

Abstract
AIMS
To catalogue the perpetrators of CYP-mediated pharmacokinetic drug–drug interactions (PK-DDIs) using clinically relevant criteria, and to compare this with an analogous catalogue.
METHODS
Candidate inhibitors and inducers of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A (‘perpetrators’) were evaluated using published clinical pharmacokinetic interaction studies. Studies were selected on the basis of ≥six human subjects, use of a validated in vivo probe substrate for the CYP enzyme, and clinically relevant dosing. Inhibitors were described according to the FDA classifications of strong, moderate or weak, whereas inducers were classified as major (≥twofold decrease in AUC) or weak (<twofold decrease in AUC). A catalogue of major perpetrators was constructed based on twofold changes in the clearance of probe substrates. Perpetrators in the clinical version of the Cytochromes P450 Drug Interaction Table (CDIT) were compared with the ‘accepted’ major perpetrators.
RESULTS
From a list of 216 candidate drugs (349 CYP-perpetrator pairs, CYP-PPs), 36 inhibitors and eight inducers were accepted as major perpetrators of PK-DDIs, resulting in 58 CYP-PPs. In comparison, the clinical version of the CDIT had a sensitivity of 33% and a positive predictive value of 68%. One hundred and ninety-nine CYP-PPs were rejected as major perpetrators, and 92 CYP-PPs had insufficient published human pharmacokinetic data for robust classification.
CONCLUSIONS
Using a criteria-based assessment, the number of drugs that are proven or likely major perpetrators of CYP-mediated PK-DDIs is relatively small. Current clinical decision support on PK-DDIs is inconsistent with the published evidence and can be improved using simple criteria.
Keywords: CYP, CYP induction, CYP inhibition, cytochromes P450, drug–drug interactions, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Many drugs inhibit or induce cytochrome P450 enzymes (CYP) to cause clinically significant changes in the concentrations of other drugs, i.e. ‘perpetrate’ pharmacokinetic drug–drug interactions (PK-DDIs).
Tables that list the substrates, inhibitors and inducers of CYP are common, but they lack consistency and are constructed from evidence of variable quality.
WHAT THIS STUDY ADDS
This is the first study to catalogue important perpetrators of PK-DDIs using objective criteria and clinical pharmacokinetic drug interaction studies. This information is intended to inform clinical decisions on PK-DDIs.
Existing tables of CYP inhibitors and inducers have low sensitivity and low positive predictive value in identifying the major perpetrators of PK-DDIs.
Several drugs were identified which potentially perpetrate CYP-mediated PK-DDIs, but quality clinical pharmacokinetic interaction studies are lacking. This information may be used to inform future research.
Introduction
Pharmacokinetic drug–drug interactions (PK-DDIs) are a major contributor to the failure of drug therapy, either from toxicity leading to patient harm or sub-therapeutic concentrations resulting in loss of efficacy. PK-DDIs are often difficult for clinicians to identify as they are not defined by the pharmacological actions of the drugs involved. However, the understanding of PK-DDIs has increased remarkably over the past three decades, primarily because of the availability of molecular and analytical techniques that have rapidly advanced our knowledge of the biochemistry of drug metabolism and transport [1]. Many drugs have been identified as inhibitors or inducers of drug metabolizing enzymes, particularly the cytochromes P450 (CYP). These are considered ‘perpetrators’ of PK-DDIs when the clearances of respective ‘object’ drugs are altered to a clinically significant extent.
Reference sources providing clinicians with information on PK–DDIs are generally presented in three ways: (i) lists of interactions within individual drug monographs; (ii) commercial software checkers based on pairs of interacting drugs; and (iii) tables of drugs as substrates, inhibitors and inducers of drug metabolizing CYP enzymes (‘CYP tables’). These reference sources are based on DDI studies using multiple methodologies of variable quality in a range of experimental models. Studies may be conducted in silico, in vitro or in vivo, the latter comprising formal pharmacokinetic interaction studies in animals and humans, or as reports of clinical observations. The assessment of these studies is seldom explicit, and there is lack of consistency and considerable disparity between resources [2–6]. For example, in a recent study of four international drug interaction compendia, between 14% and 44% of the interactions classified as major in any one compendium were not listed in other compendia [4]. Thus, interpreting the clinical relevance of a given perpetrator is often difficult, particularly for healthcare providers subject to ‘information overload’ and ‘alert fatigue’[7, 8]. Computerized DDI checkers at the point of prescribing appear no better, delivering alerts that are frequently irrelevant to clinical practice. Approximately 90% of DDI alerts are overridden by clinicians [9, 10]. Hence, there is a need for a more precise set of perpetrators to support prescribing decisions on PK-DDIs.
The primary aim of this study was to catalogue the perpetrators of CYP-mediated PK-DDIs using clinically relevant criteria. Candidate drugs were accepted or rejected as major perpetrators using the FDA-defined boundary between moderate and weak inhibition of drug metabolism (i.e. a twofold change in the clearance of the probe substrate). This was based on the premise that greater changes in object drug exposure are more likely to result in clinically important PK-DDIs (i.e. changes in safety and/or efficacy). The assessment shows that a relatively small number of drugs are either proven or likely to be major perpetrators of CYP-mediated PK-DDIs, and that the sensitivity and positive predictive value of existing CYP tables are low.
Methods
Development of the catalogue
A list of candidate inhibitors and inducers of CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A, which are generally considered to be the most important drug metabolizing CYP enzymes, was compiled from available resources: the Australian Medicines Handbook (AMH 2009, ISBN: 978-0-9757919-9-8), eMIMS (Version 5.01.0097, CMPMedica Australia Pty Ltd), the full version of the Cytochromes P450 Drug Interaction Table (CDIT, Version 5.0, http://medicine.iupui.edu/clinpharm/ddis/table.asp), Obach et al. 2006 [11], the Preferred Medicines List 11th edition (Canterbury District Health Board, ISBN: 0-473-08171-7) and personal knowledge. All drugs listed in these resources were extracted, including supplements and some foods (the exceptions were broccoli, brussel sprouts and char-grilled meat, which are listed as CYP1A2 inducers in the full version of the CDIT but were not analysed here). If drug classes were listed in original resources (e.g. fluoroquinolones), all members of the class were included for assessment. The list of 216 candidate drugs, sorted into 349 CYP-perpetrator pairs (CYP-PPs), was compiled using a spreadsheet (Microsoft Office Excel 2003). Note that the number of candidate CYP-PPs was greater than the number of candidate drugs, because some perpetrators inhibit or induce multiple CYP enzymes.
All CYP-PPs were assessed using the primary literature retrieved from Medline (Ovid 1966–2009), PubMed (1966–2009) and GoogleScholar®. Industry published product information was used to identify unpublished studies conducted by industry. Study inclusion criteria were developed by the investigators based on their combined expertise in the evaluation of PK-DDIs. These were: (i) clinical pharmacokinetic interaction studies in humans (≥six subjects); (ii) use of a validated in vivo CYP probe substrate (fraction of the probe metabolized by the CYP enzyme, fmCYP≥0.8); (iii) clinically relevant dosing of the candidate perpetrator; and (iv) dosing to steady state or for the duration of the commonly recommended clinical regimen. In most cases, studies used pre-probe dosing periods several times greater than the time required to reach steady state, i.e. pre-probe dosing of ≥1 week. The following in vivo CYP probe substrates were considered appropriate based on previous recommendations [12, 13]: caffeine, theophylline or tizanidine for CYP1A2, phenytoin, S-warfarin or tolbutamide for CYP2C9, mephenytoin or omeprazole for CYP2C19, debrisoquine, desipramine, dextromethorphan, metoprolol or sparteine for CYP2D6, and buspirone, felodipine, lovastatin, maraviroc, midazolam, triazolam, sildenafil or simvastatin for CYP3A. Studies meeting these criteria were considered Level A evidence. Information on perpetrator half-life, dosing regimen, in vivo CYP probe and interaction magnitude was collated in an electronic spreadsheet with references to the primary literature.
The magnitude of CYP-mediated PK-DDIs was assessed by changes in probe pharmacokinetics following administration of perpetrators. In order of preference: (i) area under the plasma concentration–time curve (AUC); (ii) total systemic clearance; and (iii) plasma or urinary metabolite ratios. Inhibitors were described using the FDA classifications of strong, moderate or weak[13], i.e. a strong inhibitor is one that causes a ≥fivefold increase in AUC or ≥80% decrease in clearance of probe substrate, a moderate inhibitor is one that causes a ≥twofold but <fivefold increase in AUC or ≥50% but <80% decrease in clearance, and a weak inhibitor is one that causes a ≥1.25-fold increase in AUC or ≥20% but <50% decrease in clearance. Inducers were classified as major (≥twofold decrease in AUC or ≥100% increase in clearance) or weak (<twofold decrease in AUC or <100% increase in clearance). In the absence of AUC or clearance data, phenocopying of CYP2D6 extensive metabolizers to poor metabolizers from the urinary ratios of dextromethorphan : dextrorphan, debrisoquine : 4-hydroxydebrisoquine or sparteine : dehydrosparteine was considered to indicate strong inhibition of CYP2D6. Moderate and weak inhibition of CYP2D6 were determined on results relative to other CYP2D6 inhibitors for which metabolite ratio and AUC or clearance data with another probe substrate were available. Similarly, the urinary S-mephenytoin : R-mephenytoin and plasma omeprazole : hydroxyomeprazole ratios were used to evaluate changes in CYP2C19 activity. When multiple clinical studies were available, with or without different recommended probes, the studies were assessed collectively, and when data gave conflicting results, the perpetrator was classified conservatively using the greater magnitude of effect. A representative study at the upper end of the reported magnitudes of interaction was referenced.
Figure 1 shows how CYP-PPs were classified in the catalogue according to the hierarchies of evidence. First, drugs not available in Australia or New Zealand were excluded. Candidates were then sorted into ‘accepted major perpetrators’ (strong and moderate inhibitors and major inducers) and ‘rejected major perpetrators’ (weak inhibitors and weak inducers, and drugs that do not alter the clearance of probes). The remaining perpetrators without Level A evidence were assessed using lower levels of evidence (Level B) to identify drugs as ‘likely’ or ‘unlikely’ major perpetrators, leaving a number of drugs as ‘possible’ major perpetrators. Level B evidence included clinical pharmacokinetic interaction studies not meeting the above criteria, observational reports, in vitro interaction studies and in silico assessments. Level B evidence was assessed subjectively by the investigators by comparison with similar studies of the accepted major perpetrators and by using their combined professional expertise.
Figure 1.
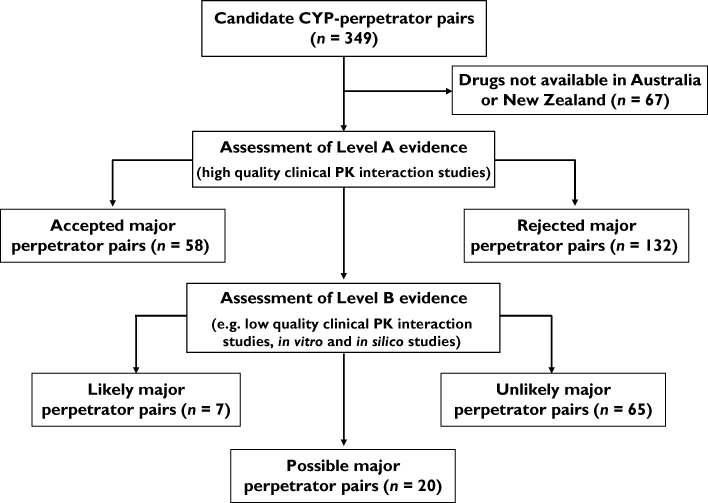
Method for classifying the perpetrators of CYP-mediated pharmacokinetic drug–drug interactions
Comparison of an existing CYP table with the accepted major perpetrators catalogue
The perpetrators identified by the clinical version of the CDIT were evaluated by comparison with the list of accepted major perpetrators. Only inhibitors indicated as strong or moderate were considered, whereas all inducers were included in the analysis as the degree of induction potency was not indicated [14]. Drugs were categorized as: identified appropriately (true positives), identified inappropriately (false positives) and omitted (false negatives). The catalogue was compared by calculating sensitivity (true positives/true positives + false negatives) and positive predictive values (true positives/true positives + false positives). Analysis of this resource was repeated with addition of the ‘likely’ major perpetrators to the comparator list.
Results
The catalogue of perpetrators of CYP-mediated PK-DDIs is presented in Tables 1–4. Fifty-five candidate perpetrators (67 CYP-PPs) were drugs not available in Australia or New Zealand (Table 1). Note that these drugs also have limited availability/use in other countries, including in Europe and North America. Using a criteria-based assessment, 36 inhibitors and eight inducers of CYP enzymes were accepted as major perpetrators of PK-DDIs (58 CYP-PPs; Table 2). Of the remaining 224 candidate CYP-PPs, there was Level A evidence to exclude 132 (59%) as major perpetrators (Table 3). For 92 CYP-PPs (41% of the remaining 224), Level B evidence identified an additional five drugs as likely major perpetrators (seven CYP-PPs) and 65 CYP-PPs as unlikely to be important. This left 18 drugs (20 CYP-PPs) as possible major perpetrators (Table 4).
Table 1.
Drugs implicated as perpetrators of CYP-mediated PK-DDIs that are not available in Australia or New Zealand
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A |
|---|---|---|---|---|
| Armodafinil | Azapropazone | Armodafinil | Clemastine | Armodafinil |
| Artemisinin | Benzbromarone | Artemisinin | Deramciclan | Chloramphenicol* |
| Clinafloxacin | Bucolome | Halofantrine | Clotrimazole* | |
| Enoxacin | Indisulam | Hydroxyzine | Conivaptan | |
| Etintidine | Lornoxicam | Levomepromazine | Diethyldithiocarbamate | |
| Furafylline | Miconazole* | Melperone | Mibefradil | |
| Idrocilamide | Nizatidine | Mepyramine | Miconazole* | |
| Mibefradil | Oxandrolone | Mibefradil | Nefazodone | |
| Nafcillin | Phenylbutazone | Midodrine | Nelfinavir | |
| Perfloxacin | Secobarbital | Perphenazine | Quinidine | |
| Phenylpropanolamine | Stiripentol | Propafenone | Stiripentol | |
| Pipemidic acid | Sulfamethizole | Quinidine | Sulfadimidine | |
| Rofecoxib | Sulfaphenazole | Ranolazine | Sulfinpyrazone | |
| Stiripentol | Sulfinpyrazone | Rofecoxib | Telithromycin | |
| Thiabendazole | Stiripentol | Tofisopam | ||
| Troleandomycin | Thioridazine | Troglitazone | ||
| Zileuton | Tripelennamine | Troleandomycin |
Systemic preparations not available. PK-DDI, pharmacokinetic drug–drug interactions.
Table 4.
Candidate perpetrators of CYP-mediated PK-DDIs evaluated using Level B evidence
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A | |
|---|---|---|---|---|---|
| Likely major perpetrators | Carbamazepine*[27]† | Amprenavir§ | |||
| Ciprofloxacin [171] | |||||
| Delavirdine§ | |||||
| Fosamprenavir [172] | |||||
| Phenobarbital[173] | |||||
| Rifabutin§ | |||||
| Possible major perpetrators | Amiodarone [174] | Capecitabine [175] | Carbamazepine[176] | Clomipramine [177] | Darunavir§ |
| Zafirlukast [178] | Fuoxetine‡ | Isoniazid [179] | Haloperidol | Dasatinib§ | |
| Phenobarbital | Phenobarbital | Methadone | Erlotinib§ | ||
| Phenytoin | Metoclopramide | Lapatinib | |||
| Prednisone | Nilotinib | Nilotinib§ | |||
| Unlikely major perpetrators | Atazanavir | Atazanavir | Chloroquine [180] | Artemether/lumefantrine | Anastrozole |
| Insulin | Eavirenz | Cimetidine [181] | Chloroquine [180] | Atenolol | |
| Methoxsalen [182] | 5-fluorouracil | Efavirenz | Chlorpheniramine | Atorvastatin | |
| Modafinil | Imatinib | Indomethacin | Chlorpromazine | Buspirone§ | |
| Isoniazid | Lansoprazole [183] | Citalopram [184] | Caffeine | ||
| Itraconazole [185] | Modafinil | Cocaine | Citalopram§ | ||
| Leflunomide | Norethindrone | Disulfiram [186] | Cyproterone | ||
| Phenytoin | Oxcarbazepine | Doxorubicin | Danazol | ||
| Ritonavir | Pantoprazole [187] | Grapefuit juice [188] | Darifenacin§ | ||
| Teniposide | Propoxyphene [189] | Hydroxychloroquine [190] | Digoxin | ||
| Rabeprazole [191] | Ibuprofen | Domperidone | |||
| Orange juice [188] | Ethosuximide | ||||
| Propoxyphene [189] | Gemfibrozil | ||||
| Ticlopidine | Metoprolol | ||||
| Verapamil [192] | Metronidazole | ||||
| Mifepristone | |||||
| Nevirapine§ | |||||
| Norfloxacin | |||||
| Omeprazole§ | |||||
| Oxcarbazepine§ | |||||
| Propoxyphene [193] | |||||
| Quinupristin-dalfopristin | |||||
| Raloxifene | |||||
| Repaglinide | |||||
| Valproate |
Drugs highlighted in italics are candidate inducers of CYP, whereas all other drugs are candidate inhibitors of CYP.
Drugs with a reference indicate that pharmacokinetic interactions studies have been reported but they do not meet the inclusion criteria (see Methods).
Drugs without a reference are implicated as inhibitors or inducers of CYP enzymes but clinical pharmacokinetic interaction studies have not been reported.
Assessment based on clinical pharmacokinetic interaction studies referred to in product information. PK-DDI, pharmacokinetic drug–drug interactions.
Table 2.
Accepted major perpetrators of CYP-mediated PK-DDIs based on Level A evidence
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A | |
|---|---|---|---|---|---|
| Strong inhibitors* | Ciprofloxacin [35]§¶ | Fluconazole [36] | Bupropion [37] | Clarithromycin [38] | |
| Fluvoxamine [39] | Fluvoxamine [40] | Fluoxetine [41] | Erythromycin [22] | ||
| Ticlopidine [42] | Paroxetine [43] | Grapefruit juice [44] | |||
| Perhexaline [45] | Indinavir** | ||||
| Itraconazole [46] | |||||
| Ketoconazole [46] | |||||
| Lopinavir/ritonavir [47] | |||||
| Ritonavir [48] | |||||
| Saquinavir [49] | |||||
| Saquinavir/ritonavir [50] | |||||
| Voriconazole [51] | |||||
| Moderate inhibitors† | Ethinylestradiol [52] | Fluconazole [53] | Clarithromycin [54] | Cinacalcet [55] | Aprepitant [56] |
| Interferon alpha-2b [57] | Fluoxetine [58] | Doxepin [30] | Atazanavir [50] | ||
| Moclobemide [59] | Duloxetine [60] | Atazanavir/ritonavir [50] | |||
| Moriconazole [61] | Flecainide [62] | Cimetidine [63] | |||
| Moclobemide [32] | Cyclosporin [64] | ||||
| Quinine [65] | Diltiazem [66] | ||||
| Terbinafine [67] | Fluconazole [68] | ||||
| Fluvoxamine [69] | |||||
| Imatinib [70] | |||||
| Posaconazole** | |||||
| Verapamil [66] | |||||
| Major inducers‡ | Phenytoin [71] | Rifampicin [72] | Lopinavir/ritonavir [47] | Bosentan [73] | |
| Rifampicin [74] | Rifampicin [75] | Carbamazepine [76] | |||
| St John's wort [77] | Efavirenz [78] | ||||
| Modafinil [79] | |||||
| Phenytoin [80] | |||||
| Rifampicin [81] | |||||
| St John's wort [82] |
≥fivefold increase in AUC or ≥80% decrease in clearance of in vivo CYP probe.
≥twofold but <fivefold AUC increase or ≥50% but <80% decrease in clearance of in vivo CYP probe.
≥twofold decrease in AUC or ≥100% increase in clearance of in vivo CYP probe.
Please see Appendix S1 for complete reference list.
In cases with multiple clinical pharmacokinetic interaction studies, references are given for a representative study rather than all studies investigating the interaction.
Assessment based on clinical pharmacokinetic interaction studies referred to in product information. PK-DDI, pharmacokinetic drug–drug interactions.
Table 3.
Rejected major perpetrators of CYP-mediated PK-DDIs based on Level A evidence
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A | |
|---|---|---|---|---|---|
| Weak inhibitors* | Cimetidine [83]‡ | Amiodarone [27] | Finasteride [84] | Amiodarone [85] | Alprazolam [86] |
| Clarithromycin [87] | Aimetidine [88] | Interferon alpha-2b [57] | Amitriptyline [89] | Amiodarone [90] | |
| Diltiazem [91] | Co-trimoxazole [92] | Ketoconazole [93] | Aspirin [94] | Aspirin [94] | |
| Echinacea [95] | Disulfiram [96] | Omeprazole [97] | Celecoxib [98] | Azithromycin [99] | |
| Erythromycin [87] | Echinacea [95] | Oral contraceptives [100] | Cimetidine [101] | Bicalutamide [102] | |
| Mexiletine [103] | Fuvastatin [104] | Roxithromycin [105] | Darifenacin§ | Co-trimoxazole [108] | |
| Moclobemide [59] | Fluvoxamine [106] | Desvenlafaxine [107] | Cyclosporin [112] | ||
| Norfloxacin [109] | Ketoconazole [110] | Diltiazem [111] | Fluvoxamine [114] | ||
| Propranolol [26] | Lovastatin [27] | Diphenhydramine [113] | Isoniazid [117] | ||
| Roxithromycin [87] | Metronidazole [115] | Escitalopram [116] | Maraviroc [121] | ||
| Ticlopidine [118] | Sertraline [119] | Felodipine [120] | Oral contraceptives [124] | ||
| Verapamil [122] | Simvastatin [27] | Fluvoxamine [123] | Ranitidine [63] | ||
| Sulfadiazine [125] | Gefitinib [126] | Roxithromycin [129] | |||
| Voriconazole [127] | Hydralazine [128] | Rimvastatin [131] | |||
| Zafirlukast§ | Imatinib [130] | ||||
| Interferon alpha-2b [57] | |||||
| Ketoconazole [132] | |||||
| Methadone [133] | |||||
| Metoprolol [134] | |||||
| Nortriptyline [135] | |||||
| Oral contraceptives [136] | |||||
| Oxprenolol [134] | |||||
| Pindolol [134] | |||||
| Propranolol [134] | |||||
| Ranitidine [137] | |||||
| Risperidone [138] | |||||
| Ritonavir [139] | |||||
| Raw palmetto [140] | |||||
| Rertraline [43] | |||||
| Timolol [134] | |||||
| Venlafaxine [141] | |||||
| Weak inducers† | Carbamazepine [71] | Aprepitant§ | Aspirin [94] | Carbamazepine [142] | Dexamethasone [148] |
| Lopinavir/ritonavir [47] | Bosentan§ | Ginkgo biloba [143] | Phenobarbitone [144] | Ginkgo biloba [150] | |
| Montelukast [145] | Carbamazepine [146] | Rifampicin [147] | Quinine¶ | ||
| Phenobarbitone [149] | Lopinavir/ritonavir [47] | Smoking [152] | |||
| Smoking [151] | Terbinafine [153] | ||||
| Valproate [71] | |||||
| No apparent change in clearance | Amodiaquine [154] | Amodiaquine [154] | Amodiaquine [154] | Amodiaquine [154] | Echinacea [95] |
| Aspirin [94] | Darifenacin§ | Levonorgestrel [155] | Dexamethasone [156] | Ezetimibe [157] | |
| Omeprazole [158] | Modafinil [159] | Risperidone [138] | Echinacea [95] | Finasteride [84] | |
| Risperidone [138] | Omeprazole [160] | Topiramate [161] | Ginkgo-biloba [162] | Fluoxetine [114] | |
| Prednisone [163] | Maraviroc [164] | Gatifloxacin [165] | |||
| St John's wort [82] | Omeprazole [97] | Methylprednisolone [166] | |||
| Smoking [167] | Paroxetine [168] | ||||
| Pioglitazone [131] | |||||
| Saw palmetto [140] | |||||
| Sertraline [169] | |||||
| Tenofovir [108] | |||||
| Tipranavir/ritonavir [50] | |||||
| Venlafaxine [170] |
≥1.25-fold but <twofold AUC increase or ≥20% but <50% decrease in clearance of in vivo CYP probe.
<twofold decrease in AUC or <100% increase in clearance of in vivo CYP probe.
In cases with multiple clinical pharmacokinetic interaction studies, references are given for a representative study rather than all studies investigating the interaction.
Assessment based on clinical PK interaction studies referred to in product information.
Reported at Clinical Trials.gov (http://clinicaltrials.gov/ct2/show/study/NCT00785486?sect=X701&view=results). PK-DDI, pharmacokinetic drug–drug interactions.
Drugs reported as inhibitors and inducers of CYP enzymes by the clinical version of the CDIT were evaluated by comparison with the list of accepted major perpetrators in Table 2. The numbers of true positives, false positives and false negatives are shown in Table 5. On this basis, the clinical version of the CDIT had a sensitivity of 33% (19/58) and a positive predictive value of 68% (19/28). There was no significant change in these results when the five additional likely major perpetrators from Table 4 were included in the analysis (data not shown).
Table 5.
Comparison of the clinical version of the CDIT with the accepted major perpetrators in Table 2
| Perpetrators | Clinical CDIT +ve | Clinical CDIT –ve |
|---|---|---|
| CYP inhibitors | ||
| Accepted major +ve | 14 | 31 |
| Accepted major –ve | 4 | 0 |
| 18 | 31 | |
| CYP inducers | ||
| Accepted major +ve | 5 | 8 |
| Accepted major –ve | 5 | 0 |
| 10 | 8 |
CDIT, Cytochromes P450 Drug Interaction Table.
Discussion
This is the first study to catalogue the perpetrators of CYP-mediated PK-DDIs using clinically relevant criteria. Assuming that ≥twofold changes in object drug exposure are more likely to cause PK-DDIs of clinical importance (see discussion below), 39 inhibitors and 10 inducers are either proven or likely major perpetrators of CYP-mediated PK-DDIs. This is a manageable list with which clinicians can become familiar, particularly when considered by class, for example most HIV protease inhibitors, macrolide antibiotics and azole antifungals.
Of note, there were several candidate drugs for which human pharmacokinetic studies were not available, and these were evaluated on the basis of Level B evidence. For example, erlotinib and dasatinib are mechanism-based inactivators of CYP3A [15, 16], a characteristic of many important inhibitors of drug metabolism [17, 18]. Product information for these drugs indicates minor impact on the clearance of CYP3A substrates [Sprycel® (dasatinib), labelling, Tarceva® (erlotinib) labelling], but these data are not in the public domain. On this basis, the potential of erlotinib and dasatinib to act as perpetrators of CYP-mediated PK-DDIs remains unclear, and they were catalogued as ‘possible’ major perpetrators (Table 4). For such drugs, urgent clinical research is needed to facilitate objective assessment of their risk.
An existing CYP table that is widely used by healthcare providers was compared with the accepted major perpetrators in Table 2. The CDIT was developed by Flockhart and colleagues at Indiana University, and is considered the ‘gold standard’ open access CYP table [14]. The ability of the clinical version of the CDIT to detect an important perpetrator was low (sensitivity); 39 major inhibitors and inducers were not included. The probability of a given drug in this resource being a major perpetrator was also low (positive predictive value of 68%), primarily as a result of false positive inducers (Table 5). Although only one existing CYP table was included for comparison, it was chosen on the basis of availability and wide use, and it is likely that other versions would perform similarly. Indeed, many ‘in-house’ CYP tables are simply unreferenced derivatives of the CDIT, while others fail to rank the severity of CYP inhibition and induction [19], and approach with very low sensitivity and positive predictive value. Although many drug, disease, patient and prescribing factors influence DDI risk, these data suggest that existing CYP tables have limited utility in supporting clinicians with decisions on PK-DDIs.
Several important issues arise with respect to the criteria that define this study.
A ≥twofold change in the clearance of in vivo CYP probe substrates was set as the boundary between the accepted and rejected major perpetrators. This should not be interpreted as a clinical relevance threshold. Rather, if PK-DDIs are considered as a probability problem, the twofold value seems a reasonable delineation to identify which perpetrators are most likely to be clinically important in both the number of object drugs affected and the interaction magnitude. However, it is emphasized that assessing actual clinical relevance is only possible if the pharmacokinetics (including fmCYP, hepatic extraction ratio and bioavailability) and pharmacodynamics of object drugs are also considered, together with individual patient physiology. In particular, the therapeutic index of an object drug is a key factor in assessing the clinical importance of a potential PK-DDI.
It is known that the sensitivity of in vivo CYP probe substrates influences the magnitude of pharmacokinetic changes in the presence of perpetrators, e.g. buspirone is approximately 1.5 times more sensitive to changes in CYP3A activity than midazolam [20, 21]. This arises from different pharmacokinetic properties, e.g. probes with very low bioavailability due to high first pass extraction are more sensitive to inhibition of metabolic clearance. Here, any clinical pharmacokinetic interaction study with a recommended probe was analysed [12, 13], and the highest interaction magnitude was selected to cover the worst case scenario (e.g. erythromycin was catalogued as a strong inhibitor of CYP3A based on its interaction with buspirone rather than with midazolam [22, 23]). The sensitivities of in vivo probes for drug metabolizing CYP enzymes other than CYP3A still require formal evaluation, particularly for CYP2C9 and CYP2C19, which together with CYP3A exhibit substrate-dependent inhibition in vitro[24, 25]. The use of objective criteria facilitates appropriate re-classification when new information emerges.
Only data from clinical pharmacokinetic interaction studies with appropriate in vivo CYP probes and sufficient subjects were accepted as Level A evidence. Other types of studies, such as observational reports and predictions from in vitro–in vivo extrapolations, were used as Level B evidence, and predominantly excluded rather than promoted candidate drugs (Table 4). Our approach in developing a clinical catalogue was to minimize the ‘noise’ generated by these data, which are sometimes very difficult to interpret. Of the 92 CYP-PPs evaluated subjectively using Level B evidence, only a further five drugs (amprenavir, delavirdine and fosamprenavir as inhibitors, and phenobarbital and rifabutin as inducers) were classified as ‘likely’ major perpetrators to add to the accepted list in Table 2.
Clinically relevant dosing of perpetrators was considered essential. Steady state concentrations are required to obtain valid interaction data. Hence a minimum pre-probe dosing of 4–5 half-lives was required (note that in most cases, drugs taken chronically had clinical studies with pre-probe dosing periods several times greater than the time required to reach steady state). Similarly, studies with unusually high or low doses of perpetrators were excluded. For example, propranolol may be classified as a moderate inhibitor of CYP1A2 based on its interaction at 720 mg day–1 with theophylline [26]. However, using a criteria-based assessment, propranolol is a weak inhibitor of CYP1A2 as typical doses have minor impact on theophylline clearance [26].
The catalogue is easily adaptable if the criteria require redefinition and as additional clinical pharmacokinetic studies become available. The perpetrator list can also be changed to suit local clinical practice.
There are several limitations in constructing catalogues of CYP inhibitors and inducers. First, the disparity in clinical relevance between selective and non-selective perpetrators is not immediately apparent. Most object drugs are metabolized by more than one CYP enzyme, and the effect of a relatively selective perpetrator (e.g. bupropion) may be modest compared with the effect of a non-selective perpetrator (e.g. fluconazole). Second, primary studies use in vivo CYP probe substrates of varying sensitivities for changes in clearance (as discussed above). Hence, caution is required when comparing studies using different but apparently selective probes. Third, in addition to drugs with only Level B evidence, the classification of some drugs with Level A evidence requires subjective decisions. For example, the inhibition of CYP2C9 by amiodarone has Level A evidence with an effect at the boundary between moderate and weak inhibition [27–29]. Likewise, when only changes in urinary or plasma metabolite ratios are available (as is the case with many older studies for CYP2D6 and CYP2C19 substrates), interactions must be assessed by comparison with other perpetrators for which both metabolite ratios and AUC or clearance data using a recommended probe are reported. For example, doxepin was classified as a moderate inhibitor of CYP2D6 based on the 2.9-fold increase in sparteine : dehydrosparteine urinary ratio [30], which is similar to that reported for moclobemide (4.3-fold) [31], an inhibitor of CYP2D6 known to increase the AUC of dextromethorphan in the moderate range (average = 3.3-fold) [32]. Fourth, the selectivity of in vivo CYP probes is not absolute, giving rise to misleading classifications for weak perpetrators. The decreases in theophylline clearance caused by erythromycin, clarithromycin, diltiazem and verapamil are described as weak inhibition of CYP1A2, but these changes probably arise via selective mechanism-based inactivation of CYP3A [33], an enzyme involved to a minor extent in the disposition of theophylline [34]. Finally, more complex aspects of CYP-mediated PK-DDIs, such as the time course of CYP activity changes, were not considered.
Despite these limitations, the catalogue uses objective criteria to classify perpetrators in a way that can be useful in screening for potential PK-DDIs during prescribing. Put simply, a drug classified as a major perpetrator is more likely to cause PK-DDIs than a drug that is not. Therefore, starting or stopping a drug from Table 2, together with the additional five ‘likely’ drugs in Table 4, should trigger further assessment of potential changes in drug effects. The classification of object drugs by their PK/PD properties is equally important and, when used together, the information about perpetrators and objects can improve the handling of PK-DDIs in clinical practice without unnecessary increases in workload. The improvement in clinical relevance is also expected to facilitate the development of more precise decision support, e.g. DDI checkers at the point of prescribing.
In conclusion, this study catalogued the perpetrators of CYP-mediated PK-DDIs using clinically relevant criteria. The number of drugs that are either proven or likely major perpetrators is relatively small, 39 inhibitors and 10 inducers. There are several potential perpetrators for which robust PK-DDI data are urgently required. Current clinical decision support related to PK-DDIs is inconsistent with the published evidence and can be improved using simple criteria.
Acknowledgments
We thank Katie A. Jessen for contributing to the analysis of the CYP3A inhibitors and inducers, and Karli N. Goodwin for referencing.
Competing Interests
There are no competing interests to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Appendix S1
Supplementary references
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
REFERENCES
- 1.Pang KS, Rodrigues AD, Peter RM. Enzyme- and Transporter-Based Drug–Drug Interaction. 1st edn. New York: Springer; 2010. [Google Scholar]
- 2.Abarca J, Malone D, Armstrong E, Grizzle AJ, Hansten PD, Van Bergen RC, Lipton RB. Concordance of severity ratings provided in four drug interaction compendia. J Am Pharm Assoc. 2004;44:136–41. doi: 10.1331/154434504773062582. [DOI] [PubMed] [Google Scholar]
- 3.Fulda TR, Valuck RJ, Vander Zanden J, Parker S, Byrns PJ. Disagreement among drug compendia on inclusion and ratings of drug-drug interactions. Curr Ther Res. 2000;61:540–48. [Google Scholar]
- 4.Vitry AI. Comparative assessment of four drug interaction compendia. Br J Clin Pharmacol. 2007;63:709–14. doi: 10.1111/j.1365-2125.2006.02809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chao S, Maibach H. Lack of drug interaction conformity in commonly used drug-interaction compendia for selected at-risk dermatological drugs. Am J Clin Dermatol. 2005;6:105–11. doi: 10.2165/00128071-200506020-00005. [DOI] [PubMed] [Google Scholar]
- 6.Vasudev A, Harrison R. Prescribing safely in elderly psychiatric wards: survey of possible drug interactions. Psychiatr Bull. 2008;32:417–8. [Google Scholar]
- 7.Mallet L, Spinewine A, Huang A. The challenge of managing drug interactions in elderly people. Lancet. 2007;370:185–91. doi: 10.1016/S0140-6736(07)61092-7. [DOI] [PubMed] [Google Scholar]
- 8.Weingart SA, Toth M, Sands DZ, Aronson MD, Davis RB, Phillips RS. Physician's decisions to override computarized drug alerts in primary care. Arch Intern Med. 2003;163:2625–31. doi: 10.1001/archinte.163.21.2625. [DOI] [PubMed] [Google Scholar]
- 9.Isaac T, Weissman JS, Davis RB, Massagli M, Cyrulik A, Sands DZ, Weingart SN. Overrides of medication alerts in ambulatory care. Arch Intern Med. 2009;169:309–11. doi: 10.1001/archinternmed.2008.551. [DOI] [PubMed] [Google Scholar]
- 10.Taylor LK, Tamblyn R. Reasons for physician non-adherence to electronic drug alerts. Stud Health Technol Inform. 2004;107:1101–05. [PubMed] [Google Scholar]
- 11.Obach RS, Walsky RL, Venkatakrishnan K, Gaman EA, Houston JB, Tremaine LM. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug interactions. J Pharmacol Exp Ther. 2006;316:336–48. doi: 10.1124/jpet.105.093229. [DOI] [PubMed] [Google Scholar]
- 12.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan LS, Grimm SW, Kao J, King SP, Miwa G, Ni L, Kumar GN, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a pharmaceutical research and manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–32. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 13.US Food and Drug Administration. Guidance for Industry Drug Interaction Studies – Study Design, Data Analysis, and Implications for Dosing and Labeling. Draft Guidance. Rockville, MD: US Food and Drug Administration; 2006. [Google Scholar]
- 14.Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine; 2007. Available at http://medicine.iupui.edu/clinpharm/ddis/table.asp (last accessed 31 January 2010) [Google Scholar]
- 15.Li X, He Y, Ruiz CH, Koenig M, Cameron MD. Characterization of dasatanib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242–50. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Kamenecka TM, Cameron MD. Cytochrome P450-mediated bioactivation of the epidermal growth factor receptor inhibitor erlotinib to a reactive electrophile. Drug Metab Dispos. 2010;38:1237–45. doi: 10.1124/dmd.109.030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polasek TM, Miners JO. In vitro approaches to investigate mechanism-based inactivation of CYP enzymes. Expert Opin Drug Metab Toxicol. 2007;3:321–29. doi: 10.1517/17425255.3.3.321. [DOI] [PubMed] [Google Scholar]
- 18.Venkatakrishnan K, Obach RS. Drug-drug interactions via mechanism-based cytochrome P450 inactivation: points to consider for risk assessment from in vitro data and clinical pharmacologic evaluation. Curr Drug Metab. 2007;8:449–62. doi: 10.2174/138920007780866861. [DOI] [PubMed] [Google Scholar]
- 19.MIMS. Emims Version 5.01.0097. Sydney: CMPMedica Australia Pty Ltd; 2010. [Google Scholar]
- 20.Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. Selection of alternative CYP3A4 probe substrates for clinical drug interaction studies using in vitro data and in vivo simulation. Drug Metab Dispos. 2010;38:981–87. doi: 10.1124/dmd.110.032094. [DOI] [PubMed] [Google Scholar]
- 21.Ragueneau-Majlessi I, Boulenc X, Rauch C, Hachad H, Levy RH. Quantitative correlations among CYP3A sensitive substrates and inhibitors: literature analysis. Curr Drug Metab. 2007;8:810–4. doi: 10.2174/138920007782798135. [DOI] [PubMed] [Google Scholar]
- 22.Kivisto KT, Lamberg TS, Kantola T, Neuvonen PJ. Plasma buspirone concentrations are greatly increased by erythromycin and itraconazole. Clin Pharmacol Ther. 1997;62:348–54. doi: 10.1016/S0009-9236(97)90038-2. [DOI] [PubMed] [Google Scholar]
- 23.Olkkola KT, Aranko K, Luurila H, Hiller A, Saarnivaara L, Himberg JJ, Neuvonen PJ. A potentially hazardous interaction between erythromycin and midazolam. Clin Pharmacol Ther. 1993;53:298–305. doi: 10.1038/clpt.1993.25. [DOI] [PubMed] [Google Scholar]
- 24.Foti RS, Wahlstrom JL. CYP2C19 inhibition: the impact of substrate probe selection on in vitro inhibition profiles. Drug Metab Dispos. 2008;36:523–28. doi: 10.1124/dmd.107.019265. [DOI] [PubMed] [Google Scholar]
- 25.Kumar V, Wahlstrom JL, Rock DA, Warren CJ, Gorman LA, Tracy TS. CYP2C9 inhibition: impact of probe selection and pharmacogenetics on in vitro inhibition profiles. Drug Metab Dispos. 2006;34:1966–75. doi: 10.1124/dmd.106.010926. [DOI] [PubMed] [Google Scholar]
- 26.Miners JO, Wing LMH, Lillywhite KJ, Robson RA. Selectivity and dose-dependency of the inhibitory effect of propranolol on theophylline metabolism in man. Br J Clin Pharmacol. 1985;20:219–23. doi: 10.1111/j.1365-2125.1985.tb05064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herman D, Locatelli I, Grabnar I, Peternel P, Stegnar M, Lainscak M, Mrhar A, Breskvar K, Dolzan V. The influence of co-treatment with carbamazepine, amiodarone and statins on warfarin metabolism and maintenance dose. Eur J Clin Pharmacol. 2006;62:291–96. doi: 10.1007/s00228-006-0104-4. [DOI] [PubMed] [Google Scholar]
- 28.Nolan PE, Erstad BL, Hoyer GL, Bliss M, Gear K, Marcus FI. Steady-state interaction between amiodarone and phenytoin in normal subjects. Am J Cardiol. 1990;65:1252–57. doi: 10.1016/0002-9149(90)90983-8. [DOI] [PubMed] [Google Scholar]
- 29.O'Reilly RA, Trager WF, Rettie AE, Goulart DA. Interaction of amiodarone with racemic warfarin and its separated enantiomorphs in humans. Clin Pharmacol Ther. 1987;42:290–94. doi: 10.1038/clpt.1987.149. [DOI] [PubMed] [Google Scholar]
- 30.Szewczuk-Boguslawska M, Kiejna A, Beszlej JA, Orzechowska-Juzwenko K, Milejski P. Doxepin inhibits CYP2D6 activity in vivo. Pol J Pharmacol. 2004;56:491–94. [PubMed] [Google Scholar]
- 31.Gram LF, Brosen K. Moclobemide treatment causes a substantial rise in the sparteine metabolic ratio. Br J Clin Pharmacol. 1993;35:649–52. doi: 10.1111/j.1365-2125.1993.tb04196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Härtter S, Dingemanse J, Baier D, Ziegler G, Hiemke C. Inhibition of dextromethorphan metabolism by moclobemide. Psychopharmacology. 1998;135:22–6. doi: 10.1007/s002130050481. [DOI] [PubMed] [Google Scholar]
- 33.Polasek TM, Miners JO. Macrolide-theophylline interactions: no role for the inhibition of cytochrome P4501A2. Br J Clin Pharmacol. 2008;66:898–900. doi: 10.1111/j.1365-2125.2008.03299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birkett DJ, Miners JO. Methylxanthines. In: Levy RH, Thummel K, Trager WF, Hansten PD, Eichelbaum M, editors. Metabolic Drug Interactions. 1st edn. Philidelphia, PA: Lippincott Williams & Wilkins; 2000. pp. 469–82. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.