

Microglial lysosomes lack ClC-7, which leads to incomplete lysosomal acidification. In quiescent microglia, ClC-7 is targeted for proteasomal degradation apparently by an endoplasmic reticulum-associated degradation (ERAD) pathway. Microglial activation recruits ClC-7 to lysosomes and causes lysosomal acidification, which leads to efficient amyloid degradation.
Abstract
Incomplete lysosomal acidification in microglia inhibits the degradation of fibrillar forms of Alzheimer's amyloid β peptide (fAβ). Here we show that in primary microglia a chloride transporter, ClC-7, is not delivered efficiently to lysosomes, causing incomplete lysosomal acidification. ClC-7 protein is synthesized by microglia but it is mistargeted and appears to be degraded by an endoplasmic reticulum–associated degradation pathway. Activation of microglia with macrophage colony-stimulating factor induces trafficking of ClC-7 to lysosomes, leading to lysosomal acidification and increased fAβ degradation. ClC-7 associates with another protein, Ostm1, which plays an important role in its correct lysosomal targeting. Expression of both ClC-7 and Ostm1 is increased in activated microglia, which can account for the increased delivery of ClC-7 to lysosomes. Our findings suggest a novel mechanism of lysosomal pH regulation in activated microglia that is required for fAβ degradation.
INTRODUCTION
Microglia are the main immune cells of the brain (Nimmerjahn et al., 2005). Under nonpathological conditions, microglia are highly ramified cells. When activated by injury or insult to the brain, microglia proliferate, undergo morphological changes, and secrete chemokines and cytokines (Lucin and Wyss-Coray, 2009; Yong and Rivest, 2009).
Studies in human Alzheimer's disease (AD) samples and mouse models of AD demonstrate that microglia are recruited to amyloid β plaques (Meyer-Luehmann et al., 2008; Yong and Rivest, 2009). Despite this extensive microglial recruitment to the plaques, little clearance by microglia is generally observed, and the plaques remain stable for long periods of time (Bolmont et al., 2008). In contrast, robust plaque clearance by activated microglia was seen in mouse models of AD when microglia were activated either by immunotherapy or by treatment with proinflammatory reagents (Schenk et al., 1999; Monsonego and Weiner, 2003; Wilcock et al., 2004; Monsonego et al., 2006; Lucin and Wyss-Coray, 2009). Plaque clearance by activated microglia also induced significant improvements in behavioral deficits in mouse models of AD when analyzed by the radial-arm water maze task (Morgan et al., 2000).
In studies with primary mouse microglia it was found that microglia internalized fibrillar Alzheimer's amyloid β peptide (fAβ) and delivered it to lysosomes, but the fAβ was only partially digested (Paresce et al., 1997; Chung et al., 1999; Majumdar et al., 2008). Opsonization of fAβ with immunoglobulin (Ig)G or complement altered receptor binding of fAβ but did not increase the ability of microglia to degrade it in lysosomes (Brazil et al., 2000). Microglia that were activated by macrophage colony-stimulating factor (MCSF) or other inflammatory stimuli showed a significantly increased ability to degrade internalized fAβ (Majumdar et al., 2007). It was found that quiescent microglia maintain their lysosomes at an unusually high pH (∼6.0), but activation induced lysosomal acidification to approximately pH 5.0 (Majumdar et al., 2007). This further acidification of the lysosomes was required for degradation of fAβ. Because lysosomal acidification induced efficient degradation of fAβ, understanding the mechanism of lysosomal pH regulation in activated microglia became important.
In general, the regulation of lysosomal pH is complex and not fully understood. Acidification of lysosomes is mediated by the V-ATPase proton pump, which pumps protons into the lysosomal lumen in an ATP-dependent manner. Transport of the protons into the lysosomal lumen generates an inside-positive membrane potential, which limits further proton transport and inhibits lysosomal acidification (Pillay et al., 2002). Parallel ion transport pathways present in the lysosomes dissipate this membrane potential and allow full lysosomal acidification (Van Dyke, 1993; Pillay et al., 2002). The nature of these parallel ion transport pathways is unclear and is an area of active research. Studies done with isolated rat liver lysosomes or with lysosomes from HeLa cells indicate that anion transport, mainly chloride ion influx, constitutes the parallel ion transport (Ohkuma et al., 1982; Graves et al., 2008), whereas recent studies of RAW cell lysosomes demonstrate that efflux of cations instead of (or along with) anion influx constitutes the major parallel ion transport pathway (Steinberg et al., 2010). Thus the nature of the parallel ion transport pathways required for lysosomal acidification remains poorly defined, and various mechanisms may operate at different levels in specific cell types to achieve a steady-state lysosomal pH.
In a previous study we provided evidence that anion transport into lysosomes may be important for lysosomal pH regulation in activated microglia (Majumdar et al., 2007). Chloride channels and transporters have been proposed to be the mediators of this anion transport pathway, and they are known to be important for the acidification of endosomes and lysosomes (Bae and Verkman, 1990). ClC-7 is the primary chloride transporter in lysosomes (Graves et al., 2008; Weinert et al., 2010). In several cell types, ClC-7 has been shown to be a lysosomal membrane protein (Jentsch, 2007; Weinert et al., 2010) that plays an essential role in lysosomal function (Wartosch et al., 2009). Loss of ClC-7 causes osteopetrosis, which is associated with improper acidification of an extracellular bone-remodeling compartment formed by osteoclasts (Kornak et al., 2001; Lange et al., 2006). ClC-7 forms a heterodimeric complex with another membrane protein, Ostm1, and formation of this complex is important for the folding, stability, and proper trafficking of ClC-7 (Lange et al., 2006). Loss of Ostm1 also causes osteopetrosis (Chalhoub et al., 2003), and in a ClC-7−/− mouse Ostm1 is absent from the lysosomes (Lange et al., 2006).
Herein we explore the role of ClC-7 in regulating the pH in the lysosomes of microglia. We show that quiescent microglia express the ClC-7 protein, but ClC-7 is not delivered efficiently to lysosomes. Instead, the ClC-7 protein in microglia is mistargeted and appears to be degraded by the endoplasmic reticulum–associated degradation (ERAD) pathway. Quiescent microglia express only low levels of Ostm1, and that impairs the lysosomal trafficking of ClC-7 protein. Activation of microglia by MCSF increases the transcription of both ClC-7 and Ostm1. The Ostm1/ClC-7 interaction allows proper lysosomal trafficking of ClC-7, leading to complete lysosomal acidification and degradation of fAβ.
RESULTS
ClC-7 in primary mouse microglia undergoes degradation by the ERAD pathway
Because the lysosomal localization of ClC-7 is reported in other cell types to be important for controlling lysosomal pH, we examined the localization of ClC-7 in microglia. As a lysosomal marker, we used fAβ, which is delivered to lysosomes after binding to type A scavenger receptors (SRAs) on microglia (Paresce et al., 1996). Using a relative colocalization index (described in Supplemental Information), ∼85% of internalized fAβ is found in lysosomes containing lysosomal-associated membrane protein 1 (LAMP-1) (Supplemental Figure S1, G–I). We found that most of the ClC-7 in microglia did not colocalize with fAβ in lysosomes (Figure 1, A–C). When the relative colocalization index was used to quantify the extent of localization of ClC-7 to the fAβ-positive lysosomes, it was found that only 20% of the ClC-7 protein localized to the lysosomes. Unlike microglia, in J774 macrophages and in U2OS-SRA cells (an osteosarcoma cell line transfected with SRA) more than 70% of the ClC-7 localized to the fAβ-containing lysosomes (Supplemental Figure S1, A–F). In microglia, most of the ClC-7 protein was found to be concentrated in a perinuclear region.
FIGURE 1:

ClC-7 in primary mouse microglia is localized in the perinuclear region. (A–C) Immunolocalization of ClC-7 (green) as compared with Cy3fAβ (red) that had been endocytosed and delivered to lysosomes. The image is a single slice from a confocal stack. Bar: 5 μm. (D) Changes in the amount of ClC-7 in primary mouse microglia after proteasome inhibition by MG-132 treatment. Microglial cells were treated with 5 μM or 10 μM MG-132 for 12 h. Cells were fixed, and total ClC-7 was quantified by immunofluorescence. Error bars represent SEM, and p values were obtained using Student's t test (two tailed). (E and F) Immunolocalization of ClC-7 (green) in primary mouse microglia after MG-132 treatment. Microglial cells were treated with 5 or 10 μM MG-132 for 12 h. The cell boundaries have been outlined. Scale bars: 3 μm. (G–I) Immunolocalization of ClC-7 (green) and vimentin (red) in primary mouse microglia. The image represents a single slice from a confocal stack. Bar: 5 μm.
The perinuclear ClC-7 in microglia does not colocalize well with markers for ER, Golgi, or lysosomes (Supplemental Figure S2). The characteristics of the ClC-7 distribution resemble distributions of misfolded membrane proteins, such as (Δ-508) cystic fibrosis transmembrane conductance regulator (CFTR) (Johnston et al., 1998), which are targeted for proteasomal degradation by the ERAD pathway. For multimeric proteins, lack of proper subunit oligomerization and quaternary structure formation is the main reason for ERAD-mediated turnover (Ellgaard et al., 1999; Vembar and Brodsky, 2008). To test whether ClC-7 in primary microglia is degraded by proteasomes, we incubated microglial cells with MG-132, a proteasome inhibitor (Lee et al., 2003), for 12 h. The amount of ClC-7 in the cells increased by ∼50% when proteasomal degradation was inhibited (Figure 1, D–F).
Collections of proteins that are being degraded by the ERAD pathway are often surrounded by a vimentin cage (Johnston et al., 1998), and the perinuclear localization of these proteins requires microtubules (Johnston et al., 1998). The perinuclear ClC-7 colocalized to a large degree with vimentin (Figure 1, G–I), γ-tubulin (unpublished data), and 20S proteasomal subunits (unpublished data). We found that nocodazole treatment (20 μM for 30 min) reduced the extent of the perinuclear ClC-7 localization, and the protein was found in smaller structures throughout the cell (Supplemental Figure S3, A–F). Detergent solubility of the ClC-7 protein was tested, and most of the ClC-7 protein in microglia was detergent insoluble and was found in the pellet fraction (Supplemental Figure S3G). Polyubiquitinylation of ClC-7 was also tested; although ClC-7 was enriched in the same part of the cell as polyubiquitinated proteins (Supplemental Figure S4, A–C), ClC-7 itself was not polyubiquitinated (unpublished data). It is clear that in quiescent primary mouse microglia ClC-7 does not traffic to lysosomes efficiently. The perinuclear accumulation resembles early aggresomes (Johnston et al., 1998), but further work would be required to precisely identify the nature of these accumulations, which appear to be associated with an ERAD pathway.
Activation of primary mouse microglia with MCSF recruits ClC-7 to the lysosomes and leads to lysosomal acidification
We have shown previously that activation of microglia with MCSF induces both lysosomal acidification and degradation of internalized fAβ (Majumdar et al., 2007). We treated cells with MCSF (25 ng/ml) for 3 d (Treatment I) or for 18 d (Treatment II) and examined the effect on ClC-7 localization. MCSF treatment, especially Treatment II, activated microglia (Majumdar et al., 2007), and they started to proliferate. MCSF Treatment II also changed the morphology of the cells; they became flat and formed ruffles on the cell surface. Activation of microglia with MCSF decreased the amount of ClC-7 in the perinuclear region and increased the lysosomal targeting of ClC-7 (Figure 2, A–F). Compared to unactivated primary microglia, the longer MCSF exposure (Treatment II) induced extensive lysosomal recruitment of ClC-7, whereas MCSF Treatment I induced partial recruitment of ClC-7 to the lysosomes. Quantification of the extent of localization of ClC-7 to the fAβ-containing lysosomes by the relative colocalization index showed that after MCSF Treatment II 75% of the total ClC-7 was recruited to the lysosomes, whereas 40% ClC-7 lysosomal recruitment was seen after Treatment I. Both treatments induced significantly higher ClC-7 lysosomal recruitment as compared with the 20% that was observed with unactivated primary microglia. The total amount of ClC-7 in the cells also increased with MCSF Treatment II (see Figure 4A later in the paper).
FIGURE 2:

Activation of primary mouse microglia with MCSF recruits ClC-7 to lysosomes and leads to lysosomal acidification and fAβ degradation. (A–C) Immunolocalization of ClC-7 (green) as compared with Cy3fAβ (red) that had been endocytosed and delivered to lysosomes in cells subject to MCSF Treatment I. (D–F) Immunolocalization of ClC-7 (green) as compared with Cy3fAβ (red) that had been endocytosed and delivered to lysosomes in cells subject to MCSF Treatment II. The images are single slices from a confocal stack. Bar: 5 μm. (G) Frequency distribution of lysosomal pH in microglia receiving MCSF Treatment II (green curve), MCSF Treatment I (blue curve), or control microglia (red curve). The values plotted are the fraction of lysosomes with pH values in the interval within ±0.25 pH units of the indicated value (e.g., 4.75–5.25 for pH 5.0). Error bars represent SEM. The horizontal bar on the figure shows the SD for the measurement of pH values of single lysosomes in cells fixed at pH 5.5. We analyzed the distribution of fluorescein/rhodamine fluorescence ratio values in (fluorescein-rhodamine)-dextran-loaded lysosomes to test for significance in the differences in the pH distributions. The distributions for both MCSF-treated cells differed from the distribution in control cells (p < 0.0001 for MCSF II and p < 0.001 for MCSF I treatments). (H) Degradation of Cy3fAβ by microglial cells receiving MCSF Treatment I or II. Cy3 fluorescence retained inside the cells 72 h after a 1-h uptake of Cy3fAβ is shown. Error bars represent SEM, and p values are obtained using Student's t test (two tailed).
FIGURE 4:

Expression of ClC-7 and Ostm1 in MCSF-treated primary mouse microglia and degradation of fAβ in primary mouse microglia after overexpression of Ostm1. (Aa) Expression of ClC-7 protein in control and MCSF Treatment II primary mouse microglia analyzed by immunoblot. (Ab) Expression of Ostm1 protein (lysosome resident form, 40 kDa) in control and MCSF Treatment II primary mouse microglia analyzed by immunoblot. (B) Relative changes in the expression of ClC-7 and Ostm1 mRNA in primary mouse microglia after MCSF Treatment II as compared with control quiescent microglia. The fold change in mRNA levels were measured by qRT-PCR using the ΔΔCT method. Error bars represent SEM.
The MCSF treatments induced a graded decrease in the lysosomal pH, with cells exposed to Treatment II having an average lysosomal pH of 5.0 (Figure 2G) as reported previously (Majumdar et al., 2007). The cells exposed to Treatment I had a broader range of lysosomal pH values, but their lysosomes were more acidic (average pH 5.5) than in untreated cells (average pH 5.9). In our previous study, we showed that lysosomal acidification was associated with increased degradation of internalized fAβ (Majumdar et al., 2007). Consistent with this finding, the two treatments with MCSF showed a graded increase in the ability of the cells to degrade fAβ (Figure 2H).
Because MCSF treatment induced lysosomal acidification along with increased ClC-7 lysosomal recruitment, we decided to monitor the counterion permeability in the lysosomes of microglia after MCSF treatment. For this purpose we used an assay (Lukacs et al., 1991; Barriere et al., 2009) that is based on the assumption that the rate of dissipation of a lysosomal pH gradient by the addition of a protonophore (20 μM CCCP [carbonyl cyanide m-chlorophenylhydrazone]) is rate-limited by the lysosomal counterion conductance in the presence of the V-ATPase inhibitor, bafilomycin A1. As shown in Supplemental Figure S5, we found that, in the presence of bafilomycin A1, the lysosomal pH gradient dissipated at a much faster rate after CCCP addition in MCSF II–treated microglia than in quiescent microglia. This result indicated that lysosomes of MCSF-treated microglia had a higher counterion conductance, which would be consistent with increased recruitment of ClC-7 to lysosomes upon MCSF treatment.
Knockdown of ClC-7 by siRNA blocks lysosomal acidification and fAβ degradation in activated primary mouse microglia
To test whether ClC-7 is required for the MCSF-dependent acidification of lysosomes, we used siRNA to knock down ClC-7 expression in primary mouse microglia. Cells that had been activated by MCSF Treatment II were extremely difficult to transfect, so we transfected unactivated microglia twice within 24 h with a 100 nM siRNA pool against ClC-7 (described in Materials and Methods) and then activated them with MCSF Treatment I. The extent of knockdown of ClC-7 mRNA and protein was monitored 72 h after the second siRNA transfection (Supplemental Figure S6), and these treatments caused an approximately 60% reduction in the expression of ClC-7 protein. We found that there was ∼70% inhibition of fAβ degradation in the ClC-7 siRNA–transfected cells compared with cells transfected with nontargeted siRNA (Figure 3A). This finding indicated that partial loss of ClC-7 expression reduces fAβ degradation in activated microglia. This impaired lysosomal degradation was accompanied by reduced acidification in the lysosomes of microglia transfected with ClC-7 siRNA (Figure 3B). These results establish ClC-7 as a regulator of lysosomal pH in microglia.
FIGURE 3:
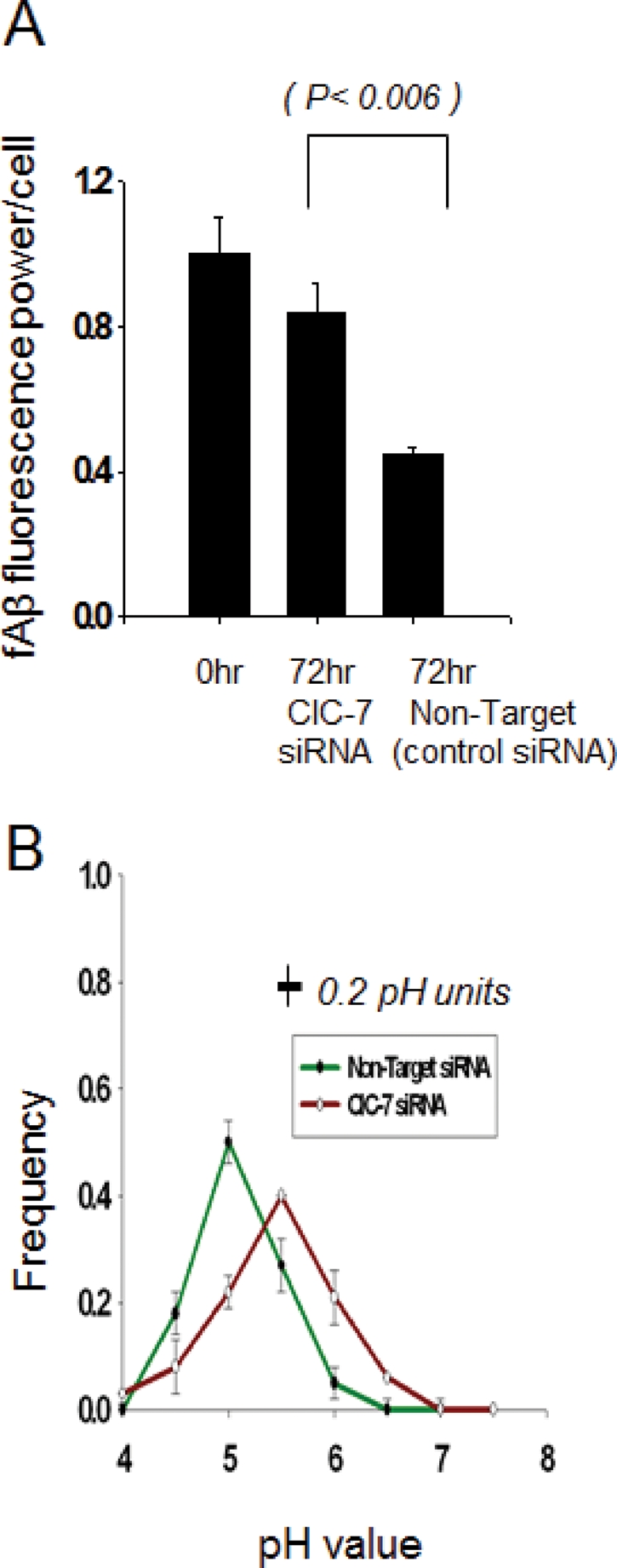
Degradation of Cy3fAβ and lysosomal acidification in MCSF I–treated primary mouse microglia after ClC-7 knockdown. (A) Degradation of Cy3fAβ by MCSF I–treated primary microglia after control and ClC-7 siRNA treatment. Cy3 fluorescence retained inside the cells 72 h after a 1-h uptake of Cy3fAβ. Error bars represent SEM, and p values are obtained using Student's t test (two tailed). (B) Frequency distribution of lysosomal pH in MCSF I–treated primary microglial cells treated with nontarget control siRNA (green) or siRNA against ClC-7 (orange). The values plotted are the fraction of lysosomes with pH values in the interval within ±0.25 pH units of the indicated value (e.g., 4.75–5.25 for pH 5.0). Error bars represent the SEM. The horizontal bar on the figure shows the SD for the measurement of pH values of individual lysosomes in cells fixed at pH 5.5. We analyzed the distribution of fluorescein/rhodamine fluorescence ratio values in (fluorescein-rhodamine)-dextran-loaded lysosomes to test for significance in the differences in the pH distributions. The distributions differed significantly (p < 0.001).
As a control for off-target effects, we transfected microglia twice within 24 h with two additional siRNAs (siRNAs 6 and 7) against ClC-7 (10 nM) and then monitored Cy3fAβ degradation in these cells after MCSF Treatment I. We found that, similar to our siRNA pool transfection experiments, individual siRNA transfections also led to a partial block in Cy3fAβ degradation (Supplemental Figure S7). We also monitored the level of ClC-7 protein by immunofluorescence in these siRNA-transfected cells and found that there was an approximately 40% decrease in the level of ClC-7 protein after the siRNA transfections (Supplemental Figure S7).
Overexpression of Ostm1 recruits ClC-7 to lysosomes and increases fAβ degradation
ClC-7 requires a partner protein, Ostm1, for proper folding and exit from the ER (Lange et al., 2006; Jentsch, 2008). Ostm1 is a lysosomal membrane protein, and in Western blots it migrates as a doublet around 40 kDa (Chalhoub et al., 2003; Lange et al., 2006). The lysosomal form of Ostm1 is derived from a precursor form that migrates around 80 kDa (Lange et al., 2006; Jentsch, 2008). When compared with J774 macrophages and U2OS-SRA cells, primary mouse microglia expressed much less Ostm1 in both the mature and precursor forms (Supplemental Figure S8A). To explore the role played by Ostm1 in the trafficking of ClC-7 in microglia, we measured the levels of Ostm1 protein and mRNA in MCSF-activated (Treatment II) and quiescent microglia. We found that activation of microglia by MCSF treatment increased the expression of the lysosome resident form of the Ostm1 protein (Figure 4, Ab). We used quantitative reverse transcription-polymerase chain reaction (qRT-PCR) to quantify the levels of ClC-7 and Ostm1 mRNA in MCSF II–treated microglial cells and compared them with the mRNA levels in control quiescent microglia. As shown in Figure 4B, we found that after MCSF II treatment there was a 12-fold increase in the level of ClC-7 mRNA and a fourfold increase in the level of Ostm1 mRNA with respect to the control.
To test whether increased expression of Ostm1 in microglia leads to increased delivery of ClC-7 to lysosomes, we transfected Ostm1 cDNA into quiescent primary mouse microglia. Transfection procedures that yielded high efficiency of transfection partially activated the microglia, and they started to partially degrade fAβ. Using a protocol (Coull et al., 2005) that gave ∼40% transfection efficiency, we transfected microglia with a plasmid encoding V5 epitope-tagged Ostm1. Seventy-two hours after the transfection, we monitored Ostm1 expression in microglia by immunofluorescence by using an anti-V5 antibody. V5-tagged Ostm1 was expressed in microglia and was partially recruited to the lysosomes that contained fAβ (Supplemental Figure S8C).
Ostm1 overexpression significantly increased the number of cells that showed recruitment of ClC-7 to lysosomes (Figure 5A). Overexpression of Ds-Red had no effect, and ClC-7 remained associated with perinuclear structures (Supplemental Figure S8D). To further substantiate this result, we coexpressed Ostm1 and green fluorescent protein (GFP) in microglia using an internal ribosome entry site (IRES) GFP expression system and monitored the colocalization of ClC-7 with the fAβ-containing lysosomes in the GFP-expressing cells. We found that the Ostm1-expressing cells (selected on the basis of GFP expression) clearly showed more colocalization between ClC-7 and Cy5fAβ (Figure 5B). Using a relative colocalization index, we quantified the ClC-7 recruitment to the lysosomes, and Ostm1 overexpression increased the ClC-7 recruitment to the fAβ-containing lysosomes (Figure 5C).
FIGURE 5:

Localization of ClC-7 after Ostm1 overexpression. (A) Immunolocalization of ClC-7 (green) and LAMP-1 (red) in primary microglia overexpressing V5-Ostm1. (Aa–Ac) Immunolocalization in mock transfected cells and (Ad–Af) immunolocalization in V5-Ostm1–transfected cells. The images are single slices from a confocal stack. Scale bars: 5 μm. (B) Immunolocalization of ClC-7 (green) and Cy5fAβ (red) in primary mouse microglial cells coexpressing Ostm1 and GFP from an IRES-GFP vector. (Ba–Bd) Immunolocalization in mock transfected cells and (Be–Bh) immunolocalization in pIRES-GFP-Ostm1–transfected cells. The images are single slices from a confocal stack. Scale bars: 5 μm. (C) Quantification of the colocalization between ClC-7 and Cy5fAβ after the coexpression of Ostm1 and GFP using an IRES-GFP expression system. The figure shows the relative colocalization index. Error bars represent the SEM, and the p value was obtained by performing a Student's t test (two tailed). (D) Degradation of Cy3fAβ by primary microglia after V5-Ostm1 overexpression. Cy3 fluorescence retained inside the cells 72 h after a 1-h incubation with Cy3fAβ is shown. Error bars represent SEM, and the p values are obtained using Student's t test (two tailed).
We also examined the effect of overexpression of the V5-tagged Ostm1 on fAβ degradation. After 24 h, the cells were loaded with Cy3fAβ, and the degradation of fAβ was monitored (Figure 5D). Ostm1-transfected cells degraded Cy3fAβ more effectively than did untransfected cells or enhanced GFP-transfected cells.
Knockdown of Ostm1 expression by siRNA blocks fAβ degradation and the lysosomal recruitment of ClC-7
Overexpression of Ostm1 protein in primary mouse microglia induced both fAβ degradation and the lysosomal trafficking of ClC-7. To further demonstrate the role of Ostm1 in regulating ClC-7 lysosomal recruitment, we transfected primary microglial cells twice with 10 nM siRNA 1 against Ostm1. Seventy-two hours after the second siRNA transfection, an ∼40% reduction of Ostm1 protein was achieved (Supplemental Figure S9B). We applied MCSF Treatment I to these Ostm1 siRNA–treated cells and used a relative colocalization index to monitor the lysosomal recruitment of ClC-7. As shown in Figure 6, A–B, knockdown of Ostm1 reduced the lysosomal recruitment of ClC-7 in MCSF-treated microglia, indicating that Ostm1 was necessary for lysosomal targeting of ClC-7. Because the recruitment of ClC-7 to lysosomes correlated with Cy3fAβ degradation, we monitored the degradation of Cy3fAβ in MCSF-treated microglia. Knockdown of Ostm1 protein reduced fAβ degradation in microglia that received MCSF Treatment I (Figure 6C). We transfected microglia with two additional siRNAs (siRNAs 2 and 3) against Ostm1 (10 nM) as a control for off-target effects. As shown in Supplemental Figure S10, we found that degradation of Cy3fAβ after MCSF treatment was blocked in the microglial cells that were transfected with these siRNA. These siRNA transfections also decreased the levels of Ostm1 mRNA and Ostm1 protein as analyzed by qRT-PCR and immunoblot, respectively (Supplemental Figure S10).
FIGURE 6:

Knockdown of Ostm1 blocks lysosomal recruitment of ClC-7 and fAβ degradation in MCSF-treated primary mouse microglia. (A) Immunolocalization of ClC-7 (green) and Cy3fAβ (red) in MCSF I–treated primary mouse microglia after Ostm1 and control nontarget siRNA transfection. (Aa–Ac) Immunolocalization in Ostm1 siRNA–treated cells. (Ad–Af) Immunolocalization in control nontarget siRNA–treated cells. The images are single slices from a confocal stack. Scale bars: 5 μm. (B) Quantification of the colocalization between ClC-7 and Cy3fAβ in MCSF I–treated primary mouse microglia after Ostm1 and control nontarget siRNA transfection. The relative colocalization index is shown. Error bars represent SEM, and the p value was obtained by Student's t test (two tailed). (C) Degradation of Cy3fAβ in MCSF I–treated primary microglia after Ostm1 and control nontarget siRNA transfection. Cy3 fluorescence retained in the cells 72 h after a 1-h incubation with Cy3fAβ is shown. Error bars represent SEM, and p values were obtained by Student's t test (two tailed). (D) Interaction between Ostm1 and ClC-7 monitored by coimmunoprecipitation. ClC-7 was immunoprecipitated from 50 μg of total protein lysate with 10 μg of antibody and 20 μl of protein A/G-Agarose (Pierce immunoprecipitation kit). Immunocomplexes were recovered, washed, and subjected to SDS–PAGE. The gels were transferred to polyvinylidene fluoride membranes and subjected to LI-COR infrared imaging followed by Western blotting with Ostm1 antibody. Tubulin was used as a loading control. The ratio of Ostm1 to ClC-7 was quantified by using the LI-COR infrared fluorescence imaging system, and the data presented are the average value from two different experiments, done on different days.
Increased interaction of ClC-7 with Ostm1 upon MCSF treatment
We explored further how MCSF treatment helps in the lysosomal delivery of ClC-7. Studies using mouse brain lysates have shown that Ostm1 interacts with ClC-7 and that this interaction is important for ER exit and lysosomal recruitment of ClC-7 (Lange et al., 2006). We monitored ClC-7/Ostm1 interaction in control and MCSF II–treated primary microglia using immunoprecipitation of ClC-7 followed by Western blotting for Ostm1. MCSF treatment increased ClC-7/Ostm1 interaction (Figure 6D), which can account for the increased lysosomal recruitment of ClC-7.
Localization of ClC-7 in microglia from Tg19959 AD mouse
Our data from primary mouse microglia show that inefficient recruitment of ClC-7 to the lysosomes leads to a block in fAβ degradation. Because the rate of fAβ degradation may be important in AD pathology (Bacskai et al., 2001), we monitored the localization of ClC-7 in microglia in a mouse model of AD, the Tg19959 mouse (Li et al., 2004). The Tg19959 mouse expresses a doubly mutated version of human amyloid precursor protein (APP), and it harbors the KM670/671NL (Swedish) and the V717F (Indiana) familial AD mutations. This mouse produces higher levels of Aβ peptide, and it starts to form plaques at the age of 2–3 mo. For our experiments, we selected plaques in tissue sections based on UV autofluorescence (Thal et al., 2002) and used CD11b staining to identify microglia around the plaques. When ClC-7 localization was observed in these microglia, it was found that ClC-7 staining was not punctate but showed a diffuse staining pattern (Figure 7, A–C). Unlike microglia, other cells around the plaques showed a punctate ClC-7 staining pattern. We imaged more than 20 dense core plaques from three different Tg19959 mice and saw that at least 70% of the CD11b-positive microglia around the plaques had a diffuse ClC-7 staining pattern. As a control we stained neurons in the CA1 region and found that the ClC-7 protein had a punctate staining pattern (Supplemental Figure S11A) similar to previous results (Kasper et al., 2005).
FIGURE 7:

Localization of ClC-7 in microglia in Tg19959 AD mouse brain. (A–C) Immunolocalization of ClC-7 (green) in CD11b (red)-labeled microglial cells around dense core plaques in a brain section from a Tg19959 mouse. The image is a single slice from a confocal stack. Bar: 25 μm. (D) Histogram showing the number of CD11b-positive microglial cells that were scored on the basis of punctate vs. diffuse staining pattern for ClC-7 and LAMP-1 immunofluorescence. (E–G) Sum projection images of ClC-7 staining in CD11b-labeled microglial cells from brain sections of the Tg19959 mouse. The images represent a range of distributions from a typical diffuse cell (scored as –2) to a cell with a mixture of diffuse and punctate staining (scored as 0). (H and I) Sum projection images of LAMP-1 staining in CD11b-labeled microglial cells from brain slices of a Tg19959 mouse. The images show a partially punctate cell (scored as +1) and a highly punctate cell (scored as +2).
To further explore the localization of ClC-7 in the microglial cells around the plaques, we imaged the microglial cells at a higher magnification so that individual CD11b-positive microglial cells were clearly identifiable (Supplemental Figure S11, B and D). Our goal was to determine whether the ClC-7 staining resembled a punctate, lysosomal distribution. As a control we also imaged LAMP-1 staining in CD11b-positive microglial cells. We provided the images to blinded investigators and asked them to compare the ClC-7 and LAMP-1 staining in microglia, following methods described in the Supplementary Information. As shown in Figure 7D, we found that ClC-7 staining in microglial cells was more diffuse than was the LAMP-1 staining pattern. This finding indicated that ClC-7 is not recruited efficiently to the lysosomes in microglial cells around plaques.
DISCUSSION
Incomplete acidification of lysosomes of microglia leads to limited degradation of internalized fAβ by these cells (Majumdar et al., 2007). In this article, we show that ClC-7 is inefficiently targeted to lysosomes in quiescent primary microglia. Instead, most ClC-7 in microglia is subject to degradation, apparently by an ERAD pathway. Transport of Cl− ions can be important for the regulation of lysosomal pH (Ohkuma et al., 1982; Bae and Verkman, 1990) because it helps to dissipate the inside-positive electrical potential that is generated by the electrogenic V-ATPase (Pillay et al., 2002; Graves et al., 2008). ClC-7 is the main Cl− transporter in lysosomes (Graves et al., 2008), and evidence presented here indicates that its low levels in lysosomes of quiescent microglia is associated with impaired lysosomal acidification.
As noted in the Introduction, our understanding of ion transport across the lysosomal membrane is incomplete, and other ion transport processes such as cation efflux have also been proposed to play an important role in regulating lysosomal pH (Steinberg et al., 2010). To examine the importance of counterion transport in facilitating acidification of microglial lysosomes, we used an experimental protocol in which the V-ATPase is inhibited by bafilomycin A1, and changes in lysosomal pH are then monitored when the proton-selective carrier CCCP is added to the cells (Lukacs et al., 1991). It is expected that the lysosomal pH should rise more quickly if there is sufficient counterion transport activity to balance the electrogenic efflux of protons mediated by CCCP. We found that addition of CCCP to quiescent microglia produced no measurable lysosome alkalinization in bafilomycin A1–treated quiescent microglia, but lysosome alkalinization was observed in the MCSF-activated microglia (Supplemental Figure S5). This result indicates that the lysosomes in MCSF-treated cells have greater counterion conduction than do lysosomes in quiescent microglia. The presence of ClC-7 could provide such an increase in counterion conductance, but other mechanisms could also contribute.
It is interesting to note that the lysosomal pH in microglia rose by ∼0.7 pH units in 2 min when the cells were treated with bafilomycin A1. This rise was observed in both quiescent and MCSF-treated microglia (Supplemental Figure S5), and it indicates that there is a significant proton leak in the lysosomal membrane in microglia. By comparison, phagosomes in thioglycolate-elicited mouse macrophages alkalinized at a rate of 0.09 pH units per minute (Lukacs et al., 1991), and lysosomes in A431 cells alkalinized by 1 pH unit in 30–40 min (Yoshimori et al., 1991). The source of the proton leak will require further investigation. A further complicating factor in understanding lysosomal pH regulation in microglia is that signal transduction pathways can alter ion conductance; we found that acidification of lysosomes in MCSF-treated microglia is reduced by inhibition of protein kinase A (Majumdar et al., 2007).
Although the mechanisms of lysosomal pH regulation are not fully understood, it is clear that activated microglia have more acidic lysosomes than do quiescent microglia (Majumdar et al., 2007). Furthermore, this increased lysosomal acidification correlates strongly with the extent of ClC-7 recruitment to the lysosomes. Additionally, siRNA knockdown of ClC-7 in microglia reduces lysosomal acidification in MCSF-activated microglia. Taken together, these results establish ClC-7 as an important regulator of lysosomal acidification in microglia.
We investigated the mechanism behind the inefficient lysosomal delivery of ClC-7 in quiescent microglia. Ostm1 is the β subunit of ClC-7 (Lange et al., 2006), and it has been shown in other cells to be important for the ER exit and lysosomal trafficking of ClC-7 (Lange et al., 2006). We found that Ostm1 plays a critical role in lysosomal trafficking of ClC-7 in microglia. Quiescent microglia express a low level of Ostm1 protein, and the absence of adequate Ostm1 is associated with inefficient delivery of ClC-7 to lysosomes.
The degradation of ClC-7 is in many ways similar to ERAD-mediated turnover of other multisubunit membrane proteins, notably the α subunit of the heptameric T-cell receptor (TCR) complex (Lippincott-Schwartz et al., 1988; Vembar and Brodsky, 2008). ERAD-mediated turnover of αTCR is mainly seen in immature T-cells, where synthesis of certain subunits of TCR is blocked. When T-cells mature, synthesis of all seven subunits of the TCR complex is initiated, which induces subunit interaction and rescues αTCR from ERAD-mediated degradation (Bonifacino et al., 1989).
In microglia, MCSF treatment increases the expression of both ClC-7 and Ostm1. Higher concentration of these two proteins enhances Ostm1/ClC-7 interaction and complex formation. Increased Ostm1/ClC-7 interaction would be expected to improve ER exit and lysosomal delivery of ClC-7. Interestingly, ClC-7 in primary microglia that is targeted for degradation becomes concentrated in the perinuclear region in a pattern similar to structures called aggresomes, which are cellular inclusions formed by misfolded proteins such as the cystic fibrosis mutant (Δ-508) CFTR (Kopito and Sitia, 2000). The perinuclear accumulations of ClC-7 are not as compact as a mature aggresome formed by the cystic fibrosis mutant (Δ-508) CFTR (Kopito and Sitia, 2000), but they appear similar to earlier stages of aggresome formation. Further work would be required to understand the detailed mechanisms for perinuclear segregation and degradation of the ClC-7 in quiescent microglia.
To demonstrate the relevance of our findings with respect to AD pathology, we observed ClC-7 localization in microglia surrounding Aβ plaques in tissue sections from a mouse model of AD. Most of the microglial cells around Aβ plaques have a diffuse staining pattern for ClC-7, which is unlike the punctate lysosomal staining pattern for ClC-7 seen in neurons. LAMP-1 staining for lysosomes showed a typical punctate pattern in the microglia that showed that ClC-7 in microglia in an AD mouse was not properly targeted to the lysosomes, similar to the observations in primary mouse microglia. Whereas ClC-7 in cultured primary microglia shows a perinuclear staining pattern, in adult microglia in brain tissue the ClC-7 distribution is diffuse and resembles an ER distribution. We do not understand the reason for this difference, but in both cases little ClC-7 gets to the lysosomes. Our finding that ClC-7 in microglia is not punctate is consistent with a previous observation of ClC-7 distribution in microglia in mouse brain slices (Wartosch et al., 2009).
Inefficient plaque degradation by microglia can explain the paradox of plaque persistence even with intense microglial recruitment (Bolmont et al., 2008). Studies in the (APPswe)/PS1 mouse models of AD have shown that treatment with MCSF leads to improvement in cognitive functions as analyzed by the water T-maze task (Boissonneault et al., 2009). Improvement of cognitive functions upon MCSF treatment was observed in mice that were at early and at late stages of the AD pathology (Boissonneault et al., 2009). Mice that showed improved cognitive functions after the MCSF treatment had a lower plaque burden and also demonstrated efficient plaque clearance by activated microglia (Boissonneault et al., 2009). Plaque clearance by activated microglia after MCSF treatment in the AD mouse model was due to the enhanced lysosomal degradation of amyloid β aggregates (Boissonneault et al., 2009), which correlates well with our studies with primary mouse microglia. This correlation suggests that the mechanism of pH regulation that we have described is likely to be relevant in vivo.
The data presented herein show that in microglia the delivery of ClC-7 to lysosomes affects lysosomal acidification. Studies with neurons, macrophages, and osteoclasts from the ClC-7−/− mice, however, show that lysosomes can acidify to a pH near 5.0 by a mechanism that is independent of ClC-7 (Kornak et al., 2001; Kasper et al., 2005; Steinberg et al., 2010). The mechanisms regulating lysosomal acidification are complex and not fully understood. Different cell types might use different mechanisms to maintain a steady-state value of the lysosomal pH. One interesting point is that the steady-state value of the lysosomal pH in quiescent microglia is different from that of neurons, macrophages, or osteoclasts. Whereas these other cell types all have a lysosomal pH close to 5.0, quiescent microglia maintain their steady-state pH at 5.9. Furthermore, there is no evidence that these other cell types alter lysosomal pH in response to physiological stimuli. Thus it appears that the mechanisms of lysosomal pH regulation can vary among different cell types.
It is not clear what purpose is served by the activation-dependent acidification of microglial lysosomes. Dendritic cells also acidify their lysosomes upon activation as part of the process leading to antigen presentation (Trombetta et al., 2003). There is evidence that microglia, like dendritic cells, can engage in antigen presentation in the lymph nodes (Hochmeister et al., 2008), so control of pH in the lysosomes of microglia might also be associated with their role in antigen processing and presentation.
The failure of ClC-7 to traffic to the lysosomes is the main cause of the reduced lysosomal acidification in microglia. Increased lysosomal acidification and consequent fAβ degradation can be achieved if ClC-7 is properly assembled and targeted to the lysosomes. Our data provide a model (Figure 8) for the regulation of lysosomal pH in microglia during activation. We show that activation of microglia induces efficient ClC-7/Ostm1 complex formation, which leads to increased lysosomal acidification through the recruitment of ClC-7 to the lysosomes. These findings describe a novel mechanism for lysosomal pH regulation and also open up a paradigm for generating new therapeutic strategies for AD.
FIGURE 8:

Model for lysosomal pH regulation in microglia. Model for the regulation of lysosomal pH in activated microglia. (A) In unactivated microglia ClC-7 is degraded by a pathway involving proteasomes that appears similar to ERAD quality control mechanisms in other systems. This degradation may be due to poor assembly of ClC-7/Ostm1 heterodimers in the ER. (B) In an activated microglial cell, the Ostm1 protein is associated with ClC-7. This complex leads to an increase in ClC-7's proper assembly and efficient lysosomal targeting. The presence of ClC-7 in lysosomes results in a lowered pH and increased degradation of fAβ by these activated microglia.
MATERIALS AND METHODS
Materials
MCSF was purchased from R&D Systems (Minneapolis, MN). MG-132 and nocodazole were purchased from Sigma (St. Louis, MO). Rabbit polyclonal antibody (lot #217354) against ClC-7 was purchased from Abcam (Cambridge, MA). Mouse monoclonal antibodies (mAbs) against protein disulfide isomerase, mannosidase II, LAMP-1, γ tubulin, 20S proteasome, and vimentin were also purchased from Abcam. Mouse monoclonal anti-V5 antibody was purchased from Invitrogen (Carlsbad, CA). The mAb against polyubiquitin was purchased from BioMol (Plymouth Meeting, PA). Rabbit polyclonal antibody raised against human Ostm1 was purchased from Atlas Antibodies (Stockholm, Sweden). Mouse mAb against CD11b was obtained from Serotec (Raleigh, NC). All fluorescent secondary antibodies were purchased from Invitrogen. Blocking buffer for Western blots and secondary antibodies tagged with infrared dyes were bought from LI-COR Biosciences (Lincoln, NE).
Cells
Primary mouse microglial cultures were isolated and maintained as described previously (Paresce et al., 1997). J774.A1 mouse macrophage-like cells (American Type Culture Collection) were maintained as described previously (Majumdar et al., 2007). U2OS-SRAs, which are human U2OS osteosarcoma cells expressing the murine scavenger receptor A, were maintained in McCoy's 5A supplemented with 1.2 g/l sodium bicarbonate, 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1 mg/ml G418.
Mice
All experiments with mice were performed in compliance with the guidelines of the Institutional Animal Care and Use Committee of Weill Cornell Medical College in accordance with the National Institutes of Health guidelines. Brain sections from at least three different Tg19959 mice (5 mo old) were analyzed for each experimental set.
Treatment of microglial cells
Microglial cells were plated 2 d before an experiment in 35-mm coverslip-bottom dishes in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. For the MCSF Treatment I of microglial cells, MCSF (25 ng/ml) was included in the culture medium of purified microglial cells plated in 35-mm coverslip-bottom dishes for 72 h. For the MCSF Treatment II of microglial cells, MCSF (25 ng/ml) was included in the culture medium during the incubation of microglia with mixed glial cultures for 10 d. Isolated microglial cells were then incubated for another 8 d in medium with MCSF (25 ng/ml). For treatment with proteasomal inhibitor MG-132, microglial cells were incubated in complete medium with 5 and 10 μM MG-132 for 12 h.
Cellular degradation of Cy3fAβ
Degradation of Cy3fAβ by microglia was measured using previously described protocols (Chung et al., 1999). Briefly, the fluorescence power per cell was determined for one set of cells after a 1-h incubation with Cy3fAβ, and similar measurements were taken at later chase times. After protein degradation, the Cy3 leaves the cell, so the remaining fluorescence is a measure of the undegraded Cy3fAβ remaining in the cells. The fields for measurement were selected at random throughout the dish. Background signal was removed from the fluorescence image by subtracting the mean pixel intensity measured in the region surrounding each cell. The cells in each image were identified using Alexa Fluor 488–labeled wheat germ agglutinin cell surface staining. The fluorescence power was then calculated as the sum of all the pixel intensities within the cell boundaries. For each Cy3fAβ degradation experiment, approximately 30 cells per condition from five different fields were analyzed. The results of the Cy3fAβ degradation experiments show the average of three different experiments done on different days.
Immunofluorescence of treated and untreated microglia
Immunofluorescence was performed as described previously (Majumdar et al., 2007).
Detergent solubility
For making the detergent-soluble (supernatant) and the pellet fractions from microglial cell lysates, cells were lysed in RIPA buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% Triton X-100 supplemented with protease inhibitors (Sigma). Lysates were centrifuged at 16,000 × g at 4°C for 20 min. The soluble (supernatant) and insoluble (pellet) fractions were used for SDS–PAGE followed by immunoblot analysis. The insoluble pellet was dissolved in 5× sample buffer, boiled at 95°C for 5 min, and used for SDS–PAGE followed by immunoblotting.
Immunoprecipitation
Immunoprecipitation was performed using the Pierce Cross-link Immunoprecipitation kit (catalog #26147; Pierce, Rockford, IL) following the manufacturer's protocols. For each immunoprecipitation reaction, 50 μg of total protein was used. Control and MCSF-treated microglial cells were lysed using a lysis buffer supplied by the manufacturer, and ClC-7 was immunoprecipitated with an anti–ClC-7 antibody. Immunocomplexes were eluted with elution buffer supplied by the manufacturer and were subjected to SDS–PAGE and immunoblotting with an anti-Ostm1 antibody. For detection of the protein bands after immunoblot, a LI-COR imaging system and appropriate infrared dye–labeled secondary antibodies were used. Image quantification and background correction were done by drawing boxes around protein bands and using the LI-COR image quantification software following the manufacturer's protocols.
Lysosomal pH measurement
The measurement of lysosomal/endosomal pH by confocal microscopy (Presley et al., 1993; Dunn et al., 1994) is based on the use of the ratio of the pH-sensitive fluorescein fluorescence to pH-insensitive rhodamine fluorescence. Cells were incubated for 16 h with 5 mg/ml dextran conjugated to both fluorescein and rhodamine (70,000 mol weight; Molecular Probes, Eugene, OR) in complete growth medium. Cells were washed thoroughly in complete medium and then incubated for another 4 h to chase the dextran to lysosomes. The cells were then washed with Medium 2 (150 mM NaCl, 20 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM KCl, 1 mM MgCl2) and kept in Medium 2 for 20 min at 37°C in air. Cells were then confocally imaged at 37°C. Confocal images were collected on an LSM 510 laser scanning confocal unit (Carl Zeiss, Thornwood, NY) attached to an Axiovert 100 M inverted microscope (Carl Zeiss) with a 63× numerical aperture (NA) 1.4 plan Apochromat objective (Carl Zeiss). Excitation on the LSM 510 laser confocal microscope was with 25-mW Argon laser emitting at 488 nm and a 1.0-mW helium/neon laser emitting at 543 nm. Emissions were collected using a 505- to 530-nm band pass filter to collect fluorescein emission and a 560- to 615-nm band pass filter to collect rhodamine emission. Typically, 0.3- to 0.5-μm vertical steps were used with axial resolution <1.0 μm. Using image-processing techniques developed previously (Dunn et al., 1994), lysosome fluorescence was quantified, and the ratio of fluorescein to rhodamine fluorescence was determined for each individual lysosome. Images were analyzed using MetaMorph image-processing software (Universal Imaging; Molecular Devices, Sunnyvale, CA). For all experimental sets, cross-talk of the fluorophores was negligible. Calibration curves were generated after fixing and equilibrating the (fluorescein-rhodamine)-dextran-loaded cells to a range of pH buffers. For the calibration curve, cells were fixed with 3% paraformaldehyde for 5 min and then they were incubated with different pH buffers in the presence of 50 mM methylamine chloride for 15 min. Buffers used contained: 50 mM HEPES, pH 7.5, 50 mM MES (2-(N-morpholino)ethanesulfonic acid), pH 6.5, 50 mM MES pH 5.5, and 50 mM sodium acetate, pH 4.5. Although the fluorescein/rhodamine ratio was sensitive to differential photobleaching of fluorescein and rhodamine fluorescence, the effects of differential photobleaching were minimized by 1) attenuating the laser intensity, 2) minimizing the cells’ exposure to the lasers, and 3) rigidly adhering to a standard protocol for image collection for both living cells and pH-standardized fixed cells. Consequently, the effect of photobleaching on fluorescein/rhodamine fluorescence was negligible and consistent for all the images.
Determination of the relative counterion permeability in the lysosomes of microglia
The relative counterion permeability was monitored following protocols described by Barriere et al. (2009). Relative lysosomal counterion permeability was measured as a function of the dissipation of the lysosomal pH after sequential treatments with a proton pump inhibitor (bafilomycin A1) and the protonophore (CCCP). For monitoring the dissipation of the lysosomal pH, cells were loaded with 5 mg/ml concentration of fluorescein and rhodamine–labeled dextran (70,000 mol weight; Molecular Probes) in complete growth medium. After incubation with the labeled dextran, the cells were washed, chased, and imaged with a confocal microscope as described earlier in the text. For this experiment, the confocal pinholes were opened such that axial resolution was close to 7.0 μm, and data were collected from a field containing ∼50 lysosomes. For each condition, four such data points from four different fields were collected, and those four fields were followed throughout the course of the experiment. Lysosomal pH dissipation was measured in the presence of 0.4 μM bafilomycin A1 and 20 μM CCCP.
RNA interference in primary mouse microglia
Microglial cells were plated at more than 95% confluence in 35-mm coverslip-bottom dishes in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Twenty-four hours after plating, the cells were transfected with a 100-nM siRNA pool directed against ClC-7 or with a 100-nM scrambled nontarget siRNA pool (Dharmacon, Lafayette, CO) with Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Transfection was allowed to occur for 16 h, after which the transfection medium was replaced with fresh medium. The first siRNA transfection was always followed with a second siRNA transfection after a gap of 24 h (Coull et al., 2005). The extent of mRNA or protein knockdown was analyzed 72 h after the second siRNA transfection. For the knockdown of Ostm1 protein in primary mouse microglia, 10 nM siRNA against Ostm1 and 10 nM control siRNA (Qiagen, Valencia, CA) were used. As a further control for off-target effects, two additional siRNAs (Qiagen) against ClC-7 and Ostm1 were used. For the pool of siRNA against ClC-7, a mixture of the following four siRNAs were used: 1) GAAUCAACCACACGGCUUU, 2) GGAAGGACCUAGCAAGAUA, 3) GAAUAUUUCCGCAGAGAUA, and 4) GGGCAGAUCCGGGUAAAUA. The sequences of the individual siRNAs used for ClC-7 and Ostm1 knockdown are provided below.
siRNA 6 against ClC-7: UAUCUUGCUAGGUCCUUCC
siRNA 7 against ClC-7: UUCAAUUUCAGUCUUCAUG
siRNA 1 against Ostm1: UACCUUUACAACAAAGCCC
siRNA 2 against Ostm1: UUUAUUGUAAAGCAUAAGG
siRNA 3 against Ostm1: UAACUUAAUGAAAUUCAU
RNA isolation and qRT-PCR
Total cell RNA was extracted and purified from cells using the RNeasy mini-kit (Qiagen). Each sample was treated with DNase I (Qiagen) and was reverse-transcribed using oligo(dT) primer and the SuperScript III RNase H- Reverse Transcriptase Kit or SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen), according to the manufacturer's instructions. Negative control samples (no first-strand synthesis) were prepared without the addition of reverse transcriptase. The cDNA (1 μl) was used for real time RT-PCR performed using the SYBR Green I kit (Qiagen). The PCR was performed using an ABI Prism thermal cycler (Applied Biosystems, Carlsbad, CA). Expression levels of genes were normalized against actin controls. The relative expression levels (fold change) were calculated by the ΔΔCT method. For each qRT-PCR reaction, validated primers (Qiagen) for the corresponding mouse genes were used. Assays were performed in triplicate, and the average value is shown in the figure. The error bars represent SEM.
Degradation of Cy3fAβ after knockdown of ClC-7 and Ostm1 with siRNA
To monitor Cy3fAβ degradation after ClC-7 and Ostm1 protein knockdown with siRNA, primary microglial cells were transfected twice with 100 nM siRNA (see RNA interference in primary mouse microglia section). Twenty-four hours after the second siRNA transfection, microglial cells were incubated for 60 min with Cy3fAβ (5 μg/ml). After 60 min, the cells were rinsed and incubated for 72 h in culture medium containing MCSF (25 ng/ml). Degradation of fAβ was determined by loss of fluorescence from the cells as described previously (Chung et al., 1999).
Lysosomal pH measurement in microglia after knockdown of ClC-7 with siRNA
To measure lysosomal pH in microglia after the knockdown of ClC-7 protein with siRNA, primary microglial cells were transfected twice with 100 nM siRNA (see RNA interference in primary mouse microglia section). After the siRNA transfections, the cells were rinsed and incubated for 72 h in culture medium containing MCSF (25 ng/ml). The pH of the lysosomes was measured as described earlier in the text.
Transfection of plasmid DNA in microglia
Microglial cells were plated at more than 95% confluence in 35-mm coverslip-bottom dishes in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Twenty-four hours after plating, the cells were transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen). Transfection was allowed to occur for 2 h, after which the transfection medium was replaced with fresh medium.
Degradation of Cy3fAβ after the overexpression of V5-epitope–tagged ClC-7 and Ostm1 in microglia
Microglial cells were transfected with plasmid DNA encoding V5-tagged Ostm1 (TOPO V5 Ostm1) or untagged Ostm1 (pIRES GFP Ostm1). Twenty-four hours after the transfection, microglial cells were incubated for 60 min with Cy3fAβ (5 μg/ml). After 60 min, the cells were rinsed and incubated for 72 h in culture medium. Degradation of Cy3fAβ was determined by loss of fluorescence from the cells as described previously (Chung et al., 1999).
Tissue preparation
Brain slices were prepared from Tg19959 mice harboring human APP695 with KM670/671NL and V717F mutations (Li et al., 2004). Mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused via the ascending aorta with 3.75% acrolein (Polyscience, Warrington, PA) and 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Vibratome-cut, 40-μm tissue sections were kept in storage buffer composed of 30% sucrose and 30% ethylene glycol in phosphate buffer at –20ºC.
Immunohistochemistry of brain slices
Free-floating sections were blocked with 10% normal goat serum in 0.1 M Tris buffer for 1 h. Sections were then incubated with primary antibodies (ClC-7 [1:100] and CD11b [1:300] or LAMP-1 [1:100] and CD11b [1:300]) in 3% normal goat serum in 0.1 M Tris buffer for 24 h at 4°C. After the incubation, the sections were rinsed and incubated with Alexa Fluor 488–labeled goat anti–rabbit IgG and Alexa Fluor 546–labeled goat anti–rat IgG (1:200; Molecular Probes) for 2 h at room temperature. Images were collected using a Leica SP5 spectral confocal microscope (Leica Microsystems, Bannockburn, IL) equipped with an HCX PL APO CS 63×/NA 1.4 oil objective.
Supplementary Material
Acknowledgments
We thank George Carlson (McLaughlin Research Institute, Great Falls, MT) for providing the Tg19959 mice. We thank Nina Pipalia for providing the U2OS-SRA cells and Haley Fraser for data analysis. This work was supported by National Institutes of Health Grants AG027140 (to G.K.G.) and R37DK27083 (to F.R.M.) and by an Appel Discovery grant (to F.R.M.).
Abbreviations used:
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- CFTR
cystic fibrosis transmembrane conductance regulator
- ERAD
endoplasmic reticulum–associated degradation
- fAβ
fibrillar Alzheimer's amyloid β peptide
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- Ig
immunoglobulin
- IRES
internal ribosome entry site
- LAMP-1
lysosomal-associated membrane protein 1
- mAb
monoclonal antibody
- MCSF
macrophage colony-stimulating factor
- MES
2-(N-morpholino)ethanesulfonic acid
- NA
numerical aperture
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- SRA
type A scavenger receptor
- TCR
T-cell receptor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-09-0745) on March 25, 2011.
REFERENCES
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bae HR, Verkman AS. Protein kinase A regulates chloride conductance in endocytic vesicles from proximal tubule. Nature. 1990;348:637–639. doi: 10.1038/348637a0. [DOI] [PubMed] [Google Scholar]
- Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, Radzioch D, Lukacs GL. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell. 2009;20:3125–3141. doi: 10.1091/mbc.E09-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009;132:1078–1092. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil MI, Chung H, Maxfield FR. Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer's disease amyloid fibrils by microglia. J Biol Chem. 2000;275:16941–16947. doi: 10.1074/jbc.M000937200. [DOI] [PubMed] [Google Scholar]
- Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A, Villa A, Vacher J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. 2003;9:399–406. doi: 10.1038/nm842. [DOI] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Park J, Semrad CE, Gelman DL, Shevell T, McGraw TE. Regulation of endocytic trafficking and acidification are independent of the cystic fibrosis transmembrane regulator. J Biol Chem. 1994;269:5336–5345. [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- Graves AR, Curran PK, Smith CL, Mindell JA. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–792. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- Hochmeister S, Zeitelhofer M, Bauer J, Nicolussi EM, Fischer MT, Heinke B, Selzer E, Lassmann H, Bradl M. After injection into the striatum, in vitro-differentiated microglia- and bone marrow-derived dendritic cells can leave the central nervous system via the blood stream. Am J Pathol. 2008;173:1669–1681. doi: 10.2353/ajpath.2008.080234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol. 2007;578:633–640. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper D, et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR, Sitia R. Aggresomes and Russell bodiesSymptoms of cellular indigestion? EMBO Rep. 2000;1:225–231. doi: 10.1093/embo-reports/kvd052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature. 2006;440:220–223. doi: 10.1038/nature04535. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Rotstein OD, Grinstein S. Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ “leak.”. J Biol Chem. 1991;266:24540–24548. [PubMed] [Google Scholar]
- Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, Maxfield FR. Degradation of fibrillar forms of Alzheimer's amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29:707–715. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, Maxfield FR. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007;18:1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, Imitola J, Petrovic S, Zota V, Nemirovsky A, Baron R, Fisher Y, Owens T, Weiner HL. Abeta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of Abeta in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2006;103:5048–5053. doi: 10.1073/pnas.0506209103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, Weiner HL. Immunotherapeutic approaches to Alzheimer's disease. Science. 2003;302:834–838. doi: 10.1126/science.1088469. [DOI] [PubMed] [Google Scholar]
- Morgan D, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ohkuma S, Moriyama Y, Takano T. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc Natl Acad Sci USA. 1982;79:2758–2762. doi: 10.1073/pnas.79.9.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid β-protein by microglial cells. J Biol Chem. 1997;272:29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Pillay CS, Elliott E, Dennison C. Endolysosomal proteolysis and its regulation. Biochem J. 2002;363:417–429. doi: 10.1042/0264-6021:3630417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Mayor S, Dunn KW, Johnson LS, McGraw TE, Maxfield FR. The End2 mutation in CHO cells slows the exit of transferrin receptors from the recycling compartment but bulk membrane recycling is unaffected. J Cell Biol. 1993;122:1231–1241. doi: 10.1083/jcb.122.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, et al. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, Grinstein S. A cation counterflux supports lysosomal acidification. J Cell Biol. 2010;189:1171–1186. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Haass C, Schultz C. UV light-induced autofluorescence of full-length Abeta-protein deposits in the human brain. Clin Neuropathol. 2002;21:35–40. [PubMed] [Google Scholar]
- Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- Van Dyke RW. Acidification of rat liver lysosomes: quantitation and comparison with endosomes. Am J Physiol. 1993;265:C901–C917. doi: 10.1152/ajpcell.1993.265.4.C901. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartosch L, Fuhrmann JC, Schweizer M, Stauber T, Jentsch TJ. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009;23:4056–4068. doi: 10.1096/fj.09-130880. [DOI] [PubMed] [Google Scholar]
- Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, Rademann J, Stauber T, Kornak U, Jentsch TJ. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science. 2010;328:1401–1403. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis. 2004;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Yong VW, Rivest S. Taking advantage of the systemic immune system to cure brain diseases. Neuron. 2009;64:55–60. doi: 10.1016/j.neuron.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.