGTATTTCGG derived from SNPs rs61937881 (SG12S16) (C), rs2660880 (G), rs6538697 (T), rs1978331 (A), rs17677715 (T), rs2247570 (T), rs2660898 (T), rs2540482 (C), rs2660845 (G), rs2540475 (G)
Two-stage analysis of LT pathway genes with haplotype-tagging SNPs
In order to comprehensively evaluate the contribution of LT pathway genes to CAD in an unbiased fashion, we next carried out a two-stage association study in Caucasian subjects. In the first stage, we used HapMap data for individuals of European ancestry (to match GeneBank) and the Tagger program to identify haplotype-tagging SNPs in 15 pathway genes that play important roles in LT biosynthesis, signaling, and degradation (Table 2). For this purpose, we chose an r 2 of 0.9 between SNPs and included the genomic region spanning 10 kb upstream and 5 kb downstream of each gene to capture potential regulatory elements. These criteria resulted in the selection of 274 haplotype-tagging SNPs (listed in Supplemental Table 1), of which 254 were successfully genotyped in stage 1. This dataset included 1,000 sequential GeneBank patients, comprised of 804 and 196 subjects with and without CAD, respectively. In order to obtain a more balanced “case–control” dataset for stage 1, we also included an additional 322 “control” subjects without CAD that were selected from GeneBank.
Based on our two-stage study design, we chose an unadjusted p value of <0.05 as the threshold for suggestive evidence of association in the stage 1 analyses. After adjustment for age and sex, 19 SNPs in the ALOX5, ALOX5AP, LTA4H, CYP4F3, MGST1, MGST2, MGST3, and PLA2G4A genes demonstrated nominal evidence of association (Supplemental Table 2). In stage 2, we genotyped these 19 SNPs in 2,425 additional Caucasian subjects (395 without CAD/2030 with CAD) and obtained evidence of association with one variant in PLA2G4A (rs12746200; p = 0.03) and another in LTA4H (rs2540477; p = 0.01) (Table 4). The direction of the associations for these two SNPs was consistent in both stages and became even more significant in a combined analysis with all subjects (Table 4). Of note, rs12746200 had a protective effect (OR = 0.7; 95% CI 0.6–0.9; p = 0.0007), whereas rs2540477 increased risk of CAD (OR = 1.2; 95% CI 1.1–1.5; p = 0.003). After full adjustment for the same covariates described above, the p values for the association of PLA2G4A rs12746200 and LTA4H rs2540477 with CAD became 0.0014 and 0.005, respectively. In the GeneBank subjects, rs2540477 was in strong linkage disequilibrium with one of the SNPs that is part of HapK (rs2540482; r 2 = 0.86), raising the possibility that the association we observed with rs2540477 was due to its linkage with HapK. Therefore, we performed a stratified analysis and observed significant association of rs2540477 with CAD even in subjects who did not carry HapK (Table 4). By comparison, a haplotype of HapK that included the rare allele of rs2540477 was not associated with increased risk of CAD, which could have been due to decreased sample size (Table 4).
Table 4.
Association of PLA2G4A and LTA4H SNPs with CAD in the two-stage analysis
| SNP (gene) | MAF | OR (95% CI) | |||
|---|---|---|---|---|---|
| rs12746200 (PLA2G4A) | Without CAD (n) | With CAD (n) | AA | AG/GG | p value* |
| Stage 1 | 0.11 (516) | 0.10 (802) | 1.0 | 0.8 (0.6–1.0) | 0.05 |
| Stage 2 | 0.11 (390) | 0.08 (1,982) | 1.0 | 0.6 (0.5–0.9) | 0.03 |
| Combined | 0.11 (906) | 0.09 (2,784) | 1.0 | 0.7 (0.6–0.9) | 0.0007 |
| SNP (gene) | MAF | OR (95% CI) | |||
|---|---|---|---|---|---|
| rs2540477 (LTA4H) | Without CAD (n) | With CAD (n) | TT | TC/CC | p value* |
| Stage 1 | 0.22 (515) | 0.26 (803) | 1.0 | 1.3 (1.0–1.7) | 0.02 |
| Stage 2 | 0.20 (382) | 0.25 (1,980) | 1.0 | 1.4 (1.1–1.7) | 0.02 |
| Combined | 0.21 (897) | 0.25 (2,783) | 1.0 | 1.3 (1.1–1.5) | 0.003 |
| HapK non-carriers | 0.08 (663) | 0.11 (1,973) | 1.0 | 1.3 (1.1–1.8) | 0.01 |
| HapK carriers | 0.57 (234) | 0.60 (810) | 1.0 | 1.7 (0.6–5.0) | 0.34 |
Sample sizes are shown based on complete genotype and phenotype data for each variant
MAF minor allele frequency, OR (95% CI) odds ratio (95% confidence intervals)
* p values were obtained from a logistic regression with adjustment for age and gender
Prospective analysis with major adverse cardiac events (MACE)
We next determined whether PLA2G4A rs12746200 and LTA4H rs2540477 were also associated with risk of future MACE (MI, stroke, or death) over 3 years of follow-up. Consistent with the results of the cross-sectional analysis, AG/GG carriers of rs12746200 had fewer numbers of MACE compared to the AA genotype group (Fig. 1a; log-rank p = 0.03), with an adjusted HR of 0.7 (95% CI = 0.5–0.9; p = 0.01; Table 5). By comparison, rs2540477 did not affect risk of future MACE (Fig. 1b; Table 5) and stratifying these analyses by HapK did not alter the results (data not shown).
Fig. 1.

Relationship between LT pathway variants and incident risk of MACE. a Kaplan–Meier survival analyses demonstrating that carriers (AG/GG) of PLA2G4A variant rs12746200 have significantly lower a rate of MACE over three years of follow-up (p = 0.03). b There is no difference in the rate of MACE between carriers and non-carriers of the LTA4H variant rs2540477. MACE were defined as the occurrence of MI, stroke, or all-cause mortality over 3 years of follow-up from the time of enrollment
Table 5.
Risk of prospective MACE in relation to PLA2G4A and LTA4H variants
| rs12746200 (PLA2G4A) | AA (n = 2,943) | AG/GG (n = 589) | p value* |
|---|---|---|---|
| Number of events (%) | 404 (14) | 61 (10) | 0.03 |
| Adjusted HR (95% CI)* | 1.0 | 0.7 (0.5–0.9) | 0.01 |
| rs2540477 (LTA4H) | TT (n = 2,095) | TC/CC (n = 1,435) | p value* |
|---|---|---|---|
| Number of events (%) | 269 (14) | 193 (13) | 0.57 |
| Adjusted HR (95% CI)* | 1.0 | 1.1 (0.8–1.3) | 0.59 |
Sample sizes are shown based on complete genotype and phenotype data for each variant
* Hazard ratios (HRs) were calculated only in Caucasians with adjustment for age, sex, medication use (statins and/or aspirin), plasma CRP levels, alcohol consumption, and Framingham ATP-III risk score
Functional characterization of associated variants
To investigate functional differences between the associated PLA2G4A and LTA4H variants, we used monocytes isolated from 105 healthy African Americans who had participated in a previous clinical intervention study to examine the effects of omega-3 fatty acid supplementation in subjects with different ALOX5 promoter variants (see “Materials and methods”). Only monocytes from the baseline visit were used for this purpose. We first performed real-time gene expression experiments, in reference to beta-glucuronidase (GUSB) as an endogenous control, which did not reveal significant differences in LTA4H mRNA levels between carriers of either LTA4H rs2540477 or HapK (Fig. 2a). However, ex vivo LTB4 production in response to stimulation with the calcium ionophore A23187 was significantly higher by approximately 50% in monocytes from subjects carrying HapK compared to non-carriers (9.2 ± 3.8 vs. 5.9 ± 3.3 nmol/L; p = 0.002) (Fig. 2b). Similarly, TC/CC carriers of rs2540477 had 33% increased LTB4 production compared to TT subjects (7.5 ± 3.6 vs. 5.9 ± 3.3 nmol/L; p = 0.03) (Fig. 2b). By comparison, LTB4 production in control cultures of monocytes incubated with DMSO was very low and not significantly different between carriers and non-carriers of either HapK (0.42 ± 0.40 vs. 0.76 ± 1.3 nmol/L, respectively; p = 0.39) or rs2540477 (0.58 ± 0.82 vs. 0.76 ± 1.4 nmol/L, respectively; p = 0.55). In addition, since these subjects were recruited based on carrying different ALOX5 promoter alleles, we also stratified the analyses of LTA4H mRNA expression and LTB4 production by ALOX5 genotype. However, this did not alter the results with rs2540477 and HapK (data not shown). Since only two subjects carried the rare allele of rs12746200, we were not able to determine whether this PLA2G4A variant affected mRNA and/or LT production in these monocyte samples.
Fig. 2.
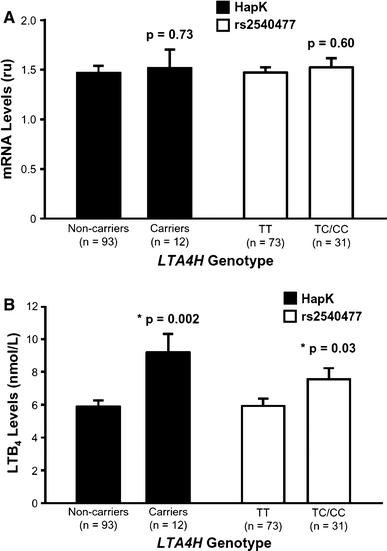
Effect of LTA4H variants on gene expression and ex vivo LTB4 production. a LTA4H mRNA levels in monocytes are not significantly different between carriers and non-carriers of the HapK or rs2540477 variants. Real-time PCR was carried out in triplicate and expression levels were normalized to GUSB as an endogenous control. b Monocytes isolated from carriers of HapK or rs2540477 produce significantly higher levels of LTB4 than non-carriers. Monocytes were isolated from healthy subjects and stimulated with the calcium ionophore A23187 for 60 min. LTB4 was measured in the supernatant by negative mode electrospray ionization tandem mass spectrometry. Data are shown as mean ± SE and the number of samples analyzed for each genotype is given in parentheses
Discussion
In the present study, we carried out a comprehensive genetic analysis of the LT pathway in a relatively large cohort of subjects ascertained through elective cardiac evaluation. Of the 15 genes studied, our results provide evidence for the contributions of ALOX5, LTA4H, and PLA2G4A to risk of CAD. Two newly discovered SNPs in LTA4H and PLA2G4A were identified using an unbiased two-stage study design with dense haplotype-tagging SNPs. While the association of LTA4H rs2540477 with prevalent CAD may have been due to linkage disequilibrium between this variant and HapK, we also observed an independent effect in subjects who did not carry HapK. These results suggest that multiple susceptibility alleles of LTA4H may exist in the population that increase the risk of CAD. By comparison, the rs12746200 variant of PLA2G4A was cardioprotective, decreasing both the likelihood of having CAD and the risk for experiencing a future MACE over 3 years of follow-up. Thus, the results from these cross-sectional and longitudinal analyses provide consistent evidence for a protective effect of this PLA2G4A variant. These latter findings are also novel since, to our knowledge, PLA2G4A was not evaluated in previous genetic studies of the LT pathway (Bevan et al. 2008; Burdon et al. 2010; Crosslin et al. 2009).
Our results also corroborate previously reported associations of CAD-related phenotypes with an ALOX5 promoter polymorphism (Allayee et al. 2008; Dwyer et al. 2004) and HapK in LTA4H (Crosslin et al. 2009; Helgadottir et al. 2006). In this regard, the shorter ALOX5 “3” and “4” promoter repeats, which lead to increased ALOX5 expression (Allayee et al. 2008; Vikman et al. 2009), were only associated with CAD in African Americans. One possibility for this observation may be due to the tenfold higher frequency of the “3” allele in African Americans compared to Caucasians (~30 vs. 1–2%). However, it should also be noted that the “3” and “4” repeats may not contribute appreciably to CAD and/or MI in Caucasians, as has been reported by other groups (Gonzalez et al. 2007; Maznyczka et al. 2008). Conversely, we observed association of HapK in Caucasians but not African Americans, which may have been due to the relatively fewer African Americans subjects in our study population. Interestingly, HapK is rare in subjects from the African continent and its presence in African Americans has been suggested to be a consequence of admixture with individuals of European descent (Helgadottir et al. 2006). Since HapK was previously associated more strongly with MI in African Americans than in Caucasians, this raises the question of whether HapK interacts with other genetic variants that are specific to or more frequent in subjects of African ancestry. As discussed above, one such variant could be the “3” repeat allele of the ALOX5 promoter and could be addressed in studies of larger African American cohorts. In this regard, it should be noted that we did not adjust for population admixture in this African American sample, which could also potentially confound the results of association studies. Lastly, we did not observe association of ALOX5AP HapA with CAD risk in either African Americans or Caucasians. The genetic contribution of HapA to cardiovascular phenotypes is not entirely clear since attempts to replicate its association have yielded both positive (Helgadottir et al. 2005; Shah et al. 2008) and negative (Koch et al. 2007; Lemaitre et al. 2009; Linsel-Nitschke et al. 2008; Tsai et al. 2009; Zee et al. 2006; Zintzaras et al. 2009) results.
Another important aspect of our study is the ex vivo functional experiments demonstrating increased LTB4 production in stimulated monocytes from subjects carrying HapK or rs2540477. These results provide a biologically plausible mechanism for the increased risk of CAD conferred by these LTA4H variants since LTB4 is a direct product of the reaction catalyzed by this enzyme and is a potent chemotactic molecule that mediates the recruitment of neutrophils, monocytes, and other leukocytes to sites of inflammation, including the arterial wall of atherosclerotic lesions. Moreover, LTB4-mediated activation of leukocytes can induce myeloperoxidase release and further contribute to the progression of atherosclerosis (Nicholls and Hazen 2005; Samuelsson 1983). Presumably, rs2540477 or the SNPs comprising HapK are not the underlying causal variant(s) since they are located in the non-coding regions of LTA4H and, based on our results, do not affect gene expression. In depth sequencing of LTA4H-coding regions in subjects carrying HapK or rs2540477 may help to identify putative amino acid substitutions that are in linkage disequilibrium with these variants and/or alternatively spliced isoforms and lead to increased LTB4 production. Additional studies will be required in order to distinguish between these possibilities as well as to functionally characterize the PLA2G4A rs12746200 variant.
One limitation of our study was not correcting for multiple comparisons, which raises the possibility that the associations we have detected are false positives. Although such an adjustment would be more appropriate for the two-stage analyses rather than the specific hypotheses we tested with previously associated variants/haplotypes, this potential problem is mitigated by the consistency of the results for rs12746200 and rs2540477 in both stages and in the longitudinal analyses with rs12746200. However, since GeneBank subjects were ascertained through cardiac evaluation at a tertiary care center, it is still important to confirm these associations in independent populations recruited through other study designs, such as traditional case–control datasets. In this regard, it is possible that the contribution of LT pathway genes to cardiovascular phenotypes are more readily detectable in the context of dietary PUFAs that serve as the substrates for LT biosynthesis. Such a concept would be analogous to our previous studies with ALOX5 and MI (Allayee et al. 2008) and it will be interesting to determine whether the LTA4H and PLA2G4A variants we identify herein exhibit nutrigenetic interactions with dietary PUFAs and CAD as well. Since dietary information was not collected as part of subject recruitment in GeneBank, we were not able to carry out such gene–dietary analyses in this study. Nonetheless, our results provide additional evidence that functional genetic variation in the LT pathway can modulate atherosclerotic processes and the risk of CAD in humans.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by National Institutes of Health grants RO1HL079353, R21AT003411, P60MD0222, the General Clinical Research Center of the Cleveland Clinic/Case Western Reserve University CTSA (UL1RR024989), and US Department of Agriculture grant CRIS Project # 5306-51530-006-00D. The Cleveland Clinic GeneBank study is supported by National Institutes of Health grants P01HL076491, P01HL098055, P01HL087018, and R01HL103866. P.A. was supported through a fellowship award from the Gustavus & Louise Pfeiffer Research Foundation. Supplies and funding for measuring fasting plasma lipid and CRP levels were provided for by Abbott Diagnostics, Inc. A portion of this work was conducted in a facility constructed with support from the National Institutes of Health Research Facilities Improvement Program (RR10600, CA62528, and RR14514) from the National Center for Research Resources.
Conflict of interest
The authors declare no conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Ahluwalia N, Lin AY, Tager AM, Pruitt IE, Anderson TJ, Kristo F, Shen D, Cruz AR, Aikawa M, Luster AD, Gerszten RE. Inhibited aortic aneurysm formation in BLT1-deficient mice. J Immunol. 2007;179:691–697. doi: 10.4049/jimmunol.179.1.691. [DOI] [PubMed] [Google Scholar]
- Aiello RJ, Bourassa PA, Lindsey S, Weng W, Freeman A, Showell HJ. Leukotriene B4 receptor antagonism reduces monocytic foam cells in mice. Arterioscler Thromb Vasc Biol. 2002;22:443–449. doi: 10.1161/hq0302.105593. [DOI] [PubMed] [Google Scholar]
- Allayee H, Baylin A, Hartiala J, Wijesuriya H, Mehrabian M, Lusis AJ, Campos H. Nutrigenetic association of the 5-lipoxygenase gene with myocardial infarction. Am J Clin Nutr. 2008;88:934–940. doi: 10.1093/ajcn/88.4.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimes TL, Knowles JW, Priest JR, Basu A, Volcik KA, Southwick A, Tabor HK, Hartiala J, Allayee H, Grove ML, Tabibiazar R, Sidney S, Fortmann SP, Go A, Hlatky M, Iribarren C, Boerwinkle E, Myers R, Risch N, Quertermous T. Common polymorphisms of ALOX5 and ALOX5AP and risk of coronary artery disease. Hum Genet. 2008;123:399–408. doi: 10.1007/s00439-008-0489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back M, Hansson GK. Leukotriene receptors in atherosclerosis. Ann Med. 2006;38:493–502. doi: 10.1080/07853890600982737. [DOI] [PubMed] [Google Scholar]
- Bevan S, Dichgans M, Wiechmann HE, Gschwendtner A, Meitinger T, Markus HS. Genetic variation in members of the leukotriene biosynthesis pathway confer an increased risk of ischemic stroke: a replication study in two independent populations. Stroke. 2008;39:1109–1114. doi: 10.1161/STROKEAHA.107.491969. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya T, Nicholls SJ, Topol EJ, Zhang R, Yang X, Schmitt D, Fu X, Shao M, Brennan DM, Ellis SG, Brennan ML, Allayee H, Lusis AJ, Hazen SL. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA. 2008;299:1265–1276. doi: 10.1001/jama.299.11.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon KP, Rudock ME, Lehtinen AB, Langefeld CD, Bowden DW, Register TC, Liu Y, Freedman BI, Carr JJ, Hedrick CC, Rich SS (2010) Human lipoxygenase pathway gene variation and association with markers of subclinical atherosclerosis in the diabetes heart study. Mediators Inflamm 2010:170153 [DOI] [PMC free article] [PubMed]
- Carlson CS, Heagerty PJ, Nord AS, Pritchard DK, Ranchalis J, Boguch JM, Duan H, Hatsukami TS, Schwartz SM, Rieder MJ, Nickerson DA, Jarvik GP. TagSNP evaluation for the association of 42 inflammation loci and vascular disease: evidence of IL6, FGB, ALOX5, NFKBIA, and IL4R loci effects. Hum Genet. 2007;121:65–75. doi: 10.1007/s00439-006-0289-8. [DOI] [PubMed] [Google Scholar]
- Crosslin DR, Shah SH, Nelson SC, Haynes CS, Connelly JJ, Gadson S, Goldschmidt-Clermont PJ, Vance JM, Rose J, Granger CB, Seo D, Gregory SG, Kraus WE, Hauser ER. Genetic effects in the leukotriene biosynthesis pathway and association with atherosclerosis. Hum Genet. 2009;125:217–229. doi: 10.1007/s00439-008-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R, Lusis AJ, Mehrabian M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med. 2004;350:29–37. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- Gonzalez P, Reguero JR, Lozano I, Moris C, Coto E. A functional Sp1/Egr1-tandem repeat polymorphism in the 5-lipoxygenase gene is not associated with myocardial infarction. Int J Immunogenet. 2007;34:127–130. doi: 10.1111/j.1744-313X.2007.00671.x. [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, Samani NJ, Gudmundsson G, Grant SF, Thorgeirsson G, Sveinbjornsdottir S, Valdimarsson EM, Matthiasson SE, Johannsson H, Gudmundsdottir O, Gurney ME, Sainz J, Thorhallsdottir M, Andresdottir M, Frigge ML, Topol EJ, Kong A, Gudnason V, Hakonarson H, Gulcher JR, Stefansson K. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–239. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Gretarsdottir S, St Clair D, Manolescu A, Cheung J, Thorleifsson G, Pasdar A, Grant SF, Whalley LJ, Hakonarson H, Thorsteinsdottir U, Kong A, Gulcher J, Stefansson K, MacLeod MJ. Association between the gene encoding 5-lipoxygenase-activating protein and stroke replicated in a Scottish population. Am J Hum Genet. 2005;76:505–509. doi: 10.1086/428066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir A, Manolescu A, Helgason A, Thorleifsson G, Thorsteinsdottir U, Gudbjartsson DF, Gretarsdottir S, Magnusson KP, Gudmundsson G, Hicks A, Jonsson T, Grant SF, Sainz J, O’Brien SJ, Sveinbjornsdottir S, Valdimarsson EM, Matthiasson SE, Levey AI, Abramson JL, Reilly MP, Vaccarino V, Wolfe ML, Gudnason V, Quyyumi AA, Topol EJ, Rader DJ, Thorgeirsson G, Gulcher JR, Hakonarson H, Kong A, Stefansson K. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat Genet. 2006;38:68–74. doi: 10.1038/ng1692. [DOI] [PubMed] [Google Scholar]
- Heller EA, Liu E, Tager AM, Sinha S, Roberts JD, Koehn SL, Libby P, Aikawa ER, Chen JQ, Huang P, Freeman MW, Moore KJ, Luster AD, Gerszten RE. Inhibition of atherogenesis in BLT1-deficient mice reveals a role for LTB4 and BLT1 in smooth muscle cell recruitment. Circulation. 2005;112:578–586. doi: 10.1161/CIRCULATIONAHA.105.545616. [DOI] [PubMed] [Google Scholar]
- Iovannisci DM, Lammer EJ, Steiner L, Cheng S, Mahoney LT, Davis PH, Lauer RM, Burns TL. Association between a leukotriene C4 synthase gene promoter polymorphism and coronary artery calcium in young women: the Muscatine Study. Arterioscler Thromb Vasc Biol. 2007;27:394–399. doi: 10.1161/01.ATV.0000252680.72734.10. [DOI] [PubMed] [Google Scholar]
- Jawien J, Gajda M, Rudling M, Mateuszuk L, Olszanecki R, Guzik TJ, Cichocki T, Chlopicki S, Korbut R. Inhibition of five lipoxygenase activating protein (FLAP) by MK-886 decreases atherosclerosis in apoE/LDLR-double knockout mice. Eur J Clin Invest. 2006;36:141–146. doi: 10.1111/j.1365-2362.2006.01606.x. [DOI] [PubMed] [Google Scholar]
- Jawien J, Gajda M, Wolkow P, Zuranska J, Olszanecki R, Korbut R. The effect of montelukast on atherogenesis in apoE/LDLR-double knockout mice. J Physiol Pharmacol. 2008;59:633–639. [PubMed] [Google Scholar]
- Koch W, Hoppmann P, Mueller JC, Schomig A, Kastrati A. No association of polymorphisms in the gene encoding 5-lipoxygenase-activating protein and myocardial infarction in a large central European population. Genet Med. 2007;9:123–129. doi: 10.1097/GIM.0b013e318030c9c5. [DOI] [PubMed] [Google Scholar]
- Lemaitre RN, Rice K, Marciante K, Bis JC, Lumley TS, Wiggins KL, Smith NL, Heckbert SR, Psaty BM. Variation in eicosanoid genes, non-fatal myocardial infarction and ischemic stroke. Atherosclerosis. 2009;204:e58–e63. doi: 10.1016/j.atherosclerosis.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima JJ, Zhang S, Grant A, Shao L, Tantisira KG, Allayee H, Wang J, Sylvester J, Holbrook J, Wise R, Weiss ST, Barnes K. Influence of leukotriene pathway polymorphisms on response to montelukast in asthma. Am J Respir Crit Care Med. 2006;173:379–385. doi: 10.1164/rccm.200509-1412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsel-Nitschke P, Gotz A, Medack A, Konig IR, Bruse P, Lieb W, Mayer B, Stark K, Hengstenberg C, Fischer M, Baessler A, Ziegler A, Schunkert H, Erdmann J. Genetic variation in the arachidonate 5-lipoxygenase-activating protein (ALOX5AP) is associated with myocardial infarction in the German population. Clin Sci (Lond) 2008;115:309–315. doi: 10.1042/CS20070468. [DOI] [PubMed] [Google Scholar]
- Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Livak KJ. SNP genotyping by the 5′-nuclease reaction. Methods Mol Biol. 2003;212:129–147. doi: 10.1385/1-59259-327-5:129. [DOI] [PubMed] [Google Scholar]
- Maznyczka A, Braund P, Mangino M, Samani NJ. Arachidonate 5-lipoxygenase (5-LO) promoter genotype and risk of myocardial infarction: a case–control study. Atherosclerosis. 2008;199:328–332. doi: 10.1016/j.atherosclerosis.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Mehrabian M, Allayee H. 5-Lipoxygenase and atherosclerosis. Curr Opin Lipidol. 2003;14:447–457. doi: 10.1097/00041433-200310000-00005. [DOI] [PubMed] [Google Scholar]
- Mehrabian M, Allayee H, Wong J, Shi W, Wang XP, Shaposhnik Z, Funk CD, Lusis AJ. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–126. doi: 10.1161/01.RES.0000028008.99774.7F. [DOI] [PubMed] [Google Scholar]
- Mehrabian M, Allayee H, Stockton J, Lum PY, Drake TA, Castellani LW, Suh M, Armour C, Edwards S, Lamb J, Lusis AJ, Schadt EE. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat Genet. 2005;37:1224–1233. doi: 10.1038/ng1619. [DOI] [PubMed] [Google Scholar]
- Mehrabian M, Schulthess FT, Nebohacova M, Castellani LW, Zhou Z, Hartiala J, Oberholzer J, Lusis AJ, Maedler K, Allayee H. Identification of ALOX5 as a gene regulating adiposity and pancreatic function. Diabetologia. 2008;51:978–988. doi: 10.1007/s00125-008-1002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Tang WH, Scoffone H, Brennan DM, Hartiala J, Allayee H, Hazen SL (2010) Lipoprotein (a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipid Res [DOI] [PMC free article] [PubMed]
- Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- Qiu H, Gabrielsen A, Agardh HE, Wan M, Wetterholm A, Wong CH, Hedin U, Swedenborg J, Hansson GK, Samuelsson B, Paulsson-Berne G, Haeggstrom JZ. Expression of 5-lipoxygenase and leukotriene A4 hydrolase in human atherosclerotic lesions correlates with symptoms of plaque instability. Proc Natl Acad Sci USA. 2006;103:8161–8166. doi: 10.1073/pnas.0602414103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568–575. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- Sanak M, Pierzchalska M, Bazan-Socha S, Szczeklik A. Enhanced expression of the leukotriene C(4) synthase due to overactive transcription of an allelic variant associated with aspirin-intolerant asthma. Am J Respir Cell Mol Biol. 2000;23:290–296. doi: 10.1165/ajrcmb.23.3.4051. [DOI] [PubMed] [Google Scholar]
- Shah SH, Hauser ER, Crosslin D, Wang L, Haynes C, Connelly J, Nelson S, Johnson J, Gadson S, Nelson CL, Seo D, Gregory S, Kraus WE, Granger CB, Goldschmidt-Clermont P, Newby LK. ALOX5AP variants are associated with in-stent restenosis after percutaneous coronary intervention. Atherosclerosis. 2008;201:148–154. doi: 10.1016/j.atherosclerosis.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanbroek R, Grabner R, Lotzer K, Hildner M, Urbach A, Ruhling K, Moos MP, Kaiser B, Cohnert TU, Wahlers T, Zieske A, Plenz G, Robenek H, Salbach P, Kuhn H, Radmark O, Samuelsson B, Habenicht AJ. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc Natl Acad Sci USA. 2003;100:1238–1243. doi: 10.1073/pnas.242716099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao K, Jala VR, Mathis S, Suttles J, Zacharias W, Ahamed J, Ali H, Tseng MT, Haribabu B. Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler Thromb Vasc Biol. 2004;24:369–375. doi: 10.1161/01.ATV.0000110503.16605.15. [DOI] [PubMed] [Google Scholar]
- Tsai AK, Li N, Hanson NQ, Tsai MY, Tang W. Associations of genetic polymorphisms of arachidonate 5-lipoxygenase-activating protein with risk of coronary artery disease in a European-American population. Atherosclerosis. 2009;207:487–491. doi: 10.1016/j.atherosclerosis.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Tymchuk CN, Hartiala J, Patel PI, Mehrabian M, Allayee H. Nonconventional genetic risk factors for cardiovascular disease. Curr Atheroscler Rep. 2006;8:184–192. doi: 10.1007/s11883-006-0072-2. [DOI] [PubMed] [Google Scholar]
- Vikman S, Brena RM, Armstrong P, Hartiala J, Stephensen CB, Allayee H. Functional analysis of 5-lipoxygenase promoter repeat variants. Hum Mol Genet. 2009;18(23):4521–4529. doi: 10.1093/hmg/ddp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee RY, Cheng S, Hegener HH, Erlich HA, Ridker PM. Genetic variants of arachidonate 5-lipoxygenase-activating protein, and risk of incident myocardial infarction and ischemic stroke: a nested case–control approach. Stroke. 2006;37:2007–2011. doi: 10.1161/01.STR.0000229905.25080.01. [DOI] [PubMed] [Google Scholar]
- Zintzaras E, Rodopoulou P, Sakellaridis N. Variants of the arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene and risk of stroke: a HuGE gene–disease association review and meta-analysis. Am J Epidemiol. 2009;169:523–532. doi: 10.1093/aje/kwn368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.