

Abstract
Cutaneous adverse drug reactions range from mild to severe and from those localized only to skin to those associated with systemic disease. It is important to distinguish features of cutaneous drug reactions which help classify the underlying mechanism and likely prognosis as both of these influence management decisions, some of which necessarily have to be taken rapidly. Severe cutaneous reactions are generally T cell-mediated, yet this immunological process is frequently poorly understood and principles for identification of the culprit drug are different to those of IgE mediated allergic reactions. Furthermore, intervention in severe skin manifestations of drug allergy is frequently necessary. However, a substantial literature reports on success or otherwise of glucocorticoids, cyclophsphamide, ciclosporin, intravenous immunoglobulin and anti-tumour necrosis factor therapy for the treatment of toxic epidermal necrolysis without clear consensus. As well as reviewing the recommended supportive measures and evidence base for interventions, this review aims to provide a mechanistic overview relating to a proposed clinical classification to assist the assessment and management of these complex patients.
Keywords: adverse drug reaction, drug allergy, skin, toxic epidermal necrolysis
Introduction
Drug hypersensitivity/allergy is an important concern for healthcare providers, as reviewed elsewhere in this series of articles. Skin manifestations are the commonest presentation of drug allergy [1, 2] and they may range from mild (two-thirds) to severe or life threatening (one-third) in a hospital setting [3]. Distinguishing these two outcomes is clearly the most important task for any clinician engaged in the management of individuals with a drug allergy. Perhaps because of the ready visibility of signs in skin, cutaneous manifestations are frequently also the earliest sign of systemic drug allergy and can therefore prove critical in providing information on the severity and prognosis of the allergic reaction. However, the reaction patterns are very variable in both mechanism and clinical features. As a result, there is no consensus on the classification of these disorders. Mechanistic, immunological classification can be very helpful in guiding clinicians and researchers in the approach to diagnosis and treatment as well as providing a scientific basis on which new diagnostic assays may be developed. Unfortunately, our current understanding of the disease processes is not sufficiently detailed to allow explanation of how apparently similar pathological mechanisms can generate remarkably different clinical patterns. Therefore, it remains useful to overlay an immunological understanding with a clinical classification. This review series includes a detailed description of the immunological mechanisms of drug allergy. Here we propose to give an overview of the mechanisms involved specific to cutaneous drug allergy with emphasis on concepts that will aid clinical diagnosis and treatment.
Mechanisms in cutaneous drug hypersensitivity reactions
The skin may be involved in drug hypersensitivity reactions either alone or as part of multi-organ involvement. A wide variety of clinical patterns occur, so it often requires an astute clinical awareness to suspect a drug or drugs in the causation. It is important for clinicians to realize that apparently similar clinical lesions in the form of erythematous raised swellings resembling weals, can be produced by very different immunological mechanisms. Careful clinical assessment of the duration of lesions and the rate at which they evolve is critical in the clinical assessment as this will be important in deciding upon treatment approaches and performance of appropriate diagnostic tests. Hypersensitivities can be classified as either those that are the result of immunological effector mechanisms, including antibodies of different classes and/or T lymphocytes, or those that do not involve immune effectors but are the result of direct chemical effects of the drug. In susceptible individuals who are ‘intolerant’, the drug may directly induce mast cell degranulation or release of inflammatory mediators such as leukotrienes which produce clinical syndromes such as urticaria, asthma or anaphylaxis that can be indistinguishable from true immunological hypersensitivities.
Regarding the fundamental mechanisms underlying cutaneous drug hypersensitivities there are a number of aspects that must be considered. These include the drug disposition and how drugs become immunogenic in certain individuals; the nature of the immune response and the immune effector mechanisms that are generated, and why the skin is involved in some reactions.
Drug disposition
The activation of the acquired immune system, that generates specific immunological memory and humoral and cellular effector mechanisms, must start with the presentation of antigen to T lymphocytes. In order for drugs with a molecular weight less than 1000 Da to become recognizable by T lymphocytes they must become ‘haptens’ by binding to a protein carrier (haptenation). Antigen presenting cells ‘process’ the hapten-modified carrier protein, degrading it to peptides which are individually loaded into the molecular groove of an MHC molecule in the classical way. The hapten:peptide:MHC complex is then displayed on the cell surface to be recognised by T cells with appropriate specific receptors. Many native drug molecules are not intrinsically protein-reactive, although some, including penicillins, cephalosporins and captopril, can bind directly to serum or cellular proteins [4, 5]. It is thought that protein-reactive haptens are generated during the process of biodegradation and detoxification. One of the main theories proposes that after phase 1 metabolism by the cytochrome P450 superfamily of enzymes, the potentially reactive intermediates are not fully detoxified by phase 2 metabolism. They therefore persist and react with ‘self’ cellular proteins which causes them to be recognized as ‘foreign’ by T cells of the immune system. Much research has gone towards identifying the proteins to which the reactive metabolites become bound but so far it has not met with much success. There are some examples which suggest the haptenated proteins may include the CYP450 enzymes involved in the phase 1 metabolism. Thus, in patients who develop drug-induced hepatitis from halothane, it appears that the critical metabolite of halothane, trifluoroacetyl halide, becomes bound to the cytochrome P450 enzyme (CYP2E1) for which halothane is a substrate, altering the structure of the enzyme: so-called substrate-mediated inhibition. This has the effect of inactivating the enzyme and presumably, targeting it for proteolytic degradation. The immune response in patients with halothane hepatitis is detected in the form of specific antibodies against the complex of trifluoroacetyl/CYP450 [6, 7]. It has yet to be resolved whether the pathogenic immune effector mechanisms are antibody or T cell-mediated. Similarly, in patients who have had ADRs to carbamazepine and other anticonvulsants, antibodies are found against the rat CYP3A family responsible for metabolism of carbamazepine [8]. T lymphocytes reactive with carbamazepine or its metabolites can also be demonstrated in patients who have suffered ADRs from that drug [9, 10]. Thus, it appears that the reactive metabolite of many drugs, generated by the actions of CYP450 enzymes, can ‘bite back’ and become covalently bound to the enzyme. This complex of haptenic metabolite and protein carrier could be what is recognised by the immune system, as in the case of halothane. However, efforts to demonstrate inadequate or defective phase 2 metabolism have been generally unsuccessful. Indeed, there is increasing evidence that for many chemicals, it is the native compound rather than a metabolite which is the immunogen [11–13].
Clearly, self-proteins are shared throughout the population and for the native drug model, the same drug would be expected to haptenate in the same way in most individuals. Therefore, the question of how native drugs may generate an immune response in certain individuals but not others is not clear. The drug still has to become a hapten and bind to proteins, but many drugs do indeed have intrinsic protein-reactive properties. The random somatic recombination critical in defining the T cell receptor (TCR) repertoire in humans has been predicted to provide 1015 potential receptors of different specificities, yet adult humans are thought to have only 107 of these in circulation [14, 15]. Therefore, it is entirely possible that drug sensitivity may also be reflected by whether certain individuals possess T cells with specific receptors for that drug.
Another theory that is attracting much attention is whether the drug molecule is sensed by the immune system as something ‘dangerous’[16]. Substantial evidence demonstrates that the immune system is designed to generate active immune responses to dangerous stimuli such as microbes or their products and cells that die by the process of necrosis, but it is designed not to react if there is no signalling of danger during the encounter with potential immunogens. The question of how drugs might give danger signals is the subject of much research but one of the likely mechanisms is the oxidative stress that occurs during drug metabolism. The activities of CYP450 enzymes and the phase 2 metabolic enzymes generates reactive oxygen species (ROS) and depletes intracellular anti-oxidant defences such as reduced glutathione. Such oxidative stress activates transcription factors including AP-1 [17], NFκB [18, 19] and NRF2 [20, 21]. These in turn induce protective anti-oxidant responses and activate the so-called innate immune response via release of cytokines which can activate the dendritic antigen-presenting cells to a high level of efficiency. It seems likely that endogenous anti-oxidant reserves and defences may be crucial in determining whether a particular drug generates danger signals in that individual which can result in an active immune response.
A further factor that is recognized but not yet understood is the association of particular drug-induced adverse reactions with particular HLA phenotypes. For example, abacavir induces a hypersensitivity reaction very preferentially in individuals of HLA-B*5701 [22], allopurinol in HLA-B*5801 [23] and in certain Chinese populations, Carbamazepine hypersensitivity is predicted by HLA-B*1502 [24]. Interestingly, this HLA association, which is extremely strong in Han Chinese populations, does not hold true in western populations. The nature of the interaction between any of these drug molecules and the MHC associated peptides is still unclear. It has been suggested that the particular amino acid structure of HLA-B*5701 may favour the binding of a key peptide in the MHC class 1 groove, but it is not clear whether abacavir is part of the bound complex. Indeed, in explanation for the lack of 100% concordance between abacavir treatment in HLA-B*5701 individuals and hypersensitivity reactions, one group has suggested that polymorphism induced changes in heat shock protein function may provide the necessary conditions for its HLA-restricted initiation of adaptive immune responses [25].
As a result of the discovery of such strong HLA associations, most regulatory agencies including the USA and UK suggest pre-screening for at risk HLA alleles prior to initiation of abacavir and for individuals with Chinese or Thai ancestry before carbamazepine.
The type of immune response
The immune system requires an initial exposure to an immunogen to initiate the response which results in the development of immunological memory - in humans this takes at least 7 days. It then requires further exposure to the immunogen to elicit the immune effector mechanisms. In the case of drug hypersensitivities, this may be a second course of the same drug at a later date, or the availability of the drug from a continuous/ongoing exposure. The immune effector components include antibodies of all classes with or without amplification cascades such as the complement system, and T lymphocytes of helper (CD4) and cytotoxic (CD8) activities. It is not known what determines the pattern of immune effectors generated by a given drug in any individual. However, the different effector mechanisms can generate very distinct clinical patterns as will be considered below.
Immunological mechanisms underlying drug hypersensitivity
The four main types of immune reaction process were originally defined by Coombs & Gell [26] and have been expanded more recently by Pichler [27]. (Table 1A,B). Types 1–3 are mediated principally by antibodies of different classes while type 4 processes are mediated by T cells but there are many different clinical syndromes that can be produced. The mechanistic aspects will be considered first after which the clinical distinctions will be explained.
Table 1.
Types of hypersensitivity reaction; mechanisms and clinical correlations
| (A) Type of hypersensitivity | Immune effector mechanisms | Clinical manifestations relevant to drug hypersensitivity |
|---|---|---|
| Type 1 Immediate or anaphylactic | IgE bound to surface of mast cells or basophils. Antigen-binding causes mast cell degranulation, release of histamine and other mediators. | Urticaria, asthma, anaphylaxis |
| Type 2 Cytotoxic | Antigenic determinants on cell surfaces are targets for antibodies, may be IgG or IgM. The antibodies damage cells/tissues by activating complement, or by binding to cells through Fcγ receptors, they activate cytotoxic killing, e.g. by K cells. | Pemphigus, Blood cell penias: haemolytic anaemia, neutropenia, thrombocytopenia |
| Type 3 Immune complex | Circulating immune complexes are deposited in vascular beds or on tissue surfaces. Complement is activated, neutrophils attracted and their products damage tissues. | Vasculitis - hypersensitivity vasculitis, Henoch-Schonlein purpura |
| Type 4 Delayed type, T cell mediated | Effector T lymphocytes, may be CD4+ or CD8+, producing different patterns of cytokines and/or cytotoxic factors. | Many clinical patterns sub-categorized in Table 1B |
| (B) | Immune mediators | Inflammation characterized by: | Clinical pattern |
|---|---|---|---|
| Type 4a | Th1/Tc1 cells: IFN-γ, TNFα | T cells, macrophages | Contact dermatitis, Tuberculin reaction |
| Type 4b | Th2 cells: IL-4/-13 IL-5 | Eosinophils | Maculopapular rash, Exanthemata with eosinophilia |
| Type 4c | Cytotoxic T cells: Perforin Granzyme B | T cells Keratinocyte apoptosis | Contact dermatitis, maculopapular rash, drug-induced exanthemata, Bullous eruptions (SJS.TEN) |
| Type 4d | T cells: CXCL8 GM-CSF | Neutrophils | AGEP (Acute generalized exanthematous pustulosis) |
Type 1 hypersensitivity involves antigen-specific IgE bound on the surface of mast cells and basophils. Interaction with antigen degranulates the cells with release of mediators including histamine, prostaglandins and leukotrienes. This results in rapid vasodilatation and increased vascular permeability producing redness and oedema in the skin (urticaria and sometimes angio-oedema), or contraction of smooth muscle resulting in bronchoconstriction (asthma) and/or intestinal cramps and diarrhoea. If the mediator release is very great, the systemic syndrome of anaphylaxis results.
Type 2 reactions involve binding of antibodies (usually IgG) to antigenic determinants on the surface of various cell types. In some drug-induced reactions the drug is part of the antigen [28]. in others, such as pemphigus foliaceous auto-antibodies target self-proteins and the precise role of the drug is unclear.
Type 3 reactions involve circulating immune complexes in which IgG antibodies of relative low affinity are complexed with the antigen in small complexes. The complexes adhere to endothelial surfaces and fix complement, which induces accumulation and activation of neutrophils resulting in damage to the endothelium sufficient to allow extravasation of erythrocytes and the clinical syndrome of allergic or hypersensitivity vasculitis with purpuric lesions in skin.
Type 4 reactions are classically referred to as ‘delayed type’ because they develop over many hours. Investigation of the reactions in skin has led to the realization that T lymphocytes can deploy a wide range of mediators and effector mechanisms (Table 1B) that generate very different clinical manifestations.
We emphasise that appreciation of this mechanistic classification is principally of importance in consideration of the very simliar clinical patterns of weal-like lesions that can be generated by both type 1 IgE-mediated mechanisms and type 4, T cell-mediated mechanisms. As will be seen below, the diagnostic and prognostic implications can be extremely different.
Clinical aspects of cutaneous drug hypersensitivity
The skin is commonly involved in drug hypersensitivity reactions, often as the only affected organ but also often as part of a multisystem disorder. Making the diagnosis of a drug-induced reaction requires familiarity with the clinical patterns while identifying the culprit drug depends largely on a very careful and detailed history of drug ingestion, documenting the temporal relationships between the onset of the reaction and the dates and doses of all drugs administered before, during and after the reaction. It should be remembered that to become allergic to a drug or any immunogen requires a minimum of 7–10 days. Therefore, when a reaction begins rapidly after the exposure, some previous exposure to the same or possibly a chemically similar and hence cross-reactive compound must have been encountered. There are exceptions which will be dealt with below (urticaria). When a drug is given for the first time in a course that lasts a week or more, the reaction can begin during that first course.
Some reaction patterns are distinctive and are readily diagnosed while others appear to have similar or overlapping clinical features and are the source of confusion among most physicians and many dermatologists (Figure 2) [29]. The biggest confusion is in the many patterns of T cell-mediated, type 4 hypersensitivities. Before they are dealt with it is important to clarify the features of the type 1 hypersensitivities which manifest as urticaria. In a recent hospital population the most frequent cutaneous reaction patterns of drug hypersensitivity were drug induced exanthemata (51.2%) and urticaria (12.2%). The other more severe reaction patterns as described below ranged from 2% to 5% of all adverse cutaneous drug reactions [30].
Figure 2.
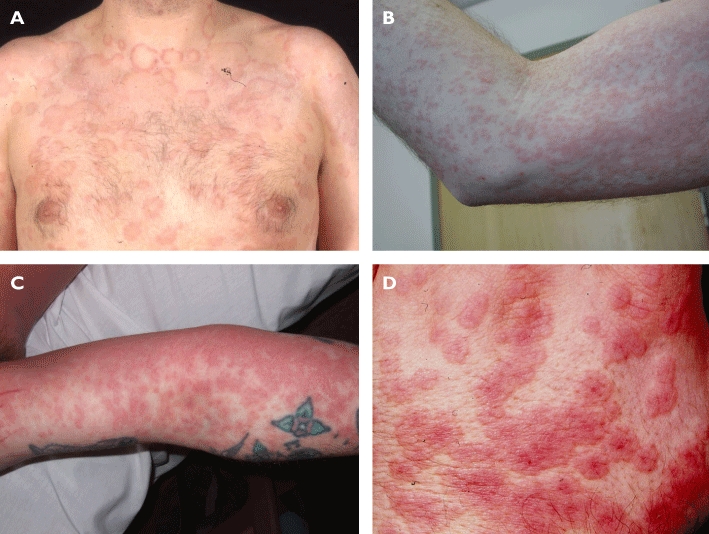
Clinical photographs depicting classic examples of four similar cutaneous drug eruption patterns. Of note, the plaques of urticarial eruptions (A) have pale centres in contrast to the uniform colour in drug-induced exanthemata (B) and drug reaction with eosinophilia and systemic symptoms (DRESS) (C) and the dark centre (target) in erythema multiforme (D)
Urticaria
Urticaria consists of individual lesions that evolve rapidly over a few hours, causing red (erythematous) swellings (weals) that are usually itchy. Careful inspection shows the brightest erythema is at the outer edges of the lesions, which may expand leaving a paler central area which can return to normal skin colour after a few hours (Figure 2). The attack may last days, but the continual appearance and disappearance of new lesions is characteristic. Patients often make the diagnosis for themselves because the reaction starts rapidly after exposure to the causative drug or agent. However, some individuals develop urticaria or anaphylaxis after the first exposure to certain drugs, commonly aspirin or non-steroidal anti-inflammatories, opiates, anaesthetic muscle relaxants and radiocontrast media. These are ‘pseudo-allergic’ reactions because they mimic the allergic symptoms but are not mediated by immune mechanisms. Instead, these are intolerance reactions in which the affected individuals are higly susceptible to the chemical effects of the drug which directly induces release of mediators such as histamine and leukotrienes from mast cells or basophils.
Drug-induced exanthemata
Drug-induced exanthemata are a group of rashes some of which can resemble urticaria but which have different temporal relationships reflecting their causation by T cell mediated mechanisms [29]. The range of rashes or exanthemata include ‘maculo-papular eruptions’ which consist of erythematous lesions varying from pin point in size up to a few millimetres across, through a wider range of rashes consisting of larger oval or round lesions. These resemble urticaria in that the brightest erythema is at the periphery of the lesions but they do not usually clear in the centres and the lesions last anything from 5 to 10 days. The attack evolves with a steady accumulation of new lesions whilst older ones enlarge slowly, increasing the density and extent of the exanthem. Many of these reactions involve only the skin but some patients manifest systemic features including fever, eosinophilia, lymphadenopathy and organ dysfunction which may prinicipally affect the liver, bone marrow and/or kidneys. The combination of a widespread eruption with systemic dysfunction is termed DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms) or DIHS (Drug-Induced Hypersensitivity Syndrome). There is now the realization that patients with DRESS often develop a worsening of the clinical picture after the initial reaction starts resolving. There may be recurrence of fever, either a leukocytosis or lymphopenia and deterioration of organ function. This is due to reactivation of members of the herpes virus family, HHV6 and HHV7 in particular, but EBV and/or CMV as well [31, 32].
Erythema multiforme, Stevens Johnson syndrome and toxic epidermal necrolysis
These syndromes can be regarded as forming a spectrum of increasing severity, but there are authors who regard them as separate [33].
Lesions of erythema multiforme (EM) can be mistaken for the above drug-induced exanthemata, but careful observation shows strikingly circular lesions with a different organization and distribution of colour (Figure 2). Lesions may be flat or raised like weals and are usually 1–2 cm in diameter. They are usually darker in the centre and paler peripherally; sometimes larger lesions form and they exhibit concentric rings of colour, so-called target or iris lesions. EM can occur in minor forms with lesions scattered peripherally on the limbs and face, a pattern usually induced by infection with herpes simplex virus. EM major is a more extensive exanthem associated with blistering of some of the lesions. There is often some systemic disturbance including fever, malaise and organ dysfunction, but there is minimal mucosal involvement. Blistering and skin loss between 1 and 10% of the body surface area (BSA) together with mucosal blistering or erosions is named Stevens Johnson syndrome (SJS). If the blistering and skin loss affect 10 -30% BSA it is called the SJS/toxic epidermal necrolysis (TEN) overlap and if the blistering and skin loss exceed 30% BSA it is TEN (Figure 3). A robust clinical scoring system (SCORTEN) has been devised which gives 1 point for each of seven clinical features (Table 2) [34]. Once the SCORTEN reaches 3 the predicted mortality is 35% and although this is the score at which it is generally recommended that patients should be admitted to the intensive care unit (ITU), we feel it would be preferrable to admit them to the ITU when the score is 2.
Figure 3.
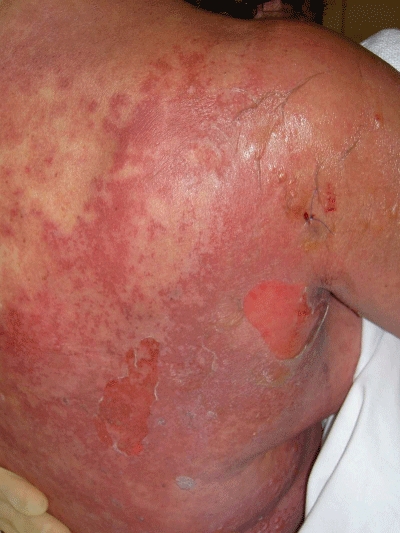
A clinical image of toxic epidermal necrolyisis (TEN) demonstrating widespread patchy eryhthema with confluence and darkening at sites of epidermal detachment (blistering)
Table 2.
SCORTEN assessment of the severity of toxic epidermal necrolysis
| Clinical parameter | Individual score | SCORTEN | Predicted mortality % |
|---|---|---|---|
| Age >40 years | Y = 1 | 0–1 | 3.2 |
| Malignancy | Y = 1 | 2 | 12.1 |
| Tachycardia >120 beats min−1 | Y = 1 | 3 | 35.3 |
| Initial area of detachment >10% | Y = 1 | 4 | 58.3 |
| Serum urea >10 mmol l−1 | Y = 1 | 5 or more | 90 |
| Serum glucose >14 mmol l−1 | Y = 1 | ||
| Bicarbonate <20 mmol l−1 | Y = 1 |
Modified from [34].
Other T cell-mediated reactions
Other T cell-mediated reactions include eczematous reactions either to skin contact with drugs (allergic contact dermatitis) or more generalized eczematous eruptions in response to ingestion of a range of culprit drugs. The fixed drug eruption is so-called because itchy or painful circular erythematous lesions develop at exactly the same sites within hours of each exposure to the culprit drug. In more severe cases, lesions can be multiple and can blister, a situation that requires careful distinction from TEN as it has a much better prognosis.
Acute generalized exanthematous pustulosis (AGEP) is a rare reaction in which extensive areas of erythema develop which are covered in sterile pustules usually 1–2 mm in diameter. Although AGEP is clearly largely a consequence of neutrophilic inflammation, it is thought to be T cell mediated, but exactly how the interaction between these two cell types arises is poorly understood. Other T cell mediated cutaneous manifestations of drug allergy including lichenoid eruptions and systemic drug-related intertriginous and flexural exanthema (SDRIFE), are less well understood and are beyond the scope of this article. The patterns of hypersensitivity mentioned above are consequences of the immune response in certain pre-disposed individuals to foreign antigens which can be derived from xenobiotics or microbes. In general the most severe reactions (DRESS and SJS/TEN) are triggered by drugs while the less severe reactions are often triggered by bugs (microbes).
The drug hypersensitivities mediated by type 2 and type 3 immune mechanisms are rather characteristic: pemphigus causes superficial fragile blisters and erosions which can occur in mucosal sites. It is accompanied by auto-antibodies that react with desmoglein proteins found in the hemisdesmosomes, structures mediating attachment of adjacent epidermal cells. There are two main patterns of drug-related pemphigus: drug-dependent pemphigus usually induced by thiol-containing drugs such as d-penicillamine or captopril and drug-triggered pemphigus triggered by non-thiol containing drugs including penicillins, cephalosporins and piroxicam. Drug-dependent pemphigus resolves once the causal drug is withdrawn, but drug-triggered pemphigus persists and exhibits the same enduring natural history as the spontaneous auto-immune form. Drug involvement in pemphigus is rare, probably being relevant in only 5% of cases of this already rare disease.
Type 3 mechanisms mediate the process of vasculitis which manifests in the skin as palpable lesions which may be erythematous at their start but which rapidly develop haemorrhage and purpura within them. Typically, vasculitic rashes exhibit a so-called ‘gravitational’ distribution in that they become more developed as one moves down the legs towards the ankles.
Management
The first principle of management in all conditions described here is withdrawal of the offending drug. Identification of the causative agent in individuals receiving multiple medications is often difficult. Although certain reaction patterns are more frequent with particular drugs (as discussed earlier) and publications highlight relative frequencies from notification registries, the most important element to this process is a detailed examination of the drug exposure timelines in relation to the event (Figure 1).
Figure 1.
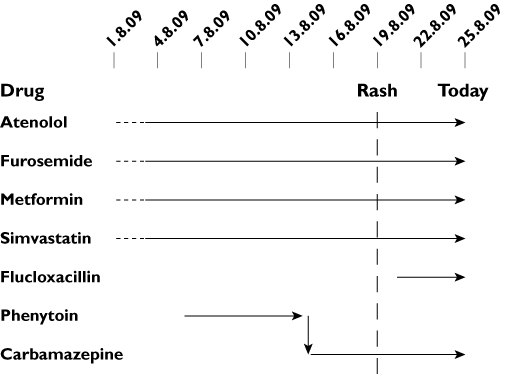
Careful documentation, with detailed dating of when drugs were started and stopped reveals the timelines which, in conjunction with knowledge of individual reported frequencies of allergy, helps to rank the drugs in order of suspicion. In a severe drug reaction such as toxic epidermal necrolysis, although this format is still very useful to identify a causative drug, it is preferable to recommend cessation of all medications. Time spent accurately drawing out this information at the first clinical encounter is considerably more effective than attempting to do this retrospectively when the condition has resolved. Horizontal arrows represent period of ingestion of listed drug. Arrow tip represents last day of ingestion. Vertical arrow represents a medication switch. Vertical dotted line represents the onset of the rash. It is important to include annotation of the day the chart is documented (Today)
General and supportive measures
In mild reactions frequently nothing more than stopping the suspected drug is required.
In severe reactions, it is first critically important to make the correct diagnosis of SJS/TEN conditions since aggressive active therapies are most likely to be successful if initiated early (see below). In addition, enhanced clearance of the drug may be considered including modalities such as haemofiltration and plasmapheresis, which may also remove pathogenic cytokines and other molecules such as soluble FasL. In a recent review of the published series (un-controlled) of plasmapheresis for the treatment of TEN the mortality was 11% suggesting a possible therapeutic benefit [35]. With extensive or inflammatory eruptions, careful consideration has to be given to fluid balance and temperature control. In severe reactions, non-specific features including pyrexia, raised white cell count and C-reactive protein are often present because of the hypersensitivity reaction and it can be difficult to resist the temptation to start antibiotics ‘in case’. Secondary bacterial infection may be more problematic if concurrent immunosuppressive treatments are employed and needs to be treated early despite the concern to avoid initiation of further medication. It is important to undertake regular swabbing of blistered sites, canulae and catheters to provide microbiological evidence of active infection and bacterial sensitivities to guide treatment. In severe eruptions, especially where blistering is involved, supportive treatment is largely analogous to that in patients with burns. Patients are likely to need management in a burns or intensive care unit [36] and survival is proportional to the time taken to reach such units [37]. These patients will need intensive emollient therapy, additional nutritional support, analgesia, warmed environments, aseptic handling and careful monitoring for systemic complications. Severe blistering disorders require involvement of ophthalmology and gynaecology teams where appropriate to prevent complications of mucosal damage. For moderate epidermal loss, liquid and white soft paraffin ointment should be applied under low-adherent or soft polymer dressings. For large areas of epidermal loss, biological dressings, epidermal allografts or xenografts have been used.
Skin directed therapy
Most inflammatory reactions are symptomatically improved by frequent emollient use. This therapy is not likely to modify the disease course but is of benefit to the patient in terms of both symptoms and limitation of complications such as scarring and infection. In cases where individual drugs have been stopped it is important and relatively straight forward to assess the progression of the reaction by examination of the skin. Itchy or uncomfortable rashes of any reaction pattern may be helped by moderate or potent topical corticosteroids. Caution must be employed in interpreting clinical improvement following use of these treatments. It is very unlikely that progression from moderate to severe reactions could be halted by topical corticosteroids but the clinical assessment of the outcome of drug cessation may be impaired.
Systemic treatments for drug allergic cutaneous reactions
The relative rarity of the most severe reaction patterns of drug hypersensitivity makes single-centre prospective randomized clinical trials very difficult. Most of the evidence regarding therapeutic efficacy of different modalities has either not been well controlled or has been retrospective against a comparable group of patients treated previously with other modalities, in the same centre. For many of the more severe reaction patterns, after withdrawal of the suspected drugs and the institution of supportive measures, the most used and generally helpful treatment is oral or IV glucocorticoids. The condition(s) which most require urgent and active treatment lie in the SJS/TEN spectrum. As indicated above, the mortality in SJS/TEN is predictable from the SCORTEN and can average 30–35%. The process of mass apoptotic death of epithelial cells cannot be stopped once it has started. By analogy, once a pin has ruptured the surface of an inflated balloon, it would be impossible to prevent the explosive fragmentation of the balloon. Therefore, prevention of the activation of the apoptosis is the only possible approach. Several classes of drug have been tried over the years, including corticosteroids, immunosuppressive anti-inflammatories (cyclophosphamide and ciclosporin), intravenous immunoglobulins (IVIG) and anti-TNF-α agents.
SJS/TEN
Corticosteroids
Originally the most widely used treatment was high dose corticosteroids. This treatment has been previously associated with increased mortality [36]. More recently in a large retrospective review of data from the European collaborative group (EuroSCAR), the same group has shown a non-statistically significant benefit from glucocorticoid treatment [38] and has suggested the subject needs re-assessment.
Cyclophosphamide
There are reports of a clear benefit from treatment with cyclophosphamide: Heng described a ‘dramatic response’ in four out of four treated patients while a fifth untreated patient died [39]. Trautmann treated eight patients with 300 mg day−1 for 3 days. There was rapid cessation of bulla formation and all eight survived [40]. This is a cheap and comparatively safe drug and it is clearly important to revisit it in a setting in which SCORTEN can be used for more accurate prognostication.
Ciclosporin
In a recent review of the literature ciclosporin has been suggested as the best evidence based intervention in TEN [35]. Mechanistically, ciclosporin inhibits CD8+ T cells and apoptotic pathways which are thought to be important in TEN pathogenesis. Arevalo et al. treated a series of 11 patients with ciclosporin (3 mg kg−1 day−1) for 2 weeks followed by a tapering period. Outcomes were compared retrospectively with six patients in a historical control group treated with cyclophosphamide (300 mg day−1) with or without corticosteroids [41]. The published clinical details demonstrate that this cohort had severe disease yet, despite 8/11 suffering from sepsis, the authors reported 100% survival.
Intravenous immunoglobulin (IVIg)
The use of intravenous immunoglobulin (IVIg) is surrounded by controversy despite strong evidence indicating therapeutic benefit. IVIg is a pooled collection of immunoglobulins from multiple donors. It is used in many auto-immune diseases to suppress synthesis of the pathogenetic auto-antibodies. The use of IVIg in TEN was promoted following the report by French's group in 1998 in Science which supported a dominant role of Fas-FasL signalling on keratinocytes in TEN. They showed that due to intrinsic anti-Fas activity of IVIg, this apoptotic signalling cascade could be blocked in vitro, and 10 cases of TEN responded to treatment in vivo[42]. Since then there have been numerous case reports and series supporting the usefulness of this intervention in TEN resulting in a lower than predicted mortality. French reviewed nine studies in which 10 or more patients were treated and seven of the nine showed highly significant reductions in mortality [43]. The largest study included in that review was a prospective study of 48 cases with SJS treated at a mean dose of 0.6 g kg−1 day−1 for 4 days with a survival of 88% [44]. However, the review suggested that in general 1 g kg−1 day−1 for 3 days is recommended. More recently a large collaborative observational series of 281 cases of SJS or TEN treated in France and Germany failed to show a benefit of IVIg at on average 1.9 g kg−1 given over 2−3 days [38]. Similarly, we have experienced both successes and failures with this treatment approach. The explanation for these conflicting results has been previously aligned to dosing regimen (insufficient quantities of IVIg or excessive delay in starting therapy) and batch to batch variation, the unknown levels of anti-FasL antibody being regarded as the possible major determinant of efficacy. The predominant weight of evidence indicates that IVIg is of benefit but there are a sufficient number of negative reports that ultimately a prospective double-blind randomized controlled trial across multiple centres will be required to help settle this controversy. It would also seem possible that differences in response may be associated with differences in immunopathogenesis between patients and future studies might aim to address the possibility of identifying IVIg responders.
Tumour necrosis factor-α (TNF-α) inhibitors and thalidomide
As well as cytotoxic lymphocytes, it has been recognized for some time that TNF-α is important in TEN pathogenesis as it is present in high levels in blister fluid and in lesional epidermis at much higher levels than seen in blistering burns [45]. On the basis of this information, the potential for blocking TNF-α in TEN has attracted significant interest. Thalidomide has been reported to inhibit TNF-αin vitro and in vivo[46]. In fact the only randomized controlled trial of therapy in TEN was a trial of thalidomide [47]. The trial had to be stopped early because of an excess mortality in the treatment group. However, it should be noted that the authors concluded that their data suggested that thalidomide did not inhibit TNF-α in that study, and that the excess mortality may have been related to other effects of thalidomide. Three case reports have shown a good effect from using a single dose of infliximab at 5 mg kg−1 (anti-TNF-α monoclonal antibody) [48–50] and in one case showed evidence that epidermal and dermal TNF-α sources were dramatically switched off by this drug [49]. In one further case, etanercept, a soluble TNF-α receptor, was reported to have good therapeutic efficacy [51].
Other treatment approaches including GM-CSF, insulin, zinc and N-acetylcysteine remain largely untested in significant numbers of patients [35, 47].
DRESS and AGEP
There are no controlled trials of treatment in DRESS or AGEP. Identification and withdrawal of the causative drug in AGEP is usually sufficient, the rash clearing with minimal desquamation. Although brief courses of oral steroids are occasionally given for severe reactions recent reports have suggested the possibility of using ciclosporin (5 mg kg−1 day−1) [52] or etanercept [53].
Published case reports and small series have pointed to oral prednisolone to treat DRESS as probably the most accepted intervention in the treatment of severe drug reactions: 1–1.5 mg kg−1 has been recommended but we have seen efficacious responses to 0.5 mg kg−1 (unpublished) with rapid resolution of rash and fever. The association with human-herpes virus-6 (HHV-6) re-activation characteristically 2 weeks after rash onset, was recognized in the late 1990s [32, 54, 55] and has been proposed as an explanation for the characteristic flaring seen in this condition [56] although precise explanation of the role of the virus in the disease remains unclear. Careful monitoring for potential viral reactivation is therefore essential, but it should be noted that sporadic reports have identified reactivation of other herpes viruses as well (EBV, CMV, HHV-6, HHV-7, HSV, VZV) [31]. Screening should be undertaken by PCR to detect viral DNA in the blood. Optimal management of viral reactivation has not been fully addressed in clinical trials, but we have had some success with the use of ganciclovir in such cases (unpublished).
Investigation
On the occasions when it may not be clear which drug is responsible for a reaction or when it is clinically desirable to re-introduce a medication required by the patient's clinical condition, as in the case of antibiotics for cystic fibrosis or anti-convulsants for epilepsy, there is the requirement for some sort of testing procedure. There is a range of possible skin tests and in vitro laboratory tests that have been tried but these are not widely available and tend to be limited to centres in which they are part of research activity. It is crucial to think of the immunopathogenic mechanisms when attempting to perform diagnostic tests. Thus type 1 reactions may be demonstrable in vivo with skin prick tests, intradermal tests (read at 15 min and 6–12 h) or in vitro by RAST tests. For a small range of drugs, commercial preparations are available, most particularly for the penicillin family. For type 4 reactions involving the skin, patch tests with the drug homogenized at 1–10% in white soft paraffin can be performed. Positive reactions at 48 h can be obtained in certain patterns of drug-induced rash including the EM/SJS/TEN spectrum and in some patterns of drug-induced exanthemata, but there is not a great confidence regarding their reliability [57]. Overall, positive responses are obtained in 30–50% of cutaneous adverse drug reactions [58]. They are safe and a positive reaction is usually clinically significant, but a negative reaction cannot be interpreted. In vitro assays to detect T cell reactivity to suspected drugs by measurement of drug-induced lymphocyte proliferation have been reported to give positive responses [59, 60]. Again it is not a completely dependable method and for uncertain reasons, it works well for some reaction patterns and drugs, but does not work for others. More recently other approaches include the detection of drug-induced cytokine release by ELISA and/or ELISPOT assays. It is critically important that those undertaking testing for drug allergy are fully aware of the limitations and pitfalls of these tests, a comprehensive discussion of which is covered in another review in this series [61, 62].
Conclusions
The skin is frequently involved in drug hypersensitivity reactions. Careful clinical observation is required to diagnose which of the many patterns of reaction is present. A clear understanding of the immunopathological mechanisms is important both for interpreting the clinical signs and anticipating the potentially effective forms of therapy. We have highlighted that pink raised weals can be caused by mast cell-derived mediators (type 1 hypersensitivity and drug intolerance) while weal-like lesions of longer duration are induced by T lymphocyte-dependent mechanisms, and consequently respond to different interventions. While there is not a universally accepted classification of the clinical patterns of drug hypersensitivity, distinction between the many patterns of drug-induced exanthemata is potentially of critical importance because the severe reactions of SJS/TEN and DRESS require urgent active therapy. Delay even of a few hours can make the difference between survival and death. There is not a standard ranked order of therapies based on therapeutic efficacy. While the new biological therapies (IVIg and anti-TNF agents) are the subjects of most current therapeutic studies, there is tantalizing evidence that much cheaper agents (cyclophosphamide and ciclosporin) may be effective. It is time for co-ordinated multi-centre studies to perform a proper evaluation of these agents. Randomized trials do not require a placebo arm and there are enough agents to be compared that a ranked order of efficacy could be established.
Conflicts of interest
There are no competing interests to declare.
REFERENCES
- 1.Bigby M, Jick S, Jick H, Arndt K. Drug-induced cutaneous reactions. A report from the Boston Collaborative Drug Surveillance Program on 15,438 consecutive inpatients, 1975 to 1982. JAMA. 1986;256:3358–63. doi: 10.1001/jama.256.24.3358. [DOI] [PubMed] [Google Scholar]
- 2.Hunziker T, Kunzi UP, Braunschweig S, Zehnder D, Hoigne R. Comprehensive hospital drug monitoring (CHDM): adverse skin reactions, a 20-year survey. Allergy. 1997;52:388–93. doi: 10.1111/j.1398-9995.1997.tb01017.x. [DOI] [PubMed] [Google Scholar]
- 3.Fiszenson-Albala F, Auzerie V, Mahe E, Farinotti R, Durand-Stocco C, Crickx B, Descamps V. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol. 2003;149:1018–22. doi: 10.1111/j.1365-2133.2003.05584.x. [DOI] [PubMed] [Google Scholar]
- 4.Park BK, Pirmohamed M, Kitteringham NR. Idiosyncratic drug reactions: a mechanistic evaluation of risk factors. Br J Clin Pharmacol. 1992;34:377–95. doi: 10.1111/j.1365-2125.1992.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merk HF, Hertl M. Immunologic mechanisms of cutaneous drug reactions. Semin Cutan Med Surg. 1996;15:228–35. doi: 10.1016/s1085-5629(96)80035-6. [DOI] [PubMed] [Google Scholar]
- 6.Kenna JG, Satoh H, Christ DD, Pohl LR. Metabolic basis for a drug hypersensitivity: antibodies in sera from patients with halothane hepatitis recognize liver neoantigens that contain the trifluoroacetyl group derived from halothane. J Pharmacol Exp Ther. 1988;245:1103–9. [PubMed] [Google Scholar]
- 7.Martin JL, Kenna JG, Pohl LR. Antibody assays for the detection of patients sensitized to halothane. Anesth Analg. 1990;70:154–9. doi: 10.1213/00000539-199002000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Leeder JS, Gaedigk A, Lu X, Cook VA. Epitope mapping studies with human anti-cytochrome P450 3A antibodies. Mol Pharmacol. 1996;49:234–43. [PubMed] [Google Scholar]
- 9.Mauri-Hellweg D, Bettens F, Mauri D, Brander C, Hunziker T, Pichler WJ. Activation of drug-specific CD4+ and CD8+ T cells in individuals allergic to sulfonamides, phenytoin, and carbamazepine. J Immunol. 1995;155:462–72. [PubMed] [Google Scholar]
- 10.Naisbitt DJ, Britschgi M, Wong G, Farrell J, Depta JP, Chadwick DW, Pichler WJ, Pirmohamed M, Park BK. Hypersensitivity reactions to carbamazepine: characterization of the specificity, phenotype, and cytokine profile of drug-specific T cell clones. Mol Pharmacol. 2003;63:732–41. doi: 10.1124/mol.63.3.732. [DOI] [PubMed] [Google Scholar]
- 11.Coulter EM, Jenkinson C, Wu Y, Farrell J, Foster B, Smith A, McGuire C, Pease C, Basketter D, King C, Friedmann PS, Pirmohamed M, Park BK, Naisbitt DJ. Activation of T-cells from allergic patients and volunteers by p-phenylenediamine and Bandrowski's base. J Invest Dermatol. 2008;128:897–905. doi: 10.1038/sj.jid.5701071. [DOI] [PubMed] [Google Scholar]
- 12.Wu Y, Sanderson JP, Farrell J, Drummond NS, Hanson A, Bowkett E, Berry N, Stachulski AV, Clarke SE, Pichler WJ, Pirmohamed M, Park BK, Naisbitt DJ. Activation of T cells by carbamazepine and carbamazepine metabolites. J Allergy Clin Immunol. 2006;118:233–41. doi: 10.1016/j.jaci.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Naisbitt DJ, Farrell J, Wong G, Depta JP, Dodd CC, Hopkins JE, Gibney CA, Chadwick DW, Pichler WJ, Pirmohamed M, Park BK. Characterization of drug-specific T cells in lamotrigine hypersensitivity. J Allergy Clin Immunol. 2003;111:1393–403. doi: 10.1067/mai.2003.1507. [DOI] [PubMed] [Google Scholar]
- 14.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 15.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–61. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 16.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 17.Devary Y, Gottlieb RA, Lau LF, Karin M. Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol Cell Biol. 1991;11:2804–11. doi: 10.1128/mcb.11.5.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreck R, Baeuerle PA. A role for oxygen radicals as second messengers. Trends Cell Biol. 1991;1:39–42. doi: 10.1016/0962-8924(91)90072-h. [DOI] [PubMed] [Google Scholar]
- 19.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–58. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–9. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, James I, Christiansen FT. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727–32. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 23.Hung SI, Chung WH, Liou LB, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS, Chen MJ, Lai PC, Wu MS, Chu CY, Wang KH, Chen CH, Fann CS, Wu JY, Chen YT. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005;102:4134–9. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, Chen YT. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 25.Martin AM, Nolan D, Gaudieri S, Almeida CA, Nolan R, James I, Carvalho F, Phillips E, Christiansen FT, Purcell AW, McCluskey J, Mallal S. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci USA. 2004;101:4180–5. doi: 10.1073/pnas.0307067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coombs RRA, Gell PGH. Classification of allergic reactions responsible for clinical hypersensitivity and disease. In: Gell PGH, Coombs RRA, editors. Clinical Aspects of Immunology. Oxford: Oxford University Press; 1968. pp. 576–96. [Google Scholar]
- 27.Pichler WJ. Drug hypersensitivity reactions: classification and relationship to T-cell activation. In: Pichler WJ, editor. Drug Hypersensitivity. 1st edn. Basel: Karger; 2007. pp. 168–89. [Google Scholar]
- 28.Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357:580–7. doi: 10.1056/NEJMra066469. [DOI] [PubMed] [Google Scholar]
- 29.Friedmann PS, Pickard C, Ardern-Jones MR, Bircher AJ. Drug-induced exanthemata: a source of clinical and intellectual confusion. European Journal Dermatology. 2010;20:255–9. doi: 10.1684/ejd.2010.0891. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez-Salazar A, Rosales SP, Rangel-Frausto S, Criollo E, Archer-Dubon C, Orozco-Topete R. Epidemiology of adverse cutaneous drug reactions. A prospective study in hospitalized patients. Arch Med Res. 2006;37:899–902. doi: 10.1016/j.arcmed.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Seishima M, Yamanaka S, Fujisawa T, Tohyama M, Hashimoto K. Reactivation of human herpesvirus (HHV) family members other than HHV-6 in drug-induced hypersensitivity syndrome. Br J Dermatol. 2006;155:344–9. doi: 10.1111/j.1365-2133.2006.07332.x. [DOI] [PubMed] [Google Scholar]
- 32.Shiohara T, Inaoka M, Kano Y. Drug-induced hypersensitivity syndrome (DIHS): a reaction induced by a complex interplay among herpesviruses and antiviral and antidrug immune responses. Allergol Int. 2006;55:1–8. doi: 10.2332/allergolint.55.1. [DOI] [PubMed] [Google Scholar]
- 33.Auquier-Dunant A, Mockenhaupt M, Naldi L, Correia O, Schroder W, Roujeau JC. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol. 2002;138:1019–24. doi: 10.1001/archderm.138.8.1019. [DOI] [PubMed] [Google Scholar]
- 34.Bastuji-Garin S, Fouchard N, Bertocchi M, Roujeau JC, Revuz J, Wolkenstein P. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149–53. doi: 10.1046/j.1523-1747.2000.00061.x. [DOI] [PubMed] [Google Scholar]
- 35.Chave TA, Mortimer NJ, Sladden MJ, Hall AP, Hutchinson PE. Toxic epidermal necrolysis: current evidence, practical management and future directions. Br J Dermatol. 2005;153:241–53. doi: 10.1111/j.1365-2133.2005.06721.x. [DOI] [PubMed] [Google Scholar]
- 36.Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med. 1994;331:1272–85. doi: 10.1056/NEJM199411103311906. [DOI] [PubMed] [Google Scholar]
- 37.McGee T, Munster A. Toxic epidermal necrolysis syndrome: mortality rate reduced with early referral to regional burn center. Plast Reconstr Surg. 1998;102:1018–22. doi: 10.1097/00006534-199809040-00014. [DOI] [PubMed] [Google Scholar]
- 38.Schneck J, Fagot JP, Sekula P, Sassolas B, Roujeau JC, Mockenhaupt M. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR study. J Am Acad Dermatol. 2008;58:33–40. doi: 10.1016/j.jaad.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 39.Heng MC, Allen SG. Efficacy of cyclophosphamide in toxic epidermal necrolysis. Clinical and pathophysiologic aspects. J Am Acad Dermatol. 1991;25:778–86. doi: 10.1016/s0190-9622(08)80969-3. [DOI] [PubMed] [Google Scholar]
- 40.Trautmann A, Klein CE, Kampgen E, Brocker EB. Severe bullous drug reactions treated successfully with cyclophosphamide. Br J Dermatol. 1998;139:1127–8. doi: 10.1046/j.1365-2133.1998.2576n.x. [DOI] [PubMed] [Google Scholar]
- 41.Arevalo JM, Lorente JA, Gonzalez-Herrada C, Jimenez-Reyes J. Treatment of toxic epidermal necrolysis with cyclosporin A. J Trauma. 2000;48:473–8. doi: 10.1097/00005373-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 42.Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, Hunziker T, Saurat JH, Tschopp J, French LE. Inhibition of toxic epidermal necrolysis by blockade of cd95 with human intravenous immunoglobulin. Science. 1998;282:490–3. doi: 10.1126/science.282.5388.490. [DOI] [PubMed] [Google Scholar]
- 43.French LE. Toxic epidermal necrolysis and Stevens Johnson syndrome: our current understanding. Allergol Int. 2006;55:9–16. doi: 10.2332/allergolint.55.9. [DOI] [PubMed] [Google Scholar]
- 44.Prins C, Vittorio C, Padilla RS, Hunziker T, Itin P, Förster J, Bröcker EB, Saurat JH, French LE. Effect of high-dose intravenous immunoglobulin therapy in Stevens-Johnson syndrome: a retrospective, multicenter study. Dermatology. 2003;207:96–9. doi: 10.1159/000070957. [DOI] [PubMed] [Google Scholar]
- 45.Paquet P, Pierard GE. Soluble fractions of tumor necrosis factor-alpha, interleukin-6 and of their receptors in toxic epidermal necrolysis: a comparison with second-degree burns. Int J Mol Med. 1998;1:459–62. doi: 10.3892/ijmm.1.2.459. [DOI] [PubMed] [Google Scholar]
- 46.Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993;177:1675–80. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolkenstein P, Latarjet J, Roujeau JC, Duguet C, Boudeau S, Vaillant L, Maignan M, Schuhmacher MH, Milpied B, Pilorget A, Bocquet H, Brun-Buisson C, Revuz J. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet. 1998;352:1586–9. doi: 10.1016/S0140-6736(98)02197-7. [DOI] [PubMed] [Google Scholar]
- 48.Fischer M, Fiedler E, Marsch WC, Wohlrab J. Antitumour necrosis factor-alpha antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707–9. doi: 10.1046/j.1365-2133.2002.46833.x. [DOI] [PubMed] [Google Scholar]
- 49.Hunger RE, Hunziker T, Buettiker U, Braathen LR, Yawalkar N. Rapid resolution of toxic epidermal necrolysis with anti-TNF-alpha treatment. J Allergy Clin Immunol. 2005;116:923–4. doi: 10.1016/j.jaci.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 50.Wojtkiewicz A, Wysocki M, Fortuna J, Chrupek M, Matczuk M, Koltan A. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420–1. doi: 10.2340/00015555-0462. [DOI] [PubMed] [Google Scholar]
- 51.Gubinelli E, Canzona F, Tonanzi T, Raskovic D, Didona B. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150–3. doi: 10.1111/j.1346-8138.2009.00616.x. [DOI] [PubMed] [Google Scholar]
- 52.Di Lernia V, Grenzi L, Guareschi E, Ricci C. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin. Clin Exp Dermatol. 2009;34:757–9. doi: 10.1111/j.1365-2230.2009.03480.x. [DOI] [PubMed] [Google Scholar]
- 53.Gencoglan G, Tosun M, Aktepe F. The molecular mechanism of etanercept, an anti-tumour necrosis factor-alpha receptor-fusion protein, in the treatment of acute generalized exanthematous pustulosis. J Dermatolog Treat. 2009;20:241–5. doi: 10.1080/09546630802683843. [DOI] [PubMed] [Google Scholar]
- 54.Descamps V, Bouscarat F, Laglenne S, Aslangul E, Veber B, Descamps D, Saraux JL, Grange MJ, Grossin M, Navratil E, Crickx B, Belaich S. Human herpesvirus 6 infection associated with anticonvulsant hypersensitivity syndrome and reactive haemophagocytic syndrome. Br J Dermatol. 1997;137:605–8. doi: 10.1111/j.1365-2133.1997.tb03795.x. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki Y, Inagi R, Aono T, Yamanishi K, Shiohara T. Human herpesvirus 6 infection as a risk factor for the development of severe drug-induced hypersensitivity syndrome. Arch Dermatol. 1998;134:1108–12. doi: 10.1001/archderm.134.9.1108. [DOI] [PubMed] [Google Scholar]
- 56.Tohyama M, Yahata Y, Yasukawa M, Inagi R, Urano Y, Yamanishi K, Hashimoto K. Severe hypersensitivity syndrome due to sulfasalazine associated with reactivation of human herpesvirus 6. Arch Dermatol. 1998;134:1113–7. doi: 10.1001/archderm.134.9.1113. [DOI] [PubMed] [Google Scholar]
- 57.Friedmann PS, Ardern-Jones M. Patch testing in drug allergy. Curr Opin Allergy Clin Immunol. 2010 doi: 10.1097/ACI.0b013e32833aa54d. May 18 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Barbaud A. Drug patch testing in systemic cutaneous drug allergy. Toxicology. 2005;209:209–16. doi: 10.1016/j.tox.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 59.Nyfeler B, Pichler WJ. The lymphocyte transformation test for the diagnosis of drug allergy: sensitivity and specificity. Clin Exp Allergy. 1997;27:175–81. [PubMed] [Google Scholar]
- 60.Kano Y, Hirahara K, Mitsuyama Y, Takahashi R, Shiohara T. Utility of the lymphocyte transformation test in the diagnosis of drug sensitivity: dependence on its timing and the type of drug eruption. Allergy. 2007;62:1439–44. doi: 10.1111/j.1398-9995.2007.01553.x. [DOI] [PubMed] [Google Scholar]
- 61.Frew A. General principles of investigating and managing drug allergy. Br J Clin Pharmacol. 2011;71:642–6. doi: 10.1111/j.1365-2125.2011.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedmann PS, Ardern-Jones M. Patch testing in drug allergy. Curr Opin Allergy Clin Immunol. 2010;10:291–6. doi: 10.1097/ACI.0b013e32833aa54d. [DOI] [PubMed] [Google Scholar]