

Abstract
Drug-induced hypersensitivity reactions can cause a variety of serious diseases by involving drug-specific T-cells. Many of these reactions have been explained by the hapten concept, which postulates that small chemical compounds need to bind covalently to proteins to be recognized by the immune system. Due to their chemical reactivity, haptens stimulate the innate immunity by binding covalently to endogenous proteins and form so called hapten-carrier complexes, which are antigenic and induce T-cell responses. In recent years, a new concept has been developed since drug-induced hypersensitivity reactions were also observed with chemically unreactive drugs. This concept implies direct and reversible interactions of the drug between T-cell receptors (TCR) and major histocompatability complex (MHC) molecules. Therefore it was termed pharmacological interactions with immune receptors (p-i concept). Early observations on drug reacting T-cell clones (TCC) let believe that drugs bind first to the T-cell receptor since HLA molecules could be exchanged without affecting the drug reactivity. However, MHC molecules were always required for full activation of TCC. According to its strong HLA-B*5701 association, recent data on abacavir suggest that a drug could first bind to the peptide binding groove of the MHC molecule. The thereby modified HLA molecule can then be recognized by specific T-cells. Consequently, two types of reactions based on the p-i mechanism may occur: on the one hand, drugs might preferentially bind directly to the TCR, whereas in defined cases with strong HLA association, drugs might bind directly to the MHC molecule.
Keywords: drug hypersensitivity, hapten, MHC complex, p-i, T-cell receptor, T-cells
Introduction
Adverse drug reactions (ADRs) present a serious human health problem and are major contributors to hospitalization and mortality throughout the world [1, 2]. ADRs can be classified into two broad categories, type A reactions and type B reactions. Approximately 80% of ADRs belong to type A reactions and are considered to be predictable, common and related to the pharmacologic activities of the drug. Typical examples are drug-induced toxicity, like gastrointestinal bleeding after non-steroidal anti-inflammatory drug (NSAID) treatment or sleepiness after treatment with some antihistamines of the first/second generation.
Type B reactions are uncommon, considered to be unpredictable and normally not related to the pharmacologic activities of the drug [3]. Another terminology would be idiosyncratic drug reactions, meaning that an unusual individual susceptibility is causal for the side effect: it includes drug intolerance reactions (e.g. asthma or urticaria exacerbations after use of NSAIDs) and immune-mediated adverse drug reactions, also known as allergic drug reactions or drug hypersensitivity reactions. The latter ones are dependent on one or more immunological mechanisms [4, 5].
Hypersensitivity reactions involve many distinct immune mechanisms and thus present different clinical manifestations. Allergic reactions are often classified according to the classification system of Gell & Coombs [6]. Type I reactions or so-called immediate-type reactions, are mediated by drug-specific IgE antibodies and mainly cause urticaria, anaphylaxis and bronchospasm. The symptoms normally occur within less than 1 h after drug administration. The less common type II reactions are based on immunoglobulin-mediated cytotoxic mechanisms, accounting mainly for blood cell dyscrasia. Type III reactions, on the other hand, are immune-complex mediated, occurring, for example, in vasculitis. Finally, type IV reactions are known as delayed hypersensitivity reactions (DHR), which are mediated by drug-specific T lymphocytes [7]. For all types of DHR an involvement of T-cells is required since T-cells, on the one hand, infiltrate the tissue and cause organ damage [8, 9] and, on the other hand, produce cytokines that mediate antibody class switching. Involvement of drug specific T-cells could be demonstrated by the isolation of drug reactive T-cell clones (TCC) from biopsies of hypersensitivity lesions [10] as well as from the peripheral blood of hypersensitive patients [11].
Type IVa-d reactions
Although the classification system according to Gell & Coombs provides a helpful diagnostic tool in clinical practice, DHR to drugs have turned out to be more complex than initially described. In particular, delayed appearing hypersensitivity reactions involve many more distinct immune mechanisms than originally conceived. Nowadays these immune mechanisms can be related to the distinct functions of drug reactive T-cells [7]. This finding seems to be plausible since T-cells can orchestrate different forms of inflammations. Consequently, type IV reactions can also be categorized according to the cytokine patterns and the preferential activation of different immunocytes [7, 12, 13]. Taking into consideration the well described heterogeneity of T-cell functions and phenotypes, T-cell-meditated type IV reactions were subclassified in IVa–IVd reactions:
Type IVa reactions correspond to Th1 immune reactions: Th1 type T-cells activate macrophages by IFNγ secretion, drive the production of complement fixing antibody isotypes, and act as co-stimulators for pro-inflammatory and CD8+ T-cell responses. An in vivo correlate would be on the one hand a monocyte activation, e.g. in skin tests to tuberculin. On the other hand, Th1 cells are known to activate CD8+ cells, which might explain the common combination of IVa (high IFN-γ values) and IVc reactions (e.g. in contact dermatitis).
Type IVb corresponds to the Th2 type immune response. Th2 T-cells secrete IL-4, Il-13 and IL-5 cytokines, which promote B cell production of IgE and IgG4, macrophage deactivation and mast cell and eosinophil responses. The high production of IL-5 leads to an eosinophilic inflammation, which is the characteristic inflammatory cell type in many drug hypersensitivity reactions [7, 14]. An in vivo correlate might be an eosinophil-rich maculopapular exanthema, but also the presence of nematode infestations or bronchial or nasal inflammation (asthma and rhinitis, respectively).
Type IVc reactions take into account the fact that T-cells are able to act as effector cells. They emigrate into the tissue and kill tissue cells, such as hepatocytes or keratinocytes in a perforin/granzyme B and/or FasL-dependent manner [15][16]. Such reactions occur in most drug-induced DHR, mostly together with other type IV reactions involving monocyte, eosinophil or polymorphonuclear cell recruitment and activation. Cytotoxic T-cells thus play a role in maculopapular or bullous skin diseases, as well as in neutrophilic inflammations and in contact dermatitis. Type IVc reactions appear to be predominant in bullous skin reactions like Stevens Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), where activated CD8+ T-cells kill keratinocytes [7, 16, 17].
Type IVd: T-cells driven by an antigen can coordinate (sterile) neutrophilic inflammation as well. Typical examples would be sterile neutrophilic inflammation of the skin, in particular acute-generalized exanthematous pustulosis. In this disease CXCL8 and GM-CSF producing T-cells recruit neutrophilic leucocytes and prevent their apoptosis via GM-CSF release [18].
Drug recognition by T-cells
Conventional T-cells recognize peptides presented on MHC molecules from antigen presenting cells (APC). Nevertheless, how small molecular synthetic compounds like drugs are recognized by T-cells and how they are able to elicit a generalized immune response remains an intriguing question.
In order to mount an immune response, the antigen/drug must, on the one hand, stimulate the innate immune system and, on the other hand, form antigenic determinants for specific immune receptors (B-cell and T-cell receptors (TCR) for antigen). The ability to activate the innate immune system and thus to initiate an immune response is called immunogenicity and supplements antigenicity, which is the provision of antigenic determinants for the specific immune receptors. A full antigen requires both, antigenicity and immunogenicity. While, as mentioned, the nature of protein antigen presentation to T-cells has been fully elucidated, the mechanism by which a chemical or drug antigen is presented is still subject to debate. Three pathways have been described for chemical stimulation on T-cells:
The hapten concept
A dogma in immunology stipulates that small molecules (<1000D) are not antigenic per se and therefore drugs would not be immunogenic. However, T-cell activation by small molecules can be explained by the hapten concept. It implies that a small molecule can become antigenic, when it binds to a high molecular weight protein. Haptens are chemically reactive small molecules that are able to undergo a stable, covalent binding to a larger protein or peptide, which modifies the side chain of the bound residue. Theoretically this modification can affect any kind of autologous protein, like soluble extracellular proteins (e.g. albumin), membrane proteins (e.g. an integrin) and intracellular proteins (e.g. enzymes) or possibly directly the peptide embedded in the MHC molecule itself. Chemical haptens often have a tendency to bind covalently to a particular amino acid. For instance it has been shown that penicillin derivatives were prone to bind covalently to lysine residues of serum albumin [19]. Consequently, many different new antigenic determinants are formed by hapten modification of different proteins resulting in a broad array of immune responses to a hapten. The type, location and abundance of the molecules modified have a big influence on the type of the arising immune response. This can lead to a great heterogeneity of immune responses and clinical pictures, based on formation of antibodies (IgG, IgE) directed to the soluble or cell bound proteins. Occasionally, an exclusive T-cell response will be formed, without antibody formation. One reason might be a modification of an MHC associated peptide itself. Alteration of the MHC-molecule directly is also possible, but data in the mouse model using trinitrophenol suggest that this is less frequently causing an immune response [20].
In order to get recognized by T-cells as foreign antigens, haptenized proteins need to be processed by APC to produce haptenized peptides presented on MHC molecules (Figure 1A). A response directed against such a neo-antigen represents a de novo immune response. Therefore it is thought that hapten-specific T-cells originate from the immunological naïve pool of lymphocytes. Haptenization itself is thought to be pro-inflammatory, since it must disturb functions of modified proteins. For quite a number of molecules it was shown that haptenization leads to the activation of dendritic cells (DC) in vitro[21, 22].
Figure 1.
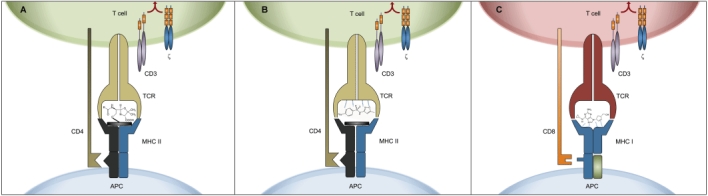
Hapten and p-i mechanism of drug recognition by T-cells. (A) Hapten model: the MHC embedded peptide is covalently modified by the drug, creating a new antigenic determinant. (B) p-i concept for CD4+ T-cell stimulations: the drug (shown as SMX in this example) may bind to the TCR. Activation of the T-cell requires the interaction with MHC-class II molecules. No strict HLA-restriction can be demonstrated [39]. (C) p-i concept for HLA-allele restricted CD8+ T-cell responses: the drug (shown as abacavir in this example) binds avidly, but non-covalently to allele specific HLA-B determinants [45]. This binding may stabilize the HLA-complex even without peptide. A CD8+ T-cell reaction against this drug-HLA complex will be developed. It can only occur if the HLA molecule binds the drug with sufficient affinity, explaining the exquisite HLA-allele association of this reaction
Allergic contact dermatitis represents a specific example of a DHR whose mechanism follows the concept of haptenization. The skin, as the outermost barrier of the body's defence system, is the first organ that encounters chemical and physical factors from the environment [23]. In some instances chemical entities can act specifically as skin allergens resulting in a T-cell mediated inflammatory reaction at the site of challenge in certain hypersensitive individuals, causing skin erythema and oedema [24]. Contact allergens are incomplete antigens that must bind to cellular and serum proteins to form stable hapten-protein complexes [25]. The ability of these hapten-protein complexes to induce sensitization relies on the activation of innate immunity. Interaction of DC with the antigen promotes maturation and migration to the draining lymph nodes [26] where the matured cells effectively present the processed antigens as anchored peptides on MHC class I and II molecules to specific naïve CD8+ and CD4+ T-cells. This results in the generation and proliferation of circulating memory T-cell populations with acquired memory for that antigen. Re-exposure of sensitized individuals to the same antigen will lead to a proliferation of the circulating memory T-cells resulting in the incidence of allergic contact dermatitis within 24–72 h of the exposure.
The pro-hapten concept
Many drugs are not chemically reactive but still able to elicit immune-mediated side effects. The pro-hapten hypothesis tries to reconcile this phenomenon with the hapten hypothesis by stating that a chemically inert drug may become reactive upon metabolism [7, 27, 28]. Sulfamethoxazole (SMX) is a prototype of such a pro-hapten. It is itself not chemically reactive but gains reactivity and thus antigenicity by intracellular metabolism. A cytochrome P450 dependent metabolism (CYP2C9) in the liver leads to sulfamethoxazole-hydroxylamine (SMX-NHOH) which can be found in the urine and which is easily converted to sulfamethoxazole-nitroso by oxidation. The latter is chemically highly reactive and easily binds to intracellular proteins creating neoantigenic determinants [29]. Moreover, toxic effects of SMX appear above a threshold level. Thus, SMX seems to have indirectly antigenic and immunogenic features. Since many different proteins might be modified, the resulting clinical picture might be as variable as with real haptens and SMX is indeed known to cause many different types of diseases affecting many organs (exanthems, anaphylaxis, SJS, hepatitis, etc.). These side effects are mediated by antibodies and/or T-cells. On the other hand, the conversion of a pro-hapten to the reactive hapten may occur exclusively in the liver or kidney and may thus cause an isolated hepatitis or interstitial nephritis [8].
An unexplained issue of the hapten theory is the fact that hapten-formation is common for given drugs like penicillin and happens in the majority of treated patients. Why only a minority of patients develops an allergic, clinically symptomatic immune reaction is unclear.
The p-i concept
A third possibility, namely the p-i concept for ‘pharmacological interactions of drugs with immune receptors’ represents a non-conventional presentation pathway that contradicts the original thought that the immune stimulatory capacity of most chemicals and drugs could be predicted by their protein reactivity [11, 30, 31]. According to the hapten and pro-hapten concept, drugs and other substances that are not chemically active and therefore incapable of coupling to a macromolecular carrier, would not be antigens and could not induce hypersensitivity reactions. However, this hypothesis has been challenged by clinical and immunological evidence that cannot be explained by hapten or pro-hapten models (Table 1, and is reviewed in [32]).
Table 1.
Evidence for the p-i concept
| In vivo: |
| A specific immune response at the first encounter, notably without sensitization phase [51] |
| DHR caused by drugs that are not known to be metabolized to a reactive compound [11, 30, 34, 35, 47, 52, 53] |
| Positive skin tests with lymphocyte infiltration caused by inert drugs unable to form hapten-carrier complexes in the skin |
| In vitro: |
| Chemically inert drugs stimulate TCC via the TCR in an MHC-dependent way [11, 30, 34, 35, 47, 52–55] |
| Glutaraldehyde fixed APCs can still present drugs [11, 30, 35, 47, 53, 56] |
| Washing of glutaraldehyde fixed APCs removes drug and prevents specific TCC activation [30, 56] |
| Ca2+ mobilization kinetics are too fast to allow antigen processing [11] |
| TCR downregulation kinetics too fast to allow antigen processing [11] |
According to the p-i concept, chemically inert drugs, unable of binding covalently to peptides or proteins, can nevertheless activate certain T-cells, if they fit with a sufficient affinity into some of the various different T-cell receptors or MHC-molecules available. This reversible interaction is similar to the one of a ligand to its receptor. In some isolated drug reactive TCC, chemical derivatives of the parental drug could even act as full antagonist or partial agonist in vitro[33]. Evidence for the p-i mechanism lies in the observations that aldehyde-fixed APC (unable to process antigen) are still able to activate specific TCC, washing steps of APC abrogates the reactivity (in the opposite of haptens) and that calcium influx in TCC happens within seconds after the addition of the drug. In contrast to haptens, p-i acting drugs, without requiring any biotransformation, are thought not to be pro-inflammatory and the innate immune system may not be stimulated [32]. Consequently, it is assumed that no generation of an own, drug (hapten) specific immune response occurs, but that the p-i-stimulated T-cells arise from previously primed effector and memory T-cells. In vivo, p-i-activated T-cells expand and subsequently infiltrate the skin and other organs resulting in a T- cell orchestrated inflammation. Effector and memory cells have a substantially lower threshold for activation than naïve T-cells, which could provide an explanation why a signal only via TCR may be sufficient. This threshold of T-cell activation might be further lowered by a concomitantly occurring massive immune stimulation of T-cells as it occurs during generalized herpes or human immunodeficiency virus (HIV) infections, but also during exacerbations of autoimmune diseases. Such immune processes go along with high cytokine levels and an increased expression of MHC- and costimulatory molecules. Consequently, T-cells are pre-activated and more ready to react to a minor signal like binding of a drug to its TCR. This would explain the high occurrence of drug hypersensitivities in these diseases. On the other hand, while the p-i concept has been documented for many drugs (SMX, lidocaine, lamotrigine, carbamazepine, p-phenylendiamine, radio contrast media) [11, 34–37], all of their metabolites are also implicated in drug hypersensitivity. Indeed, many patients react to the parent compound as well as to some metabolites simultaneously. In other words, they react with the drug bound as hapten to a protein/peptide and the ‘inert’ substance simultaneously, which is bound by Van der Waals forces/hydrogen bonds. This raises the question whether the hapten-characteristic of a drug (with all its immunostimulatory consequences on innate immunity) is a prerequisite that p-i stimulations can (also) occur.
Full T-cell activation by the drug (measured by immediate Ca2+ influx into specific T-cells, cytokine synthesis and proliferation) requires the interaction of the TCR with MHC (documented for MHC class II) on APC [11, 33]. It raises the question whether the drug binds first to the MHC molecule, modifying its structure and thus leading to specific TCR activation or whether the drug binds primarily to specific TCR, rendering the MHC interaction only a supplementing signal. Initial data suggested that the interaction of the drug happens first with the TCR, since the MHC-bound peptide could be exchanged or removed without affecting CD4+ T-cell activation [38] (Figure 1B). Moreover, some TCC reacted to the drug even in the presence of allogeneic MHC molecules, indicating that no strict HLA restriction for drug presentation exists [39]. However, this may be different for the less well analyzed CD8+ TCC. Some severe drug hypersensitivity reactions caused by certain drugs have a strikingly high HLA-B-allele association [40–42]. For example, in the case of abacavir hypersensitivity, a drug hypersensitivity syndrome strongly associated with the HLA-B*5701 allele [43], key interacting residues in the HLA-B*5701 peptide binding cleft could be identified [44], which allow the formation of non-covalent interactions with the drug abacavir [45]. Recent in silico data published by Yang et al. suggest that drugs could also fit in ‘empty’, non-peptide bearing MHC class I molecules. Thus the strong MHC allele-drug specificity can be explained by a steric complementarity together with other strong non-covalent interactions between the drug molecule and the antigen presentation groove. Indeed, it is tempting to speculate that the interaction of abacavir with ‘empty’ MHC-class I molecules may stabilize these MHC-molecules, since MHC molecules without peptide are rather unstable. On the other hand it has been hypothesized that abacavir binds irreversibly to APC proteins. This mechanism, possibly mediated through metabolism, may lead to a subsequent presentation of ‘altered self’ peptides on HLA-B*5701 alleles [46]. Indeed, analysis of various drug induced reactions revealed quite consistently the presence of hapten- and p-i mechanisms simultaneously to the parent drug and drug metabolite in the same individual [47–49]. This means that some T-cells react with the parent compound (p-i-mechanism), others to the hapten and a few to hapten and parent compound simultaneously.
The strong MHC class I associated drug hypersensitivity reactions are nowadays an interesting example of personalized medicine: HLA-B*5701 typing is done before abacavir is prescribed, which quasi eliminated severe abacavir related drug hypersensitivity reactions. Similarly, in Han Chinese, HLA-B*1502 typing drastically reduced the incidence of SJS/TEN in carbamazepine treated patients. Flucloxacillin induced hepatitis is also strongly associated with HLA-B*5701 [50]. However, the penetrance of this gene is low, meaning that only few patients would develop the disease in spite of carrying the risk allele. Thus HLA-typing is not routinely recommended.
Based on these new findings, two types of p-i mechanisms may occur. In the case of MHC-restricted drug hypersensitivity reactions, the drug may first bind to the MHC-class I molecules, and subsequently elicit a strong (CD8+ T-cell) immune response. Whether the MHC molecule remains empty and how the peptide could influence the interaction is still not clear. Nevertheless the binding to the MHC-molecule could explain the striking HLA-class I association (Figure 1C). On the other hand, a more polymorphic CD4+ T-cell response would occur if the drug interacts primarily with the TCR. Full stimulation requires an MHC interaction, probably by common determinants of the MHC structure, as various MHC-class II molecules appear to be sufficient to provide a T-cell stimulation. Therefore, no MHC-association has been found for these clinically mostly milder reactions (mainly maculopapular exanthems, rarely DRESS).
Taken together, the p-i concept represents a new way to explain drug induced hypersensitivity reactions, as it actually suggests that drug allergies are pharmacological reactions. The dogma that small chemicals are not full antigens is still valid and must not be refuted, but drugs are able to interfere with the human immune system in different ways. Connected to this surprising finding of pharmacological stimulations is the development that some of the so called ‘unpredictable’ type B drug reactions have become the most predictable drug reactions and a paradigm for personalized medicine. It seems the equation that type B reactions are ‘bizarre’ reactions is slowly becoming outdated.
Acknowledgments
We thank Thomas J. Gentinetta for his help in the elaboration of Figure 1.
This work was supported by the Swiss National Foundation, grant Nr SNF 3100AO-116113/1 and by the Swiss Center for Applied Human Toxicology (SCAHT).
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200–5. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 2.Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, Farrar K, Park BK, Breckenridge AM. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329:15–9. doi: 10.1136/bmj.329.7456.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rawlins M, Thompson W. Mechanisms of Adverse Drug Reactions. New York: Oxford University Press; 1991. [Google Scholar]
- 4.Deng X, Luyendyk JP, Ganey PE, Roth RA. Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models. Pharmacol Rev. 2009;61:262–82. doi: 10.1124/pr.109.001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Posadas SJ, Pichler WJ. Delayed drug hypersensitivity reactions – new concepts. Clin Exp Allergy. 2007;37:989–99. doi: 10.1111/j.1365-2222.2007.02742.x. [DOI] [PubMed] [Google Scholar]
- 6.Coombs PR, Gell PG. Classification of allergic reactions responsible for clinical hypersensitivity and disease. In: Gell RR, editor. Clinical Aspects of Immunology. Oxford: Oxford University Press; 1968. pp. 575–96. [Google Scholar]
- 7.Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:683–93. doi: 10.7326/0003-4819-139-8-200310210-00012. [DOI] [PubMed] [Google Scholar]
- 8.Spanou Z, Keller M, Britschgi M, Yawalkar N, Fehr T, Neuweiler J, Gugger M, Mohaupt M, Pichler WJ. Involvement of drug-specific T cells in acute drug-induced interstitial nephritis. J Am Soc Nephrol. 2006;17:2919–27. doi: 10.1681/ASN.2006050418. [DOI] [PubMed] [Google Scholar]
- 9.Mennicke M, Zawodniak A, Keller M, Wilkens L, Yawalkar N, Stickel F, Keogh A, Inderbitzin D, Candinas D, Pichler WJ. Fulminant liver failure after vancomycin in a sulfasalazine-induced DRESS syndrome: fatal recurrence after liver transplantation. Am J Transplant. 2009;9:2197–202. doi: 10.1111/j.1600-6143.2009.02788.x. [DOI] [PubMed] [Google Scholar]
- 10.Yawalkar N, Egli F, Hari Y, Nievergelt H, Braathen LR, Pichler WJ. Infiltration of cytotoxic T cells in drug-induced cutaneous eruptions. Clin Exp Allergy. 2000;30:847–55. doi: 10.1046/j.1365-2222.2000.00847.x. [DOI] [PubMed] [Google Scholar]
- 11.Zanni MP, von Greyerz S, Schnyder B, Brander KA, Frutig K, Hari Y, Valitutti S, Pichler WJ. HLA-restricted, processing- and metabolism-independent pathway of drug recognition by human alpha beta T lymphocytes. J Clin Invest. 1998;102:1591–8. doi: 10.1172/JCI3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lerch M, Keller M, Britschgi M, Kanny G, Tache V, Schmid DA, Beeler A, Gerber BO, Luethi M, Bircher AJ, Christiansen C, Pichler WJ. Cross-reactivity patterns of T cells specific for iodinated contrast media. J Allergy Clin Immunol. 2007;119:1529–36. doi: 10.1016/j.jaci.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Torres MJ, Mayorga C, Blanca M. Nonimmediate allergic reactions induced by drugs: pathogenesis and diagnostic tests. J Investig Allergol Clin Immunol. 2009;19:80–90. [PubMed] [Google Scholar]
- 14.Hari Y, Urwyler A, Hurni M, Yawalkar N, Dahinden C, Wendland T, Braathen LR, Matter L, Pichler WJ. Distinct serum cytokine levels in drug- and measles-induced exanthema. Int Arch Allergy Immunol. 1999;120:225–9. doi: 10.1159/000024271. [DOI] [PubMed] [Google Scholar]
- 15.Nassif A, Bensussan A, Dorothee G, Mami-Chouaib F, Bachot N, Bagot M, Boumsell L, Roujeau JC. Drug specific cytotoxic T-cells in the skin lesions of a patient with toxic epidermal necrolysis. J Invest Dermatol. 2002;118:728–33. doi: 10.1046/j.1523-1747.2002.01622.x. [DOI] [PubMed] [Google Scholar]
- 16.Yawalkar N, Hari Y, Frutig K, Egli F, Wendland T, Braathen LR, Pichler WJ. T cells isolated from positive epicutaneous test reactions to amoxicillin and ceftriaxone are drug specific and cytotoxic. J Invest Dermatol. 2000;115:647–52. doi: 10.1046/j.1523-1747.2000.00105.x. [DOI] [PubMed] [Google Scholar]
- 17.Schnyder B, Frutig K, Mauri-Hellweg D, Limat A, Yawalkar N, Pichler WJ. T-cell-mediated cytotoxicity against keratinocytes in sulfamethoxazol-induced skin reaction. Clin Exp Allergy. 1998;28:1412–7. doi: 10.1046/j.1365-2222.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- 18.Britschgi M, Steiner UC, Schmid S, Depta JP, Senti G, Bircher A, Burkhart C, Yawalkar N, Pichler WJ. T-cell involvement in drug-induced acute generalized exanthematous pustulosis. J Clin Invest. 2001;107:1433–41. doi: 10.1172/JCI12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider CH, De Weck AL. A new chemical aspect of penicillin allergy: the direct reaction of penicillin with epsilon-amino-groups. Nature. 1965;208:57–9. doi: 10.1038/208057a0. [DOI] [PubMed] [Google Scholar]
- 20.Martin S, Weltzien HU. T cell recognition of haptens, a molecular view. Int Arch Allergy Immunol. 1994;104:10–6. doi: 10.1159/000236703. [DOI] [PubMed] [Google Scholar]
- 21.Aiba S. Maturation of dendritic cells induced by cytokines and haptens. Tohoku J Exp Med. 1998;184:159–72. doi: 10.1620/tjem.184.159. [DOI] [PubMed] [Google Scholar]
- 22.Megherbi R, Kiorpelidou E, Foster B, Rowe C, Naisbitt DJ, Goldring CE, Park BK. Role of protein haptenation in triggering maturation events in the dendritic cell surrogate cell line THP-1. Toxicol Appl Pharmacol. 2009;238:120–32. doi: 10.1016/j.taap.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Pickard C, Louafi F, McGuire C, Lowings K, Kumar P, Cooper H, Dearman RJ, Cumberbatch M, Kimber I, Healy E, Friedmann PS. The cutaneous biochemical redox barrier: a component of the innate immune defenses against sensitization by highly reactive environmental xenobiotics. J Immunol. 2009;183:7576–84. doi: 10.4049/jimmunol.0901064. [DOI] [PubMed] [Google Scholar]
- 24.Saint-Mezard P, Rosieres A, Krasteva M, Berard F, Dubois B, Kaiserlian D, Nicolas JF. Allergic contact dermatitis. Eur J Dermatol. 2004;14:284–95. [PubMed] [Google Scholar]
- 25.Park BK, Pirmohamed M, Kitteringham NR. Role of drug disposition in drug hypersensitivity: a chemical, molecular, and clinical perspective. Chem Res Toxicol. 1998;11:969–87. doi: 10.1021/tx980058f. [DOI] [PubMed] [Google Scholar]
- 26.Cavani A, De Pita O, Girolomoni G. New aspects of the molecular basis of contact allergy. Curr Opin Allergy Clin Immunol. 2007;7:404–8. doi: 10.1097/ACI.0b013e3282ef6923. [DOI] [PubMed] [Google Scholar]
- 27.Naisbitt DJ, Gordon SF, Pirmohamed M, Park BK. Immunological principles of adverse drug reactions: the initiation and propagation of immune responses elicited by drug treatment. Drug Saf. 2000;23:483–507. doi: 10.2165/00002018-200023060-00002. [DOI] [PubMed] [Google Scholar]
- 28.Griem P, Wulferink M, Sachs B, Gonzalez JB, Gleichmann E. Allergic and autoimmune reactions to xenobiotics: how do they arise? Immunol Today. 1998;19:133–41. doi: 10.1016/s0167-5699(97)01219-x. [DOI] [PubMed] [Google Scholar]
- 29.Sanderson JP, Naisbitt DJ, Farrell J, Ashby CA, Tucker MJ, Rieder MJ, Pirmohamed M, Clarke SE, Park BK. Sulfamethoxazole and its metabolite nitroso sulfamethoxazole stimulate dendritic cell costimulatory signaling. J Immunol. 2007;178:5533–42. doi: 10.4049/jimmunol.178.9.5533. [DOI] [PubMed] [Google Scholar]
- 30.Schnyder B, Mauri-Hellweg D, Zanni M, Bettens F, Pichler WJ. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T cell clones. J Clin Invest. 1997;100:136–41. doi: 10.1172/JCI119505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichler WJ. Pharmacological interaction of drugs with antigen-specific immune receptors: the p-i concept. Curr Opin Allergy Clin Immunol. 2002;2:301–5. doi: 10.1097/00130832-200208000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Pichler WJ. Direct T-cell stimulations by drugs – bypassing the innate immune system. Toxicology. 2005;209:95–100. doi: 10.1016/j.tox.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 33.Yerly D, Gerber B, Lochmatter P, Pichler WJ. Partial and full agonisitic and antagonistic effects of drugs on T cell receptors. Manuscript. 2010 in preparation. [Google Scholar]
- 34.Sieben S, Kawakubo Y, Al Masaoudi T, Merk HF, Blomeke B. Delayed-type hypersensitivity reaction to paraphenylenediamine is mediated by 2 different pathways of antigen recognition by specific alphabeta human T-cell clones. J Allergy Clin Immunol. 2002;109:1005–11. doi: 10.1067/mai.2002.123872. [DOI] [PubMed] [Google Scholar]
- 35.Naisbitt DJ, Britschgi M, Wong G, Farrell J, Depta JP, Chadwick DW, Pichler WJ, Pirmohamed M, Park BK. Hypersensitivity reactions to carbamazepine: characterization of the specificity, phenotype, and cytokine profile of drug-specific T cell clones. Mol Pharmacol. 2003;63:732–41. doi: 10.1124/mol.63.3.732. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Farrell J, Pirmohamed M, Park BK, Naisbitt DJ. Generation and characterization of antigen-specific CD4+, CD8+, and CD4+CD8+ T-cell clones from patients with carbamazepine hypersensitivity. J Allergy Clin Immunol. 2007;119:973–81. doi: 10.1016/j.jaci.2006.12.617. [DOI] [PubMed] [Google Scholar]
- 37.Keller M, Lerch M, Britschgi M, Tache V, Gerber BO, Luthi M, Lochmatter P, Kanny G, Bircher AJ, Christiansen C, Pichler WJ. Processing-dependent and -independent pathways for recognition of iodinated contrast media by specific human T cells. Clin Exp Allergy. 2010;40:257–68. doi: 10.1111/j.1365-2222.2009.03425.x. [DOI] [PubMed] [Google Scholar]
- 38.Burkhart C, Britschgi M, Strasser I, Depta JP, von Greyerz S, Barnaba V, Pichler WJ. Non-covalent presentation of sulfamethoxazole to human CD4+ T cells is independent of distinct human leucocyte antigen-bound peptides. Clin Exp Allergy. 2002;32:1635–43. doi: 10.1046/j.1365-2222.2002.01513.x. [DOI] [PubMed] [Google Scholar]
- 39.von Greyerz S, Bultemann G, Schnyder K, Burkhart C, Lotti B, Hari Y, Pichler WJ. Degeneracy and additional alloreactivity of drug-specific human alpha beta(+) T cell clones. Int Immunol. 2001;13:877–85. doi: 10.1093/intimm/13.7.877. [DOI] [PubMed] [Google Scholar]
- 40.Tassaneeyakul W, Jantararoungtong T, Chen P, Lin PY, Tiamkao S, Khunarkornsiri U, Chucherd P, Konyoung P, Vannaprasaht S, Choonhakarn C, Pisuttimarn P, Sangviroon A, Tassaneeyakul W. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics. 2009;19:704–9. doi: 10.1097/FPC.0b013e328330a3b8. [DOI] [PubMed] [Google Scholar]
- 41.Man CB, Kwan P, Baum L, Yu E, Lau KM, Cheng AS, Ng MH. Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia. 2007;48:1015–8. doi: 10.1111/j.1528-1167.2007.01022.x. [DOI] [PubMed] [Google Scholar]
- 42.Hetherington S, Hughes AR, Mosteller M, Shortino D, Baker KL, Spreen W, Lai E, Davies K, Handley A, Dow DJ, Fling ME, Stocum M, Bowman C, Thurmond LM, Roses AD. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359:1121–2. doi: 10.1016/S0140-6736(02)08158-8. [DOI] [PubMed] [Google Scholar]
- 43.Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, James I, Christiansen FT. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727–32. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 44.Chessman D, Kostenko L, Lethborg T, Purcell AW, Williamson NA, Chen Z, Kjer-Nielsen L, Mifsud NA, Tait BD, Holdsworth R, Almeida CA, Nolan D, Macdonald WA, Archbold JK, Kellerher AD, Marriott D, Mallal S, Bharadwaj M, Rossjohn J, McCluskey J. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822–32. doi: 10.1016/j.immuni.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Chen J, He L. Harvesting candidate genes responsible for serious adverse drug reactions from a chemical-protein interactome. PLoS Comput Biol. 2009;5:e1000441. doi: 10.1371/journal.pcbi.1000441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin AM, Almeida CA, Cameron P, Purcell AW, Nolan D, James I, McCluskey J, Phillips E, Landay A, Mallal S. Immune responses to abacavir in antigen-presenting cells from hypersensitive patients. AIDS. 2007;21:1233–44. doi: 10.1097/QAD.0b013e3280119579. [DOI] [PubMed] [Google Scholar]
- 47.Naisbitt DJ, Farrell J, Wong G, Depta JP, Dodd CC, Hopkins JE, Gibney CA, Chadwick DW, Pichler WJ, Pirmohamed M, Park BK. Characterization of drug-specific T cells in lamotrigine hypersensitivity. J Allergy Clin Immunol. 2003;111:1393–403. doi: 10.1067/mai.2003.1507. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Sanderson JP, Farrell J, Drummond NS, Hanson A, Bowkett E, Berry N, Stachulski AV, Clarke SE, Pichler WJ, Pirmohamed M, Park BK, Naisbitt DJ. Activation of T cells by carbamazepine and carbamazepine metabolites. J Allergy Clin Immunol. 2006;118:233–41. doi: 10.1016/j.jaci.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 49.Castrejon JL, Berry N, El-Ghaiesh S, Gerber B, Pichler WJ, Park BK, Naisbitt DJ. Stimulation of human T cells with sulfonamides and sulfonamide metabolites. J Allergy Clin Immunol. 2010;125:411–8. doi: 10.1016/j.jaci.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 50.Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A, Daly MJ, Goldstein DB, John S, Nelson MR, Graham J, Park BK, Dillon JF, Bernal W, Cordell HJ, Pirmohamed M, Aithal GP, Day CP. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–9. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 51.Brockow K, Romano A, Aberer W, Bircher AJ, Barbaud A, Bonadonna P, Faria E, Kanny G, Lerch M, Pichler WJ, Ring J, Rodrigues Cernadas J, Tomaz E, Demoly P, Christiansen C. Skin testing in patients with hypersensitivity reactions to iodinated contrast media – a European multicenter study. Allergy. 2009;64:234–41. doi: 10.1111/j.1398-9995.2008.01832.x. [DOI] [PubMed] [Google Scholar]
- 52.Zanni MP, Mauri-Hellweg D, Brander C, Wendland T, Schnyder B, Frei E, von Greyerz S, Bircher A, Pichler WJ. Characterization of lidocaine-specific T cells. J Immunol. 1997;158:1139–48. [PubMed] [Google Scholar]
- 53.Zanni MP, von Greyerz S, Hari Y, Schnyder B, Pichler WJ. Recognition of local anesthetics by alphabeta+ T cells. J Invest Dermatol. 1999;112:197–204. doi: 10.1046/j.1523-1747.1999.00484.x. [DOI] [PubMed] [Google Scholar]
- 54.Christiansen C. Late-onset allergy-like reactions to X-ray contrast media. Curr Opin Allergy Clin Immunol. 2002;2:333–9. doi: 10.1097/00130832-200208000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Christiansen C, Pichler WJ, Skotland T. Delayed allergy-like reactions to X-ray contrast media: mechanistic considerations. Eur Radiol. 2000;10:1965–75. doi: 10.1007/s003300000543. [DOI] [PubMed] [Google Scholar]
- 56.Depta JP, Altznauer F, Gamerdinger K, Burkhart C, Weltzien HU, Pichler WJ. Drug interaction with T-cell receptors: T-cell receptor density determines degree of cross-reactivity. J Allergy Clin Immunol. 2004;113:519–27. doi: 10.1016/j.jaci.2003.11.030. [DOI] [PubMed] [Google Scholar]