

Abstract
Accounting for enzyme-mediated active sliding, disassembly, and sequence-dependent positioning of nucleosomes, we simulate nucleosome occupancy over cell-cycle-scale times using a stochastic kinetic model. We show that ATP-dependent active nucleosome sliding and nucleosome removal processes are essential to obtain in vivo-like nucleosome positioning. While active sliding leads to dense nucleosome filling, sliding events alone cannot ensure sequence-dependent nucleosome positioning: Active nucleosome removal is the crucial remodeling event that drives positioning. We also show that remodeling activity changes nucleosome dynamics from glassy to liquid-like, and that remodeling dramatically influences exposure dynamics of promoter regions.
Keywords: ATP-dependent nucleosome removal, ATP-dependent nucleosome sliding, protein–DNA interactions, chromatin dynamics
DNA in each eukaryotic cell is folded and packaged, with the help of many proteins, into chromatin. In the first level of packaging, 147-base-pair (≈50 nm) stretches of DNA are wrapped around histone protein octamers into nucleosomes (1). To a first approximation, chromatin can be considered as a linear array of nucleosomes, and a body of experimental evidence indicates that primary DNA sequence controls the position of at least a fraction of nucleosomes: Histone–DNA affinities vary with sequence over a roughly 8 kBT range (2). Nevertheless, the overall degree to which nucleosomes are positioned by DNA sequence is a subject of ongoing debate (3–5). Transcription, replication, recombination, and DNA repair all need access to bare DNA and hence the organization of nucleosomes must be accomplished in a way that allows rapid, localized access to DNA (6). However, disrupting nucleosomes requires passage of energy barriers of tens of kBT per nucleosome (7, 8). In vivo this is facilitated by ATP-consuming “chromatin remodeling complexes” (9, 10), which catalyze nucleosome repositioning and disassembly (11–17).
Previously, Teif and Rippe have studied models for the influence of remodeling enzymes on the equilibrium distribution of nucleosomes (18). Nucleosomes by themselves cannot reach thermal equilibrium on cell-cycle time scales, and no one has yet explored the kinetics of nucleosome filling in the presence of remodeling factors. Here we study the kinetics of nucleosomes in the presence of chromatin remodeling complexes in a stochastic model, focusing on assembly and positioning dynamics of nucleosomes. In addition to thermal fluctuation dynamics (always present as a “background” to any active remodeling machinery) we consider two general types of remodeling machines that: (i) slide nucleosomes along the DNA to cause repositioning, and (ii) remove (eject) histone octamers from DNA. These two processes have been implicated to be catalyzed by remodelers in the ISWI [e.g., yeast ISWI (14), ACF (17)] and SNF2 [e.g., RSC (11), yeast SWI/SNF (12, 15)] subfamilies, respectively. Using experimentally determined thermal and ATP-dependent enzyme rates we show that active sliding is necessary to achieve biological densities of nucleosomes within biologically relevant time scales, but that active nucleosome removal is also needed, to obtain nucleosome positioning at biological densities.
Model
We consider DNA as a one-dimensional (1D) lattice of N base pairs (molecule length L = N·0.34 nm) along which nucleosomes can adsorb, desorb, and slide. Adsorption of a nucleosome requires a region of empty DNA k bp long, which is filled by the adsorbed nucleosome. We account for sequence effects with the nucleosome positioning potential energy Vi determined by (2). In thermal equilibrium the Boltzmann factor exp(-Vi/kBT) is proportional to the probability of finding a nucleosome starting at base pair i in the limit of low nucleosomal coverage of DNA.
In our model we consider three kinetic processes: (i) histone octamer binding to DNA to form nucleosomes (nucleosome adsorption), (ii) histone octamer release (nucleosome desorption), and (iii) lateral displacement of histone octamers along the DNA (nucleosome sliding). Nucleosome adsorption is known to occur efficiently in vivo by ATP-independent histone chaperone-mediated pathways; here we consider only ATP-independent nucleosome adsorption. On the other hand, thermally excited desorption and sliding are very slow, and ATP-dependent enzymes are known to assist nucleosomes in these processes (11–17). We consider both ATP-dependent and ATP-independent desorption and sliding processes.
ATP-Independent Kinetics.
Nucleosomes can adsorb at any sterically permitted location on the DNA at rate ron. Thermally excited removal of a nucleosome depends on its sequence location; a nucleosome at position i is removed with rate roffi,
| [1] |
where koff is the intrinsic removal rate per nucleosome. The ATP-independent adsorption and desorption rates must satisfy the Boltzmann distribution
| [2] |
The sliding rate of the nucleosome also depends on the DNA sequence. The ratio of rates for sliding of a nucleosome from DNA site i to site j (di→j), and that of the reverse event from j to i, must also satisfy the Boltzmann relation:
| [3] |
Because this sliding transition involves many microscopic intermediate steps, it will likely depend primarily on the potential difference between the initial and final state—i.e., as
| [4] |
Because Vi - Vj has an average value of zero, D can be taken as a global estimate of the nucleosome diffusion constant.
ATP-Dependent Desorption and Sliding Kinetics.
We presume that there is no change in adsorption rate due to ATP-dependent enzymes; nucleosomes continue to be adsorbed at any sterically permitted location on the DNA at rate ron. However, ATPases do affect nucleosome desorption and sliding. We suppose that ATPases enhance nucleosome disassembly such that the effective nucleosome removal rate per nucleosome is given by:
| [5] |
The enhancement of off-rate by the nucleosome-removing enzymes is accounted for through a positive shift in the effective free energy by an amount Va, which can be considered to be the amount of chemical energy (from ATP hydrolysis) coupled into active nucleosome removal. Thus, rates of nucleosome removal can be described using the potential shift Va. Below we will specify the shift Va in terms of the average effective nucleosome binding free energy, Veff = Va + 〈V〉, where 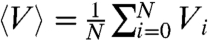 .
.
Different families of ATPases that slide nucleosomes appear to work in different ways. Recent in vitro experiments have indicated that repositioning by remodeling complexes depends on the flanking DNA length: Some remodelers cannot move nucleosomes when the flanking length is very small, while others can (13, 14, 17). Here we adopt a minimal model for active sliding: Each nucleosome is acted on a rate αp, with each sliding event moving a nucleosome in a randomly chosen direction until it sterically interacts with a neighbor. Based on experimental data (13), we assume that this active pushing is independent of DNA sequence. A more detailed model could easily account for variability of remodeling with flanking DNA, but here this level of detail is not necessary.
Methods
We obtain the sequence-dependent energy Vi using the model of Kaplan et al., who provide the static probability, Pi, of finding a nucleosome starting at base pair i (2) for any specified overall nucleosome density. The probability Pi at low nucleosome density determines the potential Vi = -kBT ln Pi, up to an overall constant. To determine the absolute value of Vi under cellular conditions, we use the value  determined experimentally from nucleosome assembly experiments in ATP-depleted Xenopus egg extracts (19).
determined experimentally from nucleosome assembly experiments in ATP-depleted Xenopus egg extracts (19).
We computed the dynamics of nucleosomes for the above model, using continuous time stochastic simulations (20) according to the methods used in ref. 19. In brief, at each step of the computation we use the rates of the events that are possible to compute the time interval until the next stochastically determined on-, off-, or slide event. The event is then implemented, and the time updated. Successive events are considered to be uncorrelated. We note that ATP hydrolysis, while not explicitly included in the kinetic model, is implicitly included through the nonthermal remodeling (removal and sliding) processes.
Results
In our simulations we focused on a heavily studied region of DNA near position 187000 on Saccharomyces cerevisiae chromosome XIV (GenBank accession number NC001134). The on-rate ron = 12 s-1 determined experimentally in cell-extract experiments (19) was used. We take the “hard core” size of nucleosomes to be k = 147 bp.
We first investigate how active sliding and nucleosome removal affects filling of initially naked DNA by nucleosomes. For yeast, it is known that close to 88% of gene-rich DNA is covered with nucleosomes (21, 22). Because the yeast cell cycle is ≈90 min, it is necessary for the cell to fill its DNA with nucleosomes in appreciably less than this amount of time. Two key questions are: (i) What should be the active (ATP-dependent) nucleosome pushing rate so that 88% of DNA is covered by nucleosomes within 1 hour, and (ii) what should be the active (ATP-dependent) nucleosome removal rate so that the steady-state filling density is ≈88%?
Active Pushing Drives Filling of DNA by Nucleosomes.
Fig. 1 shows how ATP-driven pushing accelerates the packing of nucleosomes onto initially naked DNA. Nucleosome coverage is shown as a function of time, for thermal nucleosome addition and removal rates (ron = 12 s-1 and Veff = -42 kBT). Nucleosome coverage, which specifies the percentage of DNA that is covered by nucleosomes, is given by (nak/N) × 100, where na is the number of adsorbed nucleosomes. At early times (< 1 s), the dynamics in all cases is dominated by random adsorptions, and the DNA is rapidly filled with nucleosomes (nucleosome coverage reaches 75% in about 1 sec). However, for slow sliding rates (red and green curves), filling stalls when “jamming” occurs, at about 75% filling (23) (here, jamming is the phenomenon of being unable to add nucleosomes beyond roughly 75% density by random deposition without substantial rearrangement of previously bound nucleosomes, and is characteristic of any one-dimensional deposition process; see the SI Appendix).
Fig. 1.

Nucleosome filling dynamics with and without active nucleosome sliding. The two curves at the bottom are results of the dynamics with only thermal fluctuations (αp = 0). The first curve from the bottom (red) is for D = 10-17 cm2/s (24, 25) and the second curve (green) is for D = 10-15 cm2/s (19). The top three curves are results of the dynamics with active sliding having sliding rates 0.24 s-1 (cyan), 0.024 s-1 (pink) αp = 0.0024 s-1 (blue) respectively. The horizontal line (black) corresponds to 90% filling, and the X-axis spans close to four hours. For all curves in this plot nucleosome removal occurs only thermally—i.e., Veff = -42 kBT. Simulations were performed on a 72-kb region of yeast DNA (chromosome XIV 135000-207000) and averaged for 10 realizations.
To examine the ATP-independent case we computed the dynamics with two different thermal nucleosome diffusion rates estimated previously: D = 10-17 cm2/s (0.01 bp2/s) estimated theoretically (24, 25) and D = 10-15 cm2/s as estimated experimentally for ATP-depleted Xenopus egg extracts (19). As shown by red and green curves in Fig. 1, the filling dynamics for either of these estimates for thermal nucleosome diffusion (i.e., with no ATP) is extremely slow. Even after t ≈ 4 hours, the nucleosome coverage is well below 88%; purely thermal dynamics are incapable of reaching biological fillings of DNA over biologically reasonable times. This is well known experimentally, in that in vitro nucleosome assembly reactions without ATP-dependent factors present cannot achieve nucleosome coverages > 70%.
However, with active pushing, filling is greatly accelerated (upper curves of Fig. 1). When the pushing rate is αp = 0.0024 s-1, one gets to 90% filling within ≈50 minutes (blue curve). Faster pushing of αp = 0.24 s-1 generates 90% filling in less than one minute. We note that active pushing alone leads to “overfilling” of DNA by nucleosomes; the strong binding affinity of histones for DNA permits this to occur once kinetic pathways that allow packing of nucleosomes to occur are present. The intermediate rate of αp ≈ 0.0024 s-1 is comparable to rates observed in biochemical experiments with active nucleosome remodeling enzymes (17).
Active Nucleosome Removal Controls Nucleosome Density.
We now keep the active pushing rate constant at the level needed to generate biological filling of DNA by nucleosomes over a roughly 1 hr time scale, αp = 0.0024 s-1 (comparable to rates of nucleosome sliding generated by chromatin remodeling enzymes) (17), and we add active nucleosome removal (Fig. 2). As before, nucleosome density initially increases, but then nucleosome assembly is slowed down, and the nucleosome density approaches a steady state. For a nucleosome removal rate corresponding to Veff around -7 kBT (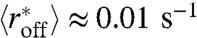 , pink curve in Fig. 2) this steady-state filling reaches the ≈88% observed in vivo (22) in about 2,000 s. Thus, combining realistic rates of active nucleosome sliding (roughly one sliding event per nucleosome every 400 sec) with a moderate amount of nucleosome removal (each nucleosome is ejected once per every 100 sec) generates a realistic and stable nucleosome level along DNA in roughly 2,000 s (≈33 min), less than the yeast cell-cycle time.
, pink curve in Fig. 2) this steady-state filling reaches the ≈88% observed in vivo (22) in about 2,000 s. Thus, combining realistic rates of active nucleosome sliding (roughly one sliding event per nucleosome every 400 sec) with a moderate amount of nucleosome removal (each nucleosome is ejected once per every 100 sec) generates a realistic and stable nucleosome level along DNA in roughly 2,000 s (≈33 min), less than the yeast cell-cycle time.
Fig. 2.

Nucleosome filling dynamics for different ATP-dependent nucleosome removal rates. Bottom to top curves are computed with 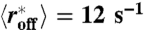 (Veff = 0 kBT, red), 1.62 s-1 (Veff = -2 kBT, green), 0.08 s-1 (Veff = -5 kBT, blue), 0.01 s-1 (Veff = -7 kBT, pink), 0.002 s-1 (Veff = -9 kBT, cyan). For all curves in this plot active pushing occurs at rate αp = 0.0024 s-1. Simulations were performed on the same 72-kb DNA region as in Fig. 1.
(Veff = 0 kBT, red), 1.62 s-1 (Veff = -2 kBT, green), 0.08 s-1 (Veff = -5 kBT, blue), 0.01 s-1 (Veff = -7 kBT, pink), 0.002 s-1 (Veff = -9 kBT, cyan). For all curves in this plot active pushing occurs at rate αp = 0.0024 s-1. Simulations were performed on the same 72-kb DNA region as in Fig. 1.
Active Remodeling Enhances Nucleosome Positioning.
Having determined rates for active chromatin remodeling enzymes suitable for generating filling of DNA by nucleosomes in biologically relevant times, we analyze the distribution of nucleosomes along DNA (i.e., probability of occupation of sequence position by nucleosomes) to determine under what conditions positioning of nucleosomes as observed in vivo (2) occurs in our model.
We first compare the nucleosome occupation when there are only thermal kinetics to that with active remodeling kinetics (Fig 3). For purely thermal kinetics, the removal rate is very small (Veff = -42 kBT), and active pushing is absent (αp = 0). This gives an average density close to the “jamming density” (≈75%) in the time scale of interest (< 1 hour). Because removal is slow, once a nucleosome is adsorbed it will remain bound indefinitely, with only very slow diffusion from side to side. When averaged over many realizations, this leads to poorly positioned nucleosomes along the DNA after 60 min (Fig. 3, green curve, T−). However, with active remodeling, nucleosomes are more rapidly removed and slid, with the result that much stronger positioning occurs after 60 min (Fig. 3, red curve, T+). The active parameters chosen correspond to the sliding rate used above (αp = 0.0024 s-1), with nucleosome removal adjusted to reach the same average density as that obtained for thermal kinetics (Veff = -3.5 kBT). Thus, active remodeling enhances nucleosome positioning, essentially by providing kinetic pathways for nucleosomes to equilibrate in the DNA-sequence-generated positioning potential.
Fig. 3.

Comparison of coverage probability (probability that a given DNA position is occupied by a nucleosome) when there are only thermal (ATP-independent) events (T−, green; Veff = -42 kBT, αp = 0), and when there is nonthermal (ATP-dependent) activity (T+, red). The parameters for the nonthermal case (αp = 0.0024 s-1, Veff = -3.5 kBT) are chosen such that the average density is the same as the thermal case. Curves are obtained by averaging over 100 simulations. Each simulation is performed on a 20-kb region of the yeast chromosome; a representative 3-kb window is shown here.
Efficient Positioning Requires Active Nucleosome Removal.
We next examine the importance of nucleosome removal to positioning. Fig. 4 shows nucleosome profiles with (green curve, Veff = 0 kBT) and without (blue curve, Veff = -42 kBT) active nucleosome removal, after 1 hour (for all curves, ron = 12 s-1 and αp = 0.0024 s-1). Without active nucleosome removal, there is strong filling, but an absence of strong positioning. With active removal, the nucleosome density is suppressed, and strong positioning is observed, in good agreement with in vivo data (black curve) and the model (red curve) of Kaplan et al. (2).
Fig. 4.

Nucleosome occupation after 60 minutes. Blue curve shows result for ATP-dependent sliding but no ATP-dependent nucleosome removal (Veff = -42 kBT). Green curve shows effect of including ATP-dependent nucleosome removal (Veff = 0 kBT). Black curve shows in vivo data (YPD) (2); red curve shows model prediction of Kaplan et al. (2). All curves are computed for αp = 0.0024 s-1, and are averaged over 100 simulations. Each simulation is performed on a 20-kb region of the yeast chromosome; a 3-kb window is shown here.
Nucleosome Positioning at Realistic Nucleosome Densities.
From consideration of final nucleosome density (Figs. 1 and 2) we obtained reasonable kinetic parameters to reach a filling of ≈88% after ≈1 hour. The nucleosome distribution obtained in this case (Veff = -7 kBT, αp = 0.0024 s-1) is shown in Fig. 5 (blue curve): Well-defined positioning with a high density of nucleosomes occurs. Fig. 5 also shows profiles for more nucleosome removal: The green curve shows steady-state nucleosome distributions for Veff = -5 kBT with a slightly lower nucleosome density but still strong positioning, while the red curve shows the distribution for Veff = 0 kBT, with a significantly lower nucleosome coverage.
Fig. 5.

Nucleosome occupation after t = 1 hour, for different active nucleosome removal rates given by Veff = 0 kBT (red, bottom curve a), Veff = -5 kBT (green, middle curve b), Veff = -7 kBT (blue, upper curve c). The red (Veff = 0 kBT) curve is similar to the data of ref. 2. The curves are obtained after averaging over 100 simulations. Each simulation is performed on a 20-kb region of the yeast chromosome; a 3-kb window is shown here.
Strikingly, in vivo nucleosome sequencing data (Fig. 4, black curve) (2) are closest to the lowest-coverage case (Fig. 5, red line), which is far below consensus values for overall nucleosome density. The resolution of this paradox is a problem for the future; it is possible that current nucleosome-sequencing experiments significantly undercount total nucleosome numbers, or alternately the currently available estimates of average nucleosome coverage ≈88% are appreciably too large.
We note that our model is not sensitive to the steric size of nucleosomes chosen (see Figs. S1–S3 in SI Appendix). Also, we have found that our model positions nucleosomes in accord with experimental data for a variety of types of genomic regions (centromeric, telomeric, replication origins; Figs. S2 and S4–S6 in SI Appendix). Finally we note that our model produces positioning in accord with in vivo experimental data over long stretches of sequence (Figs. S6 and S7 in SI Appendix).
Chromatin Remodeling Melts Frozen Nucleosomes.
The active processes considered in our model have a profound effect on nucleosome dynamics; thermal processes alone lead to frozen, “glassy” behavior, and addition of active remodeling leads to “liquid” behavior. To see this we define a “profile function” pi(m,t) for a given configuration of nucleosomes i, which is zero if site m is not covered by a nucleosome, and nonzero otherwise. Inside a nucleosome that starts at sequence position q, the nonzero values of pi(m,t) are
 |
[6] |
Note 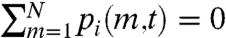 . Using pi(m,t) we can define the “overlap” for two different nucleosome organizations i and j, at different times t1 and t2 as
. Using pi(m,t) we can define the “overlap” for two different nucleosome organizations i and j, at different times t1 and t2 as
 |
[7] |
where N is the total number of base-pairs. If all nucleosomes remain frozen (no movement for all times) the overlap parameter for the same realization will be Φii(t1,t2) = 1. Thus, large Φii(t1,t2) indicate nucleosomes that are “frozen” in a given realization, or unable to significantly change their organization over times t1 to t2, while small Φii(t1,t2) indicate that nucleosome positions are liquid, or able to fluctuate. Nonzero values of Φij indicate positioning correlations that appear in different realizations; Φij = 0 indicates a lack of reproducible positioning. The negative contributions to pi ensure that Φ → 0 for uncorrelated nucleosome configurations (an alternate definition of pi that is 0 or 1 for open and covered base pairs, respectively, is discussed in the SI Appendix; see Figs. S8–S10).
We computed Φij for thermal and active kinetics (Fig. 6; t1 = 30 min, 30 min ≤ t2 ≤ 160 min). For thermal motions (a), we find that Φii(t1 = 30 min ,t2) decreases slowly as a function of time, indicating slow, glassy dynamics whereby nucleosome distributions only slowly change their organization: After 130 min, less than 50% of the initial nucleosome position correlation has been lost. Furthermore, different realizations are uncorrelated at all times, indicated by Φij(t1 = 30 min ,t2) which is nearly zero: Positioning is not able to occur reproducibly in different realizations.
Fig. 6.

Overlap correlations computed (A) without and (B) with ATPase activity (for all cases t1 = 30 min). Red filled squares show Φii, green filled circles show Φij. In the -ATP case (A), nucleosomes diffuse slowly (D = 10-15 cm2/s, Veff = -42 kBT) and the same-realization overlap Φii decays slowly, indicating glassy behavior; different-realization correlations are near zero. In the +ATP case (B), nucleosomes are pushed with a rate αp = 0.0024 s-1 and nucleosomes are actively disassembled (Veff is taken as -4 kBT so that nucleosome coverage is ≈80%); now Φii rapidly decays to a nonzero value indicating liquid behavior, but with positioning; Φij is nearly constant at the same level of correlation, indicating positioning that is reproducible from realization to realization. The calculation was performed on a 20-kb region of yeast chromosome XIV (187000-207000), averaged over 25 realizations.
By contrast, with active remodeling, there is a rapid decay of Φii indicating that nucleosomes are highly mobile; the nucleosomes are liquid (Veff = -4 kBT is used so that the nucleosome density is comparable to that encountered in the thermal case). Importantly, Φii decays to ≈0.2, indicating correlations in nucleosome positioning that persist over time; consistent with this Φij is nearly constant at a similar level ≈0.15. Thus about 1/6 of the nucleosomes are strongly positioned across realizations and over time by the action of Vi; the remainder of the nucleosomes are highly mobile. Feng et al. and Nikolaou et al. compared different available nucleosome-positioning data and found that only a small percentage of nucleosome positions are invariant among the different experimental datasets they considered (5, 4). Our simulation results are consistent with these findings (see Fig. S11 in SI Appendix).
Promoter Accessibility.
Nucleosome positioning can regulate gene expression because positioning of a nucleosome over a promoter can block transcription initiation (26). Active remodeling of transcription-blocking nucleosomes can therefore trigger activation of genes, and our model can give some insight into the kinetics of this type of gene activation. We consider a 10-bp-wide region centered on the TATA box in the GAL1 promoter (yeast chromosome II, position 278876) and we ask how active remodeling affects the “exposure” of this region. Exposure is defined as lack of nucleosome–DNA contacts along all of the 10-bp region. As nucleosomes move, exposure events start and then end; we compute the number of such events and their duration. In a simulation of a one hour period, we compute the distribution of exposure durations that occur in the last 30 min (Fig. 7). The three curves show results for fixed active sliding, for three different nucleosome removal rates: Veff = -6 kBT (red filled squares); Veff = -7 kBT (green filled circles); Veff = -9 kBT (blue open circles).
Fig. 7.

Main figure: Distribution of exposure time, computed from exposure events in a 30-minute window, for different nucleosome removal activity Veff = -6 kBT (red filled squares), Veff = -7 kBT (green filled circles), Veff = -9 kBT (blue open circles). The distributions have a bin size of 1/12 s. Inset: Average exposure time per exposure event in seconds 〈te〉 (red filled squares), total number of exposure events Ne (blue open circles), and the product Ne × 〈te〉 in seconds (green filled circles). Data computed for αp = 0.0024 s-1, averaged over 100 realizations, yeast chromosome II position 270000-290000.
The inset of Fig. 7 shows average exposure time per exposure event 〈te〉 (red filled squares) in seconds, which decreases with increasing nucleosome removal activity, because when nucleosomes are forced to move around an exposed site will soon be covered. However, the total number of exposure events (Ne, inset, blue circles) increases with nucleosome removal activity, causing a net increase in the total time the sites are exposed (Ne × 〈te〉, inset, solid green circles). For biologically reasonable rates (Veff ≈ -7 kBT, αp = 0.0024 s-1) the average exposure time is ≈4 seconds, and the average exposure rate is about 4 min-1. The average exposure time is significantly longer than the initial decay in Fig. 7, due to a long tail on the distribution associated with relatively slow rearrangements of groups of adjacent nucleosomes.
As Veff becomes strongly negative, nucleosome removals become rare, and the averages approach constant values determined by the sliding rate. Qualitatively similar behavior is obtained when nucleosome sliding is varied, although as might be expected increased nucleosome removal more strongly drives site exposure. Finally we note that we have observed similar site exposures in simulations of other TATA- and non-TATA-containing promoter regions (see Figs. S12 and S13 in SI Appendix).
Concluding Remarks.
We have presented a simple kinetic model of nucleosome positioning, based on known kinetic rates of thermally excited nucleosome assembly, disassembly, and diffusion, and including active nucleosome sliding and removal processes similar to those driven by chromatin remodeling enzymes in the ISWI and SNF2 subfamilies. We have shown that active remodeling strongly accelerates assembly of positioned nucleosomes at realistic densities along DNA. The acceleration is mainly achieved by sliding events, while the active nucleosome removal ensures sequence-dependent positioning. In the absence of active remodeling, the remaining thermally driven motions of nucleosomes along DNA are too slow for positioning to occur, or for nucleosome coverages to reach the ≈88% levels seen in vivo.
We have also shown that the jammed or frozen nucleosome patterns associated with thermally excited dynamics are converted into liquid dynamics by active remodeling. This liquid behavior of nucleosomes under the influence of remodeling drives more frequent exposure of DNA sites, facilitating binding of transcription factors. Therefore increased chromatin remodeler activity can be expected to enhance gene expression and to suppress cell-to-cell fluctuations in regulated gene expression. The nucleosome coverage and the liquid dynamics we obtained also suggest that remodelers are using the energy of ATP to maintain a chromatin state that is far from equilibrium.
We emphasize that our results have been obtained using current models for nucleosome positioning preferences of chromosomal DNA. It is likely that these models will appreciably change over the next several years; since 2006 models for positioning have qualitatively changed as sequencing technologies have improved. The remarkably low overall nucleosome densities obtained in current nucleosome-sequencing studies suggest that many nucleosomes may be somehow being missed, indicating that there is room for improvement of measurement of in vivo nucleosome profiles, the main starting point for the nucleosome-positioning free energy models. More complete measurements would allow more precise knowledge of overall nucleosome coverages, and more precise knowledge of the appropriate in vivo value of the active nucleosome removal rate (controlled here by Veff). However, the statistical properties of nucleosome-positioning models have not changed significantly, so we anticipate that the main conclusions of this paper, most importantly that active chromatin remodeling machinery is essential to obtaining well-packed and DNA-sequence-programmed nucleosome positioning on cell-cycle-like time scales, will be unchanged.
Supplementary Material
Acknowledgments.
We thank J. Widom, D. Grilley, G. Narlikar, S. Ghosh, and E. Segal for helpful discussions. Work at Northwestern University was supported by the National Science Foundation under Grants DMR-0715099 and PHY-0852130, and by National Institutes of Health Grant U54-CA143869-01. Work at Indian Institute of Technology Bombay was supported by Department of Biotechnology India through Innovative Young Biotechnologist Award.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. T.T. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015206108/-/DCSupplemental.
References
- 1.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein A, Takasuka TE, Collings CK. Are nucleosome positions in vivo primarily determined by histone–DNA sequence preferences? Nucleic Acids Res. 2010;38:709–719. doi: 10.1093/nar/gkp1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng J, et al. New insights into two distinct nucleosome distributions: Comparison of cross-platform positioning datasets in the yeast genome. BMC Genomics. 2010;11:33. doi: 10.1186/1471-2164-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikolaou C, Althammer S, Beato M, Guig R. Structural constraints revealed in consistent nucleosome positions in the genome of S. cerevisiae. Epigenetics & Chromatin. 2010;3:20. doi: 10.1186/1756-8935-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberts B, et al. Molecular Biology of the Cell. 4th Ed. New York: Garland Science; 2002. [Google Scholar]
- 7.Brower-Toland BD, et al. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc Natl Acad Sci USA. 2002;99:1960–1965. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan J, et al. Micromanipulation studies of chromatin fibers in xenopus egg extracts reveal ATP-dependent chromatin assembly dynamics. Mol Biol Cell. 2007;18:464–474. doi: 10.1091/mbc.E06-09-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusser A, Kadonaga JT. Chromatin remodeling by ATP-dependent molecular machines. BioEssays. 2003;25:1192–1200. doi: 10.1002/bies.10359. [DOI] [PubMed] [Google Scholar]
- 11.Lorch Y, Maier-Davis B, Kornberg RD. Chromatin remodeling by nucleosome disassembly in vitro. Proc Natl Acad Sci USA. 2006;103:3090–3093. doi: 10.1073/pnas.0511050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitehouse I, et al. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature. 1999;400:784–787. doi: 10.1038/23506. [DOI] [PubMed] [Google Scholar]
- 13.Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;13:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- 14.Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
- 16.Leschziner AE, et al. Conformational flexibility in the chromatin remodeler RSC observed by electron microscopy and the orthogonal tilt reconstruction method. Proc Natl Acad Sci USA. 2007;104:4913–4918. doi: 10.1073/pnas.0700706104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Racki LR, et al. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature. 2009;462:1016–1021. doi: 10.1038/nature08621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teif VB, Rippe K. Predicting nucleosome positions on the DNA: Combining intrinsic sequence preferences and remodeler activities. Nucleic Acids Res. 2009;37:5641–5655. doi: 10.1093/nar/gkp610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranjith P, Yan J, Marko JF. Nucleosome hopping and sliding kinetics determined from dynamics of single chromatin fibers in Xenopus egg extracts. Proc Natl Acad Sci USA. 2007;104:13649–13654. doi: 10.1073/pnas.0701459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- 21.Wang JP, et al. Preferentially quantized linker DNA lengths in saccharomyces cerevisiae. PLoS Comput Biol. 2008;4:e1000175. doi: 10.1371/journal.pcbi.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee W, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–44. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 23.Ranjith P, Marko JF. Filling of the one-dimensional lattice by k-mers proceeds via fast power-law-like kinetics. Phys Rev E. 2006;74:041602. doi: 10.1103/PhysRevE.74.041602. [DOI] [PubMed] [Google Scholar]
- 24.Schiessel H, Widom J, Bruinsma RF, Gelbart WM. Polymer reptation and nucleosome repositioning. Phys Rev Lett. 2001;86:4414–4417. doi: 10.1103/PhysRevLett.86.4414. [DOI] [PubMed] [Google Scholar]
- 25.Schiessel H, Widom J, Bruinsma RF, Gelbart WM. Erratum: Polymer reptation and nucleosome repositioning [Phys. Rev. Lett. 86, 4414 (2001)] Phys Rev Lett. 2002;88:129902. doi: 10.1103/PhysRevLett.86.4414. [DOI] [PubMed] [Google Scholar]
- 26.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: Advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.