

Abstract
Lack of knowledge of three dimensional structures of small and large subunits of ADP- glucose pyrophosphorylase (AGPase) in wheat has hindered efforts to understand the binding specifities of substrate and catalytic mechanism. Thus, to understand the structure activity relationship, 3D structures were built by homology modelling based on crystal structure of potato tuber ADP-glucose pyrophosphorylase. Selected models were refined by energy minimization and further validated by Procheck and Prosa-web analysis. Ramachandran plot showed that overall main chain and side chain parameters are favourable. Moreover, Z-score of the models from Prosa-web analysis gave the conformation that they are in the range of the template. Interaction analysis depicts the involvement of six amino acids in hydrogen bonding (AGP-SThr422-AGP-LMet138, AGP- SArg420-AGP-LGly47, AGP-SSer259-AGP-LSer306, AGP-SGlu241-AGP-LIle311, AGPSGln113- AGP-LGlu286 and AGP-SGln70-AGP-LLys291). Fifteen amino acids of small subunit were able to make hydrophobic contacts with seventeen amino acids of large subunit. Furthermore, decrease in the solvent accessible surface area in the amino acids involved in interaction were also reported. All the distances were formed in between 2.27 to 3.78Å. The present study focussed on heterodimeric structure of (AGPase). This predicted complex not only enhance our understanding of the interaction mechanism between these subunits (AGP-L and AGP-S) but also enable to further study to obtain better variants of this enzyme for the improvement of the plant yield.
Keywords: AGPase, ADP-glucose pyrophosphorylase, AGP-S, AGP-L, modelling, molecular docking, hydrogen bonding, hydrophobic contacts, accessible surface area
Background
Wheat (Triticum aestivum L.) is one of the most important staple crops worldwide, with a total production of over 600 million tonnes annually. The seed number and seed weight are critical yield components of wheat. Starch, which accounts 65-75% of wheat seed weight, is a major determinant wheat yield. Starch is known to be an important carbohydrate and the primary energy source for plants, having numerous industrial applications [1, 2]. It is generally accepted that four enzymes may play a key role in starch biosynthesis: ADPglucose pyrophosphorylase, starch synthase, starch branching enzyme and starch debranching enzyme [3]. ADP-glucose pyrophosphorylase (AGPase) is regarded as rate- limiting enzyme in starch biosynthesis. This enzyme plays a key role in the modulation of photosynthetic efficiency in source tissues and also determines the level of storage starch in sink tissues, thus influencing overall crop yield potential. It catalyzes the first step of starch biosynthesis by generating the sugar nucleotide ADP-glucose and inorganic pyrophosphate (Pi) from glucose-1-phosphate and ATP. ADP-glucose functions as the glucosyl donor for glucan synthesis by starch synthase. AGPase from higher plants is heterotetrameric, consisting of two large subunits (AGP-L) and two small subunits (AGP-S) encoded by two distinct genes [4]. Seed yield and plant biomass increases are conferred by deregulation of endosperm AGPase and thus, AGPase has attracted wide interest for potential crop improvements [5]. In the present study the 3D structures of the subunits (large and small) from wheat were modelled by the in silico homology techniques using the crystal structure of small subunit from potato tuber as a template. Moreover, molecular docking between these subunits was also performed to gain the insight of interaction.
Methodology
Amino acids sequences of large subunit (P12299) and small subunit (P30523) were retrieved from Swiss-Prot database [6]. Template was searched by BLAST-P analysis against PDB database. The crystal structure of potato AGPase (1YP2) available at PDB was used as template for modelling. Sequence alignment between the model sequence and template was done with ClustalW [7]. Homology models of large and small subunits were built by Modeller [8] version 9v7. One model from each subunit was selected using PROCHECK [9] and Prosa-web [10]. Further, models were subjected to energy minimization using GROMOS96 implemented via Swiss-pdb viewer [11]. RMSD of the model structures of AGP-L and AGP-S were evaluated form its template using the SUPERPOSE [12]. Docking of model structure was performed by GRAMM-X [13]. Model structure was further evaluated by Patchdock [14]. Best docked structure based on RMSD and Patchdock score was chosen for further analysis. Hydrogen and hydrophobic interactions between these subunits were analyzed by Ligplot [15]. Figures representation was generated with Discovery studio visualizer programme [16]. Accessible surface area was calculated by Mark Gerstein's calc-surface programme implemented in Chimera [17].
Results and Discussion
Model building and quality assessment
BLAST analysis of gene sequences showed highest homology with the small subunit of ADP-glucose pyrophosphorylase (AGPase) from potato. The alignment between the sequence of small subunit of ADP-glucose pyrophosphorylase (AGP-S) from wheat and potato tuber revealed at least 90% identity (Figure 1), which allowed for a predictable homology modelling approach. However, the alignment between sequences of small and large subunits revealed 52% identity (Data not shown). Template (1YP2) from potato AGPase was used for modelling the small and large subunits of wheat with Modeller 9v7. The final models were selected based on DOPE score. Evaluation of the selected models was done by Procheck and Prosa-web server. The analysis of the Ramachandran plot and Prosa-web of the template (1YP2) were used to compare the overall stereochemical quality of AGPase subunits (AGP-L, AGP-S). Ramachandran analysis showed 89% and 90% amino acids of AGP-L and AGP-S, respectively, in most favourable region (Figure 2 & Figure 3). The main chain conformation within the favoured or allowed region of Ramachandran plot and G-factor indicated the accuracy of generated models. Analysis of AGP-L and AGP-S models revealed the Z-score value of -9.56 and -8.57, respectively, which is in the range of nature conformation of the template (Table 1 see Table 1). Furthermore root mean square deviation of AGP-S and AGP-L models with respect to the equilibrated structure of AGPS (1YP2) from potato and was found to be 1.08 and 1.30, respectively. Thus overall quality assessment of the selected models assisted us to use these models for further analysis.
Figure 1.

Sequence alignment between the small subunit, large subunit of wheat and corresponding sequence of small subunit of potato (PDB: 1YP2).
Figure 2.

Model structure of AGP-S and quality assessment evaluated by Procheck and Prosa-web.
Figure 3.

Model structure of AGP-L and quality assessment evaluated by Procheck and Prosa-web.
In silico docking
We used GRAMM-X programme for docking purpose. This programme perform a rigid-body docking using Fast Fourier Transformation methods (FFT) by applying smoothed Lennard-Jones potential, knowledge-based and refinement stage scoring, which give rise to best surface match. Simulation of structural flexibility is a computational expensive process for protein- protein docking. Thus, computationally docking is difficult for putting two proteins in a complementary manner. High computational complexity restricts the flexible docking algorithms and is rarely applicable to practical protein docking at present. This problem can be overcome by using Rigid body docking algorithm. Rigid body algorithm assumes two proteins as rigid bodies. The conformation change is tolerated by allowing certain degree of penetration between proteins. This assumption will limit the problem to a six-dimensional (three for translation and three for rotation) search space [18].
GRAMM-X generated a PDB file containing the structures of ten models ranked according to the scoring function. We selected best docked complex for analyzing the interaction mechanism (Figure 2 & Figure 3). For accuracy of our result we checked our model by Patchdock [14] which uses molecular docking algorithm based on shape complimentary principle. Complementary patches are matched in order to generate candidate transformation which further evaluated by scoring function, considering both geometric fit and atomic desolvation energy [19]. For each complex top 15814 score from the Patchdock was retained for further consideration, while other were rejected due to lower score. Thus, best result from two algorithm not only increase our confidence level but also provide strength in accuracy for our output data. Ligplot analysis depicted the involvement of six amino acids in hydrogen bonding (Figure 4 & Table 2 see Table 2). Amino acids (Gln70, Gln113, Glu241, Ser259, Arg420, Thr422) of AGP-S were found to form hydrogen bonds with amino acids (Gln47, Met138, Glu286, Lys291, Ser306, Ile311) of AGP-L (Figure 4). Six hydrogen bonds were also formed in complex of small and large subunits of AGPase from potato [20]. The hydrogen bond lengths between AGP-L and AGP-S were found to be shorter then 3.3Å. Moreover, seventeen amino acids of AGP-L were involved in seventy five hydrophobic contact with fifteen amino acids of AGP-S (Table 3 see Table 3). This shows that complex is stabilized by both hydrogen bonding and hydrophobic interactions. Out of these amino acids two amino acids were found to be crucial. Amino acid Gln113 of small subunit donate the hydrogen bond to Glu286 of large subunit with 1.96Å distance which was the smallest in the complex. This amino acid also made five hydrophobic contacts with Glu286 in the distance of 2.3Å to 3.7Å. Amino acids Ile311 of AGP-L made a hydrogen bond with Glu241 of AGP-S with a distance of 3.1Å. Eight hydrophobic contacts were formed by this amino acid (Ile311-AGP-L) with Pro244 of AGP-S within the range of 2.5Å to 3.5Å. Moreover, decrease in accessible surface area in the docking complex has been observed. Change of accessible surface area of Gln113 AGP-S was found to be significant and it changed from 109.75 to 17.85 in complex (Table 4 see Table 4). As mention previously this amino acids was also found to be critical for hydrogen bonding and hydrophobic interaction (Table 3 see Table 3). Similarly accessible area of other amino acids of AGP-L; Lys291 (167.82 to 36.53), Thr300 (203.14 to 104.67) and Pro303 (102.86 to 20.94) were also found critical in the complex (Table 5 see Table 5). Thus these outcomes suggest that the interaction complex is feasible and useful for understanding the binding mechanism between AGP-L and AGP-S subunits of ADP-glucose pyrophosphorylase in wheat.
Figure 4.
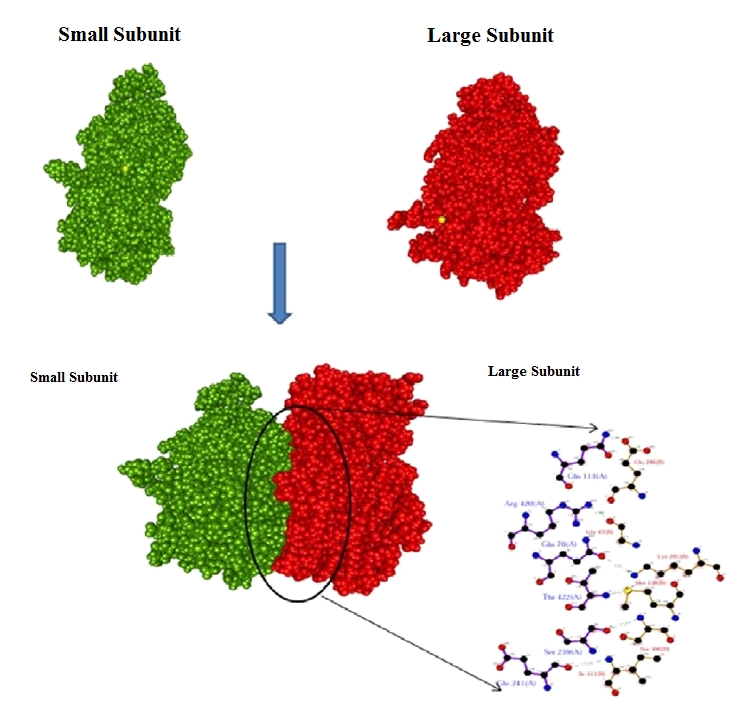
Model of AGP-S and AGP-L complex interaction and hydrogen bonding pattern.
Conclusion
ADP-glucose pyrophosphorylase (AGPase) is a major enzyme controlling starch synthesis, and has been demonstrated in many different plant species. The crystal structure of AGPase (small and large subunits) from wheat has not yet been solved. Thus, the rationale in building the AGPase subunits models and performing in silico docking study was to gain the details of interaction between two subunits (AGP-L and AGP-S). This docking study revealed the important residues involved in formation of the complex. Six amino acids of AGP-S and AGP- L were involved in hydrogen bonding where as fifteen amino acids of AGP-S were involved in hydrophobic interaction with seventeen amino acids of AGP-L. Importantly, all the amino acids that were involved in hydrogen bonding were found to be crucial as they were also caught up by hydrophobic interactions. Thus, this study hypothesise the model which can be used for further study to elucidate the role of AGPase gene in starch biosynthesis to increase the starch content in wheat and thus, grain yield.
Supplementary material
Acknowledgments
The financial support for Agri-Bioinformatics Promotion Program provided by Bioinformatics Initiative Division, Department of Information Technology, Ministry of Communications & Information Technology, Government of India, New Delhi is gratefully acknowledged. The authors are thankful to Project Director, DWR, Karnal for providing facilities for this work.
Footnotes
Citation:Danishuddin et al Bioinformation 6(4): 144-148 (2011)
References
- 1.CJ Slattery, et al. Trends Plant Sci. 2000;5:291. doi: 10.1016/s1360-1385(00)01657-5. [DOI] [PubMed] [Google Scholar]
- 2.A Tuncel, et al. Biophys J. 2008;95:3628. doi: 10.1529/biophysj.107.123042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.C Martin, AM Smith. Plant Cell. 1995;7:971. doi: 10.1105/tpc.7.7.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.PR Salamone, et al. FEBS Lett. 2000;482:113. doi: 10.1016/s0014-5793(00)01985-2. [DOI] [PubMed] [Google Scholar]
- 5.JM Cross, et al. Plant Physiol. 2004;135:137. doi: 10.1104/pp.103.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.E Boutet, et al. Methods Mol Biol. 2007;406:89. doi: 10.1007/978-1-59745-535-0_4. [DOI] [PubMed] [Google Scholar]
- 7.JD Thompson, et al. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.A Sali, TL Blundell. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 9.RA Laskowski, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 10.M Wiederstein, MJ Sippl. Nucleic Acids Res. 2007;35:407. [Google Scholar]
- 11.N Guex, MC Peitsch. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 12.R Maiti, et al. Nucleic Acids Res. 2004;32:W590. [Google Scholar]
- 13.A Tovchigrechko, IA Vakser. Nucleic Acids Res. 2006;34:W310. [Google Scholar]
- 14.D Schneidman-Duhovny, et al. Nucleic Acids Res. 2005;33:W363. [Google Scholar]
- 15.AC Wallace, et al. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 16. http://accelrys.com/products/discovery-studio/visualization.
- 17. http://www.cgl.ucsf.edu/chimera/
- 18. http://www.comp.nus.edu.sg/~leowwk/hyp/huangwenfan.pdf.
- 19.C Zhang, et al. J Mol Biol. 1997;267:707. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 20.I Baris, et al. Plos Comput Biol. 2009;5:e1000546. doi: 10.1371/journal.pcbi.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.