

Abstract
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease of unidentified aetiology, chiefly affecting the synovial membranes of joints, cartilage, bone, bursa and tendon sheath. Osteoarthritis (OA) is a degenerative disorder and encompass different sets of pathogenic pathways than RA; however, it shows a medley of clinical manifestations or symptoms resembling RA. Hence, we intend to identify more disease specific biomarkers through the meta-analysis of microarray datasets that can be crucial in the differential diagnosis, disease specific treatment as well as management of both RA and OA in a typical clinical setting.
Keywords: Rheumatoid Arthritis, Osteoarthritis, Synovial Membrane, Microarrays, Biomarkers
Background
Autoimmune diseases arise from an overactive immune response wherein the immune system mistakes some parts of the body as pathogen and attacks its own cells. Systemic autoimmune disease such as Rheumatoid arthritis (RA) affects the synovial membranes of multiple joints, cartilage and bone as well as bursa and tendon sheaths [1]. Osteoarthritis (OA), on the other hand, is a degenerative disorder of the joints showing similar symptoms such as RA. Although both the diseases share similar symptoms, it has been proposed that RA follows an inflammatory pathway of pathogenesis, thus, differentiating it from OA. Diagnosis and assessment of RA and OA depend to a great extent on the symptoms, joint damage and physical function which are semi quantitative methods of diagnosis. Till date no definite cure has been proposed for RA, management of this disease depends upon early detection and aggressive treatment. Several studies have been undertaken to identify and validate biomarkers that would help in early detection and better monitoring of these diseases. Studies have revealed biomarkers for RA including genetic association of RA with HLR-DR family (DR4, DR1, DR6 AND DR10) alleles within class II MHC region within chromosome 6 [2, 3], increased level of pro inflammatory cytokines (TNF-α,IL-1β and IL-6) [1, 4], elevated level of SPHK1 in synovium membrane of RA [5– 7]. Similarly, investigation of gene signatures of OA provided significant results including presence of MMPs, collagenase -1, MMP-3, IL-6 and TNF-α in the synovial fluid of arthritis patients [8, 9, 10]. Despite these findings and the on-going intensive investigation of the genetic pattern of these diseases, we are yet to reach an integrated outlook. Microarrays [11] have been used to study the gene expression patterns associated with various diseases models in both basic and translational research in the past decade [12– 24]. It helps to probe into the key biomarkers implicated in the pathogenesis and may provide clues for the treatment or management of human diseases. Thus, for our investigation, we applied the bioinformatics approach on publicly available microarray datasets of RA and OA in an attempt to identify key biomarkers specific to each of these diseases.
Methodology
For our analysis, all the datasets used were Affymetrix CEL files. Using the Gene Expression Omnibus ( http://www.ncbi.nlm.nih.gov/geo/) and search criteria based upon our aim of the experiment, three datasets were selected out of the available 77 datasets related to RA and OA. These datasets were obtained from experiments which were performed with cell lines or tissue, using different technology and platforms (see Table 1). Affymetrix CEL files were uploaded into GeneSpring GX 10.0 for statistical analysis. Normalization was performed using RMA and baseline transformation algorithm. Quality check was performed to remove unreliable data if any from each of the datasets. Statistical analysis was performed to determine differentially expressed genes. In case of all the datasets the fold change for every condition was calculated keeping Normal Donor expressions as the control condition. The cut off was set at 2.0 for all the datasets. The resulting fold change list was then subjected to statistical analysis. For all the datasets, the entity list selected was the one obtained after filtration by Fold Change (> = 2.0). The statistical test selected was one way ANOVA for data sets GSE1919 & GSE 12021, since there were more than two groups in each of these datasets and the cut off was adjusted at p< = 0.05. Student t-test was used for GSE10500 (since there were only two groups to be compared). Multiple testing, Benjamini Hochberg FDR was used for data set GSE1919. Hierarchical clustering was run for each of the resulting statistically significant entity lists. Cluster on both entities and condition was selected for all the datasets and the distance metric selected for all the datasets was Euclidean.
Results and Discussion
Cluster analysis of the selected datasets
Statistical analyses performed on the datasets showed encouraging results (see Table 2) and hence, hierarchical clustering (HCL) was applied to each of the datasets to visualize the significant gene clusters and their specific pattern of expression in normal as well as test samples. As expected, the HCL (Figure 1) uncovered the differential gene expression patterns for each of the dataset analyzed when compared with the corresponding control groups. The differential expression of genes in normal control against RA patients showed regions where there was down regulation of genes in the normal control whereas up regulation at that region can be observed in RA genes, indicating the difference in gene expression patterns in diseased and normal states. Similar observations can be made for each of the other datasets.
Figure 1.
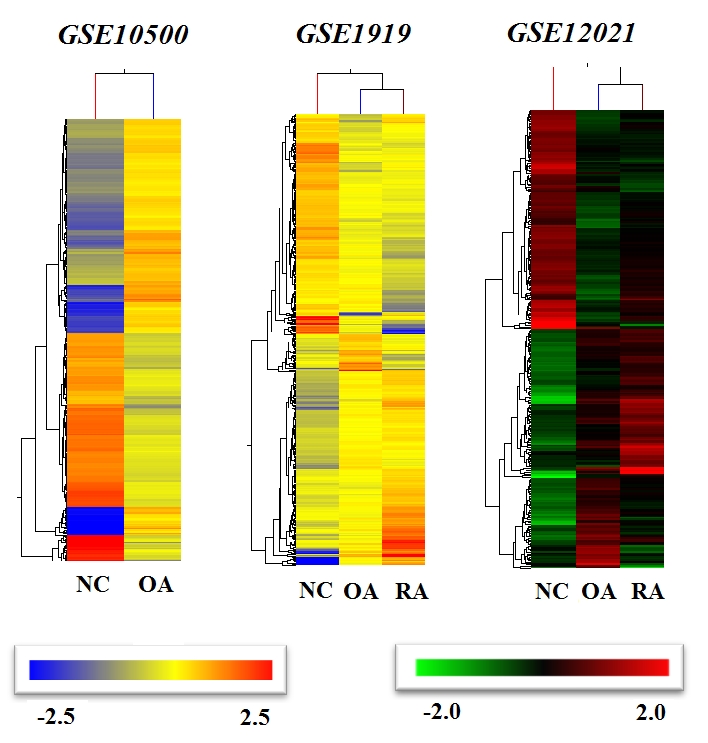
Dendrograms generated by hierarchical clustering algorithm using GeneSpring GX10.0 for each dataset (GSE10500, GSE1919, GSE12021) showing the gene signatures of RA and OA compared with NC (NC-Normal Control, RA- Rheumatoid Arthritis, OA- Osteoarthritis).
Gene expression analysis in RA
Data set GSE10500 was used in understanding the pathogenesis of RA. Pro inflammatory genes such as TNF, NFAT, cJUN, IL15 showed increased expression in diseased state (see Table 3). On the other hand, anti-inflammatory cytokines such as Interleukin 10 (IL 10) and its receptor (IL10RB) showed decreased expression. Additionally, there was increased expression of chemokines such as IL8 and CCL7. Up regulation of genes related to signal transduction such as GEM, NR4A2 were also reported. Genes regulating proteases were also found to be up regulated (MMP1, MMP3). Additionally, one of the interesting observations was the expression of transcription factor ATF4 associated with Endoplasmic reticulum (ER) stress pathway and VEGF associated with angiogenesis.
Gene expression profile of RA and OA
The datasets GSE1919& GSE12021 were used to distinguish between OA and RA. These datasets showed interesting results (see Table 4). The expression of genes related to pro inflammatory genes were up regulated in case of both OA and RA indicating inflammation as a condition in both the diseased states. Chemokines (IL8, CXCL12, CCL5, CCR5) that aid in mounting an increased immune response were also found to be expressed. Other Immunoregulatory genes such as FCGR1A, FCGR3A were found to be up regulated in both RA and OA. Additionally, certain genes involved in regulation of Phospholipase C (PLCG2) were seen to be up regulated in both OA and RA. Though activity of PLC in RA has been reported earlier, in case of OA, it was something unusual. Cytokines have been found to be playing a major role in RA. They promote autoimmunity by maintaining chronic inflammatory synovitis leading to the destruction of adjacent joint tissues(1). Cytokines have therefore, been found to combine the immune regulatory and tissue destructive events that lead to progression of RA. In our metanalysis, we have found that there was an up regulation of genes coding for proinflammatory cytokines IL-1β, TNF-α, IL-15 etc., (Table 3). IL-1β plays a key role in triggering the up-regulation of pro-inflammatory factors (Okusawa S et.al 2005).
Role of T cells in RA
In all the datasets analysed, there was an up regulation of T-cell receptor alpha chain (TRAα) (Table 3 & Table 4) indicating that T-cells play pivotal role in the pathogenesis of RA. Another indication of involvement of T cells is the up regulation of IL-15 which is one of the key growth factor for the synovial Tcells [25]. Some of the studies have shown that IL-15 exhibited pro inflammatory activity in vitro [25] as well as in the animal models of arthritis [26] suggesting that IL15 plays an important role in RA. Interleukin 8 (IL8) a chemokine, showed increased activity in RA samples in two out of three datasets (GSE10500, GSE1919). It can be seen IL8 might possibly play an important role in the pathogenesis of RA. It helps in up regulating the expression of genes involved in angiogenesis (VEGF), as well as tumor invasions (MMP2 and MMP9). IL8 triggers inflammation in cells like neutrophils leading to chemotaxis, respiratory burst, granule release, increased cell adhesion and mobilization of Ca2+ in the cell, thus, helping initiation of expression of Ca2+ dependent transcription factors like NFAT. It thus, becomes evident that IL8 plays an important role in the pathogenesis of RA patients. Our results on RA datasets have shown the differential regulation of most of these genes already found in the literature and strongly supports the involvement of proinflammatory mediators released from the auto-reactive immune cells in the pathogenesis of RA.
Role of B cells in RA
The potential involvement of B cells in the development of RA is also indicated by the up regulation of some of the specific genes (CXCL13, CCL21, MMP1, MMP3) (Table 3 & Table 4). Mature dendritic cells in synovial membranes ectopic germinal centres produce increased level of B-cell survival factor (tumor necrosis superfamily member TNFSF13) [27, 28]. Increased expression of Tumor Necrosis Factor Superfamily (TNFSF) members (Table 4) indicates the B cell survival during the pathogenesis of RA. The lymphocyte infiltrate in the RA synovium has various patterns of structural organizations. A study has shown that that the formation of ectopic germinal centres aids in the improper regulation of self-reactive B cells, due to local affinity maturation and receptor editing [28]. In addition, the cytokines and chemokines contribute to this lymphocyte organization. For example, the CXC-chemokine ligand 13 (CXCL13) and CCL21 help the formation of synovial germinal centres [29]. In our results, although expression of CXCL13 was not observed, another chemokine family member, CXCL12 (Table 4) was found to be up regulated in RA samples. Moreover, the chemokine CCL5, MMP1and MMP3 were up regulated in patients with RA (Table 4) which are expressed by synovial fibroblasts and are shown to support the recruitment and activation of T cells [1]. Production of vascular endothelial growth factor (VEGF) was reported by samples from dataset GSE1919 & GSE10500 (Table 3 & Table 4). The potential function played by VEGF in pathogenesis of RA is angiogenesis. It has been reported that several pathological conditions including RA are characterized by excessive angiogenesis where vessels develop in an uncontrolled or disorganized manner [30]. Together, these data indicate a central role played by cytokines in integrating the inflammatory and destructive phases in RA. Certain genes such as GTP binding protein overexpressed in skeletal muscle, Nuclear receptor subfamily 4, group A, member 2 (GEM & NR4A2) belonging to the functional group of signal transduction showed up regulation in RA. During mast cell activation, many signaling molecules are engaged in diverse activities including generation of lipid derived pro inflammatory mediators and the production of cytokines and chemokines [31]. Hence, the up regulation of the above mentioned genes indicate an inflammatory state that is typical in patients with RA. Thus, it was observed that across all datasets, the samples from RA patients showed the differential expression of genes involved in the inflammatory response, which aids in the pathogenesis of the disease.
Association of RA with Atherosclerosis
An interesting finding from the analysis of the dataset GSE10500 was the increased expression of genes related to the pathogenesis of atherosclerosis such as IL8, MMP1, and TNFSF14. Our findings support several other recorded data that the above mentioned genes play a role in promoting inflammatory response in atherosclerosis [32]. It can be seen that pro inflammatory cytokine producing genes IL1 and IL8 have been increased across the datasets. Additionally, tissue factor (TF) and MMP3 are also up regulated indicating an inflammatory condition. This condition stimulates Smooth Muscle Cells (SMC) proliferation and initial migration towards the lesion ultimately forming the stable plaque which is large lipid core covered by a thick fibro-muscular cap with SMC and extra cellular matrix (EMC). Under conditions of chronic inflammation, this stable plaque transforms into a vulnerable plaque. This is aided by a cascade of events that ultimately lead to rupturing of the vulnerable plaques resulting in thrombosis and possible myocardial infarction. As can be seen, a condition of chronic inflammatory response, such as in case of RA, simply helps in initiation and deterioration of atherosclerosis. There is evidence that cardiovascular disease is the leading cause of mortality in patients with chronic inflammatory disease [33]. Additionally, it has been reported that RA patients have increased prevalence to subclinical atherosclerosis [34]. This observation suggests that patients with chronic inflammatory disorders, such as RA, are at an increased risk of cardiovascular disease due to shared pathogenic pathways.
Association of ER pathway and RA
An important observation was up regulation of gene ATF4 (Table 4). This particular gene has previously been associated with Endoplasmic stress reticulum pathway [35].This suggests that there is a correlation between patients suffering from RA and the development of stress due to the disease. ER-initiated cell death pathways have been recognized for several diseases such as hypoxia, ischemia, heart disease, neurodegeneration and diabetes [36]. Although no data have been published on the involvement of stress pathway in RA, our finding suggests a possibility that the stress might also play a pivotal role in the aggravation of RA or vice versa.
Role of PLCγ in OA
An interesting finding was the differential regulation of genes Phospholipase C gamma (PLCγ) (Table 4). PLCγ signaling has been previously associated with disorders related to cellular development and cell signaling. Although there have been reports of PLC in rheumatoid arthritis, no records have been published related to involvement of PLC in OA. Expression of PLCγ in patients with RA is expected as PLC γ is activated by TCR, BCR, the high affinity IgE receptor and IgG receptors. In RA, there is an increased expression of TCR and BCR, thus, aiding the activation of PLCγ. Previous studies have shown that RA can be associated with increased expression of PLCγ in synovial tissues [37]. Activation of PLCγ leads to hydrolysis of PIP2 which releases second messengers DAG and IP3, out of which, IP3 stimulates the release of stored Ca2+ from ER. This released Ca2+ aids in the activity of transcription factor NFAT. A study by the same group showed that PLCγ does not play any role in osteoarthritis in human [37]. However, from our results, PLCγ can be seen to be up regulated even in case of OA, suggesting a similar potential role played by it in the pathogenesis of RA. The number of genes up regulated in both RA and OA was more when compared to the genes uniquely expressed by each of them (Figure 2). This indicates that both RA and OA share the key pathway responsible for the pathogenesis to some extent. The differentially expressed genes in the pathogenesis of RA and OA from the dataset GSE1919 showed up regulation of genes linked to the proinflammatory pathways. In case of RA, this was very well expected. However, increased expression of these genes in OA was very interesting; since it is not an inflammatory disorder, but a degenerative disorder. However, it may be assumed that in case of OA, after the initial cartilage damage due to physical/mechanical stress, the infiltration of immune cells leading to the release of inflammatory effectors as a part of the innate immune response. This could explain the differential expression levels of these proinflammatory genes in OA. However, the observation of the fold change of the genes in RA and OA indicates that there is a much higher up regulation of the genes in RA than OA.
Figure 2.
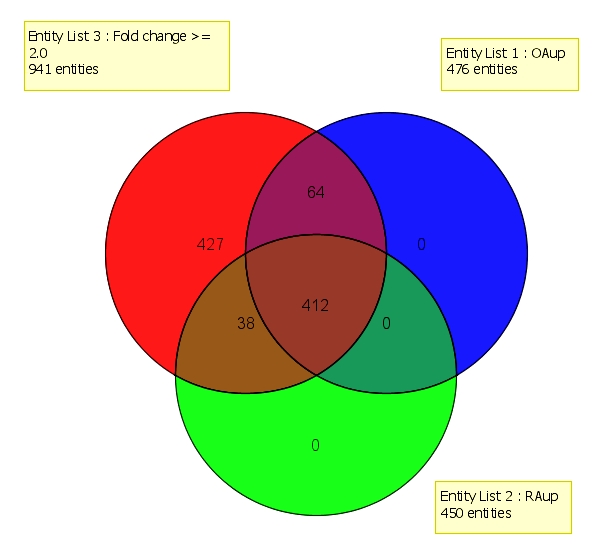
Representation of number of genes up regulated in RA and OA (GSE1919) using Venn diagram. Entity List 1 represents up regulated genes associated with OA. Entity List 2 represents up regulated genes associated with RA. Entity List 3 represents all the up regulated genes obtained after filtration by fold change (cut off >> 2.0).
Conclusion
This study is an attempt to identify potential biomarkers for RA and OA as well as to differentiate between the two disorders with respect to biomarkers. As it is observed from the results, the expression of cytokines plays an important role in the pathogenesis of these two debilitating diseases. Understanding the cytokine signaling cascade might aid in targeting these pathways during early stages of these diseases to induce tolerance. Apart from the pro inflammatory genes, other genes that help in mounting an increased immune response were also identified across all the datasets. Our study also identified some unique gene signatures such as ATF4 in RA (ER stress pathway), PLCγ signaling in OA, IL8 signaling in RA from our datasets. These pathways can be further investigated in detail for their roles in the disease prognosis to design personalized medicines or therapeutics based on the disease type and stage in individual patients. Moreover, this study highlights how inflammatory cardiovascular disease like Atherosclerosis and RA are co-related in terms of their pathogenic pathways. Hence, it can be concluded that patients with RA run a higher risk of atherosclerosis and therefore, should be screened regularly for cardiovascular disorders.
Supplementary material
Acknowledgments
Authors would like to thank Prof.Iain McInnes for providing access to the Genespring GX10 software (Agilent Technologies, Santa Clara, CA, USA) for performing the meta-analysis of high throughput gene expression data from GEO.
Footnotes
Citation:Biswas et al, Bioinformation 6(4): 153-157 (2011)
References
- 1.I McInnes, G Schett. Nat Rev Immunol. 2007;7:429. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 2.M Coenen, P Gregersen. Genes Immun. 2009;10:101. doi: 10.1038/gene.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D Mattey, et al. J Rheumatol. 2001;28:232. [PubMed] [Google Scholar]
- 4.FM Brennan, IB McInnes. J Clin Invest. 2008;118:3537. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WQ Lai, et al. J Immunol. 2008;181:8010. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- 6.WQ Lai, et al. J Immunol. 2009;183:2097. doi: 10.4049/jimmunol.0804376. [DOI] [PubMed] [Google Scholar]
- 7.L Zhi, et al. J Cell Physiol. 2006;208:109. doi: 10.1002/jcp.20646. [DOI] [PubMed] [Google Scholar]
- 8.N Ishiguro, et al. Arthritis Rheum. 1999;42:129. doi: 10.1002/1529-0131(199901)42:1<129::AID-ANR16>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.K Naito, et al. Rheumatology. 1999;98:510. [Google Scholar]
- 10.DJ Hunter, et al. Arthritis Res Ther. 2007;9:R108. doi: 10.1186/ar2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.M Jayapal, AJ Melendez. Clin Exp Pharmacol Physiol. 2006;33:496. [Google Scholar]
- 12.A Pachiappan, et al. Toxicon. 2005;46:883. [Google Scholar]
- 13.CH Koh, et al. J Cell Physiol. 2007;211:63. doi: 10.1002/jcp.20912. [DOI] [PubMed] [Google Scholar]
- 14.B Jiang, et al. J Neurosci Res. 2008;86:3481. doi: 10.1002/jnr.21800. [DOI] [PubMed] [Google Scholar]
- 15.MJ Chen, et al. Free Radic Biol Med. 2001;50:736. [Google Scholar]
- 16.A Hegde, et al. Mol Med. 2010;16:188. [Google Scholar]
- 17.RL Gurung, et al. Genome Integr. 2010;1:5. doi: 10.1186/2041-9414-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ZF Peng, et al. J Cell Mol Med. 2011 [Google Scholar]
- 19.MS Choy, et al. J Cell Physiol. 2011;226:494. doi: 10.1002/jcp.22359. [DOI] [PubMed] [Google Scholar]
- 20.MJ Chen, et al. J Cell Physiol. 2011;226:1308. doi: 10.1002/jcp.22459. [DOI] [PubMed] [Google Scholar]
- 21.X Dai, et al. Blood. 2009;114:318. [Google Scholar]
- 22.JP Newman, et al. J Cell Physiol. 2008;214:796. doi: 10.1002/jcp.21276. [DOI] [PubMed] [Google Scholar]
- 23.B Banerjee, et al. Integr Cancer Ther. 2007;6:242. doi: 10.1177/1534735407306214. [DOI] [PubMed] [Google Scholar]
- 24.R Reghunathan, et al. BMC Immunol. 2005;6:2. [Google Scholar]
- 25.IB McInnes, et al. Nat Med. 1997;3:189. [Google Scholar]
- 26.S Ferrari-Lacraz, et al. J Immunol. 2004;173:5818. doi: 10.4049/jimmunol.173.9.5818. [DOI] [PubMed] [Google Scholar]
- 27.TM Seyler, et al. J Clin Invest. 2005;115:3083. doi: 10.1172/JCI25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.S Takemura, et al. J Immunol. 2001;167:1072. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 29.T Dorner, PE Lipsky. Curr Top Microbiol Immunol. 2006;305:213. doi: 10.1007/3-540-29714-6_11. [DOI] [PubMed] [Google Scholar]
- 30.MJ Cross, L Claesson-Welsh. Trends Pharmacol Sci. 2001;22:201. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 31.M Jayapal, et al. BMC Genomics. 2006;7:210. doi: 10.1186/1471-2164-7-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.WA Boisvert, et al. J Clin Invest. 1998;101:353. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.A Naranjo, et al. Arthritis Res Ther. 2008;10:R30. doi: 10.1186/ar2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.YB Park, et al. Arthritis Rheum. 2002;46:1714. doi: 10.1002/art.10359. [DOI] [PubMed] [Google Scholar]
- 35.J Ye, C Koumenis. Curr Mol Med. 2009;9:411. doi: 10.2174/156652409788167096. [DOI] [PubMed] [Google Scholar]
- 36.C Xu, et al. J Clin Invest. 2005;115:2656. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.S Shahrara, et al. Arthritis Res Ther. 2007;9:R112. doi: 10.1186/ar2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.