

Abstract
Endoplasmatic reticulum aminopeptidase 1 (ERAP1) is a multifunctional enzyme involved in trimming of peptides to an optimal length for presentation by major histocompatibility complex (MHC) class I molecules. Polymorphisms in ERAP1 have been associated with chronic inflammatory diseases, including ankylosing spondylitis (AS) and psoriasis, and subsequent in vitro enzyme studies suggest distinct catalytic properties of ERAP1 variants. To understand structure-activity relationships of this enzyme we determined crystal structures in open and closed states of human ERAP1, which provide the first snapshots along a catalytic path. ERAP1 is a zinc-metallopeptidase with typical H-E-X-X-H-(X)18-E zinc binding and G-A-M-E-N motifs characteristic for members of the gluzincin protease family. The structures reveal extensive domain movements, including an active site closure as well as three different open conformations, thus providing insights into the catalytic cycle. A K528R mutant strongly associated with AS in GWAS studies shows significantly altered peptide processing characteristics, which are possibly related to impaired interdomain interactions.
Keywords: antigen presentation, ERAP1 mechanism, MHC restriction
Endoplasmic reticulum aminopeptidase 1 (ERAP1) is a multifunctional enzyme involved in regulation of immune and inflammatory responses. Functions attributed to ERAP1 include peptide trimming for antigen presentation on MHC class I molecules (1, 2), ectodomain shedding of cytokine receptors such as TNFR1(3), IL-6Ralpha, and IL-1RII (decoy IL-1 receptor) (4, 5), as well as regulation of blood pressure through inactivation of angiotensin II and conversion of kallidin to bradykinin in the kidney (6).
Major histocompatibility complex (MHC) class I molecules are expressed on the surface of most cells and present peptide fragments representing the cell protein repertoire to cytotoxic T lymphocytes. Display of abnormal (e.g., pathogen-derived) peptides may lead to the recognition and killing of infected cells. The peptides presented by MHC I molecules are 8–9 amino acid residues long and are derived from proteolytic fragments generated by the proteasome and other cytosolic proteases prior to transport to the endoplasmatic reticulum through TAP1/2 (7). Here, the final N-terminal trimming to the correct size occurs in the endoplasmatic reticulum by ERAP1 (1, 8–11). Epitope peptide precursor molecules of 9–15 amino acids in length are rapidly trimmed by ERAP1, while its activity is greatly reduced toward shorter peptides. The underlying mechanism for this remains unclear but has been suggested to be a “molecular ruler” mechanism (2). Importantly, ERAP1 and MHC I molecules appear to synergize in the generation of peptide repertoires that are appropriate for each of the polymorphic MHC I molecules (12). Lack of structural data for ERAP1 has thus far greatly hampered mechanistic studies. However, based on biochemical studies a model of ERAP1 activity was suggested that involves the binding of a 9–15-mer peptide to the active site of ERAP1, where successively N-terminal residues are cleaved off until the N-terminal part of the peptide is no longer reaching the active site. The rate at which this process occurs appears to be peptide sequence specific (13, 14).
Recent genome-wide association studies (GWAS) have revealed numerous ERAP1 polymorphisms to be associated with ankylosing spondylitis (AS)—a chronic inflammatory disease (15, 16) strongly associated with the HLA-B27 MHC I allele. Interestingly, recent GWAS and replication studies (17) reveal a clear association of the rs30187 SNP in ERAP1, encoding a K528R variant, with reduced AS risk, and, importantly, demonstrate a haplotype of this SNP exclusively in the context of HLA B27. This is suggestive of an influence of ERAP1 in the development of AS by a mechanism involving peptide trimming. Further, it has recently been reported that ERAP1 associations in psoriasis, an inflammatory skin disease commonly associated with AS, are also strongly correlated with MHC genotype (18). To understand the mechanism of ERAP1 catalytic activity and, possibly, to provide a structural explanation for the altered activity observed in this variant, we determined crystal structures of human ERAP1 (wild-type allele) in several open forms as well as an inhibitor bound closed conformation.
Results and Discussion
Structure Determination of Human ERAP1.
ERAP1 crystallized in two different space groups, containing either one (P622) or three molecules (P212121) in the asymmetric unit resulting in datasets with resolution limits of 2.7 and 3.0 Å, respectively (data collection and refinement statistics are given in Table S1). Each of these four molecules was present in a distinct conformation, which we postulate to represent snapshots of the enzyme during the catalytic cycle. The structure of the P622 crystal form was solved by single isomorphous replacement with anomalous scattering from a mercury derivative (SIRAS), whereas the structure of the second one (P212121) was determined by molecular replacement, using individual domains of the previously solved monomer as search models. Native crystals of space group P622 diffracted initially beyond 2.3 Å; however, the onset of radiation damage limited the final resolution to 2.7 Å. Nevertheless, the electron density map after SIRAS phasing and solvent flattening was of exceptional quality (an exemplary region of the electron density map is given in Fig. S1). Although the general metalloprotease inhibitor bestatin (N-(3R-amino-2S-hydroxy-1-oxo-4-phenylbutyl)-L-leucine) was not added to any crystallization trials, we found it bound to the active site of the first, but not the second, crystal form. The apparent occupancy of the molecule is low as judged from its increased thermal motion factors (Baverage protease domain: 14.9 Å2, Baverage bestatin: 58.6 Å2) and the patchy electron density. The most probable explanation for this observation is that bestatin was present in the general protease inhibitor mix added during purification and was thereby bound to the protein and copurified.
Overall Structure of the Closed State.
In space group P622 ERAP1 is present in a closed conformation; i.e., the active site of the molecule is occluded from the solvent. In contrast, space group P212121 contains three molecules in the asymmetric unit, and each molecule is present in slightly different open conformations (Fig. 1). The overall four-domain architecture of ERAP1 is remarkably similar to previously solved structures of Tricorn interacting factor F3 (PDB ID code 1Z5H) (19), aminopeptidase N from Escherichia coli (ePepN, PDB ID code 2ZXG) (20, 21), and Leukotriene A4 hydrolase/aminopeptidase (LTA4H, PDB ID codes 1HS6 and 3B7U) (22, 23), the latter being the closest structurally characterized eukaryotic ortholog (39% overall homology) that does not contain Domain III (Fig. S2; for comparison of domain similarities cf. Table S2). Domain I (residues 46–254) of ERAP1 (Fig. 1A) is composed of a central, saddle-shaped eight-stranded β-sheet (β1, 2, 4, 5, 8 and β11, 13, 14). This central motif is on one end flanked by a three-stranded β-sheet (β3, 6, 7) and on the opposite end by a four-stranded β-sheet (β9 + 10 and β12 + 15) that packs against the catalytic domain and interacts with domain IV via an elongated loop connecting strands 9 and 10. Domain II (residues 255–529), the thermolysin-like catalytic domain, consists of an N-terminal subdomain composed of one alpha-helix (α4) and a five-stranded β-sheet (β16–20). The exo-peptidase specific GAMEN motif (24) serves as the outermost strand of this sheet and creates one edge of the substrate binding cleft. Helix α6 is the linker to the alpha-helical subdomain and contributes the two histidine residues, His353 and His357, which form part of the canonical zinc-binding motif (H-E-X-X-H-X18-E). Moreover, this helix also forms the bottom of a central channel that runs along the surface of the catalytic domain. In contrast to thermolysin, and similar to ePepN, F3, and LTA4, the N-terminal substrate binding region of this cleft is obstructed by domain I and therefore provides a steric block, which is crucial for the exo-peptidase activity. ERAP1, like its structural homologues, lacks the calcium-binding loop of thermolysin on the C-terminal side of the bound substrate so that the cleft expands toward this side of the domain. The catalytic domain and the large C-terminal domain are connected by a small domain III (residues 530–614), which is composed of two β-sheets that form a beta-sandwich (β20–26). Domain IV (residues 615–940) consists solely of alpha-helices. It displays a bowl-like shape and in the closed state arches over the catalytic domain and thereby forms a large central cavity that completely seals off the active site. Like in F3 and ePepN, the helices adopt the form of a superhelix with an antiparallel topology. The internal cavity has an approximate volume of  and is therefore considerably larger than the one found in ePepN (
and is therefore considerably larger than the one found in ePepN (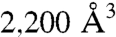 ) and LTA4 (
) and LTA4 ( ). Two disulfides anchor alpha-helices to one another, one within the thermolysin domain and the other within domain IV. Electron density interpreted as carbohydrate moieties were found attached to the following N-glycosylation sites: Asn70, Asn154, and Asn414.
). Two disulfides anchor alpha-helices to one another, one within the thermolysin domain and the other within domain IV. Electron density interpreted as carbohydrate moieties were found attached to the following N-glycosylation sites: Asn70, Asn154, and Asn414.
Fig. 1.

Overall structure and different conformational states of human ERAP1. (A) Closed form of ERAP1. ERAP1 consists of four domains (I: green, II: orange, III: yellow, IV: teal). The first crystal form showed one molecule where the active site of domain II is shielded by domain IV. N-glycosylation sites are colored in magenta. (B) Open form of ERAP1. A second crystal form contained three molecules of ERAP1 and revealed that it is indeed a highly dynamic molecule. Domain IV of molecule 2 moves away from the protease domain (II) and permits substrate access to the active site. Note that parts of domain IV, facing toward the catalytic domain, were disordered. (C) A distance plot shows the Cα-Cα distance of equivalent residues of the closed and the three open molecules. Conformational changes are mostly confined to domain III and IV. Regions that were not defined by electron density in the open molecules are blank. The insets show superpositions of the closed state (gray) and each open molecule. The different rotary motion for the individual molecules is indicated by arrows.
The Open States of ERAP1.
The second crystal form of ERAP1 contains three molecules of ERAP1 in the asymmetric unit, which all show the molecule in different open conformations. Except for specific parts of the structure (discussed below) we do not observe any significant conformational changes with respect to the closed state; the differences are confined to en bloc movements of domains III and IV (Fig. 1C). Domain III undergoes an approximately 15 ° rotation away from the core of the protein during opening, using the region around Gly529 at the beginning of the domain as a hinge (Fig. 1B). As a result of this movement, the C-alpha atom of Asp614, which marks the end of domain III and the beginning of domain IV, has shifted by around 7 Å. The relative movement of domain IV is different for the three molecules in the open crystal form (Fig. 1C). In molecule 1 domain IV swings 28 ° away from the protease domain, followed by a 12 ° lateral rotation, which leads finally to a 10-Å shift of the center of the domain away from the core. The different rotational movements for molecules 2 and 3 are highlighted in Fig. 1C. It should be noted that the tips of the helices that interact with domain I and II in the closed state show different degrees of disorder as they are not stabilized by crystal contacts, whereas the part of domain IV that interacts with domain III is ordered. Hence it is domain III that acts as a lever and pulls domain IV away from the active site and thereby makes it accessible to substrates. The observed flexibility of the C-terminal domain is a common theme among similar proteins like ePepN, LTA4H, and F3. To our knowledge, ERAP1 is so far the only one where both extremes—i.e., either open or completely closed states—have been shown. APN was reported in closed conformations, whereas F3 was reported in different open states that were postulated to be inactive (see below).
Interestingly, crystals of ERAP1 in the open state were obtained by cocrystallizing the protein in the presence of a substrate peptide. However, there is no evidence for a peptide bound to the protein, and there is also no evidence for the presence of a bestatin molecule, which was found in the active site of the closed conformation. The functional implications for this observation will be discussed below.
The Active Site of the Closed State.
ERAP1 belongs to the gluzincin family of metalloproteases and shares the common H-E-X-X-H-X18-E zinc-binding motif (24). The catalytic zinc ion is coordinated by His353, His357, Glu376 (Fig. 2), and additionally by the internal hydroxyl and carboxyl groups of bestatin in the closed state (Fig. 2A and Fig. S3). The terminal amino group of the inhibitor forms hydrogen bonds to Glu183 and Glu320, which form the N-terminal anchor for any peptide substrate. Bestatin binding to ERAP1 is similar to the one observed for ePepN (20) where the main-chain carbonyl group of the inhibitor, besides interacting with the zinc ion, also forms a hydrogen bond to the hydroxyl group of Tyr438, which in turn is involved in polarizing the carbonyl group during the catalytic cycle (Fig. S4). Glu354 is the equivalent of Glu298 in ePepN and Glu266 in F3 and is responsible for activation of a water molecule, which in turn performs a nucleophilic attack on the carbonyl carbon of the scissile bond and thereby triggers the catalytic mechanism (Fig. S4). The N-terminal phenyl ring of the bestatin moiety points into the S1 specificity pocket (Fig. 2A), where it stacks against the side chain of Phe433. The pocket is further bordered by the side chain of Met319, the aliphatic part of Glu183 and the carboxamide group of Gln181. A previous study showed that mutating Gln181 to glutamate increased the affinity of ERAP1 for basic N-terminal residues (25). The end of the pocket is formed by the aliphatic part of the side chain of Arg430 and its bottom is sealed via an electrostatic interaction of the guanidinium group of Arg430 with the carboxylate of Glu865 of domain IV, which stacks against the imidazole ring of His160 from domain I. Further interactions of ERAP1 with bestatin are shown in Fig. S3. In summary, ERAP1 forms less of a well-defined pocket, but rather a delimited area that may be instrumental for the protein to degrade peptides with different N termini (20).
Fig. 2.

Active site and internal cavity of ERAP1. (A) Bestatin binding to the active site of ERAP1. Bestatin is colored in orange and the surrounding electron density is an Fo-Fc map calculated in the absence of the molecule and drawn at 3.0σ. (B) Superposition of open (gray) and closed (colored) state of ERAP1. The movement of Tyr438 is highlighted and Phe433, which forms part of the S1 specificity pocket in the active protease is disordered in the open state. (C) Putative C-terminal substrate binding site within the internal cavity of ERAP1.
ERAP1 Is Inactive in the Open Form.
The region consisting of residues 425–434 of the catalytic domain forms part of the sidewall of the S1 pocket and is wedged between domains I and IV in the closed state. But as domain IV moves away in the open forms of ERAP1, these residues become dynamic and are no longer defined by electron density. Additionally, Tyr438, which forms a hydrogen bond to the carbonyl group of bestatin in the closed form via its hydroxyl group, is liberated and the side chain undergoes a 6-Å movement and now points away from the active site (Fig. 2B). Because the homologous residue in ePepN is involved in stabilizing the tetrahedral transition state during proteolysis (20), this argues for the inactivity of the open states. The observed conformation of Tyr438 is similar to the one observed in F3, which was also postulated to be inactive (19). Based on these observations we postulate that the binding affinity of bestatin or short peptides must be low in the open state due to lack of a structured binding pocket. The initial binding energy for longer peptides could be conferred by a secondary binding site, presumably located within the internal cavity. However, we cannot exclude an induced-fit mechanism where an interaction of the phenyl ring of the bestatin moiety with the side chain of Phe433 helps to stabilize the closed conformation. The presence of bestatin may therefore be due to altered affinities of the inhibitor in the two different crystallization conditions, which differ significantly in pH and composition. Recently, Thunnissen et al. proposed such a mechanism for the yeast ortholog of LTA4H (26), although the described structural rearrangements do not apply to ERAP1 and the resulting domain movements are very different for the two proteins. Either way, the presence of an additional site of interaction would favor longer peptide substrates by an increase in the overall binding energy and/or by increasing the local concentration of free N termini, thus increasing their binding propensity into the active site. Taken together, ERAP1 is devoid of protease activity in the open state due to a nonfunctional proteolytic site, and its closure is mandatory for proteolysis to take place.
Structural Evidence for a Molecular Ruler Mechanism.
It has been shown that ERAP1 has a strong preference for peptide substrates that are 9–16 residues long and that those substrates are degraded in a nonprocessive manner (2). Moreover, it was postulated that ERAP1 functions as a molecular ruler because cleavage efficacy is significantly reduced for peptides shorter than eight residues. The open conformations of ERAP1 revealed that the S1 pocket is not properly formed in the closed state, resulting most likely in reduced substrate binding affinities. In the absence of a cocrystal structure of ERAP1 with a peptide, it is speculated that additional binding energy is conferred by a secondary binding site within the internal cavity where the C-terminal part of the substrate is bound and hence defines the minimal product length. We identified a putative C-terminal binding pocket at the intersection of helices α21, 31, and 35 of domain IV. We modeled a phenylalanine residue into the pocket and a tripeptide into the active site in order to illustrate a possible binding mode of the C terminus and the resulting length constraints on a bound peptide (Fig. 2C). The distance of the C-terminal Cα-atom to the Cα of the P2′ residue of the modeled AAR-peptide is approximately 15 Å. This distance corresponds to about four residues and together with the residues bound to the active site adds up to a 7 mer. However, such an extended peptide conformation is unlikely, and we therefore assume that the peptide needs to encompass at least eight or nine residues to reach the active site, depending on the primary structure of the peptide. Longer peptides can easily be accommodated due to the large volume of the internal cavity. Though our modeling is tentative and the specific interactions may depend on the nature of a certain substrate, such a model would explain substrate binding in case of a disordered active site. Such a mechanism would also explain the marginal cleavage of shorter peptides, although it has been shown that ERAP1 is able to hydrolyse synthetic substrates (25), albeit with greatly reduced affinity.
It is instrumental for proteolysis to take place that domain IV swings back over the catalytic domain and in doing so activates the protease. After the N-terminal residue is cleaved, the cavity must open in order to release the products, which can then bind for another round of proteolysis. This is supported by occurrence of peptide intermediates when monitoring the shortening of ERAP substrates (see below, Fig. 3, and ref. 2). The “molecular ruler” model introduced earlier (2) is fully supported by our structural data: The non-processive nature of peptide trimming is a result of N- and C-terminal anchoring, requiring active site closure, and subsequent release of the N-terminally cleaved amino acid residue. For a new round of cleavage to occur, the trimmed peptide needs also to be released to allow another round of productive binding due to the necessity to attach the free amino-terminus at the S1 binding site. As shown in Fig. 3, all trimming intermediate products can be observed with different maxima at successive time points. That observation can only be explained by a bind-cleave-release mechanism in which only one N-terminal residue is removed at the time before the substrate peptide is released, thus supporting previous data (2). Furthermore, our data suggests that the 8-mer is still able to bind to ERAP1 but is not further processed and acts as a competitor for the 9-mer.
Fig. 3.

Altered N-terminal peptide trimming by the AS associated ERAP1 K528R mutant. (A) Extracted ion chromatograms (EIC) of the peptide cleavage product TANRELIQQEL after trimming of QITANRELIQQEL by ERAP1 wild type and the K528R mutant. Peptide precursor and enzymes were incubated for different sampling times revealing differential kinetic activity between both variants. (B) Relative quantitation of the EICs for the substrate peptide QITANRELIQQEL shows higher activity of the ERAP1 wild type. The intermediate product TANRELIQQEL is further processed by ERAP1 wild type while the K528R variant stops processing this peptide at the 11-mer stage. (C) Relative quantitation of the EICs for all observed peptide intermediates. While the wild type reaches equilibrium at the 8- and 9-mer, the K528R variant stops processing the 11-mer.
The simple model of protease activation by cavity closure provides a straightforward explanation why peptides above a certain size limit cannot be degraded because they prevent domain IV from sealing the proteolytic chamber, hence preventing the necessary activation events to happen. For peptides below this size limit, successful cleavage will depend on whether they can reach the active site from the point in the internal cavity where they are anchored.
The AS Protective ERAP1 Variant K528R Suggests a Defect in Open-Close Transitions.
An ERAP1 variant (K528R) associated with lower risk of AS was purified and tested in N-terminal trimming experiments using a peptide precursor containing the HLA-B27 restricted Chlamydia derived epitope NRELIQQEL (27). A precursor peptide with a four amino acid N-terminal extension was subjected to digestion with wild-type ERAP1 and the K528R variant in a time-dependent fashion, and the product peptide fragments were analyzed by quantitative mass spectrometry (Fig. 3). Wild-type ERAP1 protein was able to degrade the 13-mer peptide immediately in a manner that the starting peptide substrate was barely detected within a time frame of 30 min. However, the ERAP1 K528R variant was less efficient in peptide processing suggesting a defect in the catalytic process. As with other reported polymorphisms, the Lys528 residue is distant to the active site (Fig. 4A), located on the surface of domain III, where it forms polar interactions in the closed state with surrounding residues Asn414, Ser415, and Ser416 (Fig. 4B) from the catalytic domain which are further stabilized by Arg906, Gln910 and Glu913 from domain IV. In the open conformation, Lys528 interacts only with Asn414 and Ser416 (Fig. 4C). It is possible that substitution of Lys with Arg at this position results in additional formation of hydrogen bonds or polar interactions with surrounding amino acids, or through affecting interactions due to increased side-chain size. Both perturbations could result in defective transitions from the open to the closed form, thus affecting catalysis.
Fig. 4.

Polymorphism associated with ankylosing spondylitis. (A) Surface representation of ERAP1 Mutations associated to AS are colored in red. Localization of Met349Val in the substrate pocket is only visible in the open conformation. (B and C) Interactions of Lys528 with surrounding amino acids in the closed state (B) and the open state (C). The side chain of Asn414 was disordered in the open state, as were interactions with domain IV. Parts of the surface carbohydrate moiety (NAG1414) attached to Asn414 is depicted in the closed form (B).
The differential activity patterns observed for the reported ERAP1 variants (28), ranging from no obvious change in activity as determined in the Chlamydia peptide assay to severe processing defects as observed with the K528R variant (Fig. 3), supports the hypothesis that reduced trimming of peptides and consequent altered antigen presentation on MHC class I molecules is a mechanism involved in AS development. These structure-activity relationships and association to disease are currently being further investigated in our laboratories.
Materials and Methods
Cloning and Purification of Human ERAP1.
Full-length recombinant ERAP11–941 (MGC collection) was amplified and cloned into the FastBac vector containing a tobacco etch virus (TEV) protease cleavable C-teminal 10x-histidine tag. Mutants were produced using Quick Change Mutagenesis kit from Stratagene. Generation of recombinant baculoviruses, insect cell culture, and infections were performed according to the manufacturer instructions (Invitrogen). The cultures were collected 120 h postinfection, and cells were removed by centrifugation, the supernatant was used as a source of protein. Supernatants were supplemented with Tris buffer pH 8.0 to a final concentration of 50 mM, NaCl to a final concentration of 300 mM, and NiSO4 to a final concentration of 1 mM. This solution was supplemented with PMSF to a final concentration of 1 mM and 1 tablet of EDTA-free protease inhibitors (Roche) per 2 L of solution. After 4 hr of protein adsorption on the Ni-NTA resin, the suspension was loaded on a gravity column and washed with 20 bed volumes of washing buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol and 10 mM imidazole). The protein was eluted with elution buffer containing 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol and 250 mM imidazole. Fractions containing protein were combined and applied to a Superdex 200 16/60 (GE Healthcare) gel-filtration column equilibrated in 10 mM HEPES (pH 7.5), 500 mM NaCl, 5% glycerol and 1 mM TCEP. Fractions containing ERAP1 were analyzed by SDS/PAGE. The purified protein was concentrated to 17 mg/ml and used for crystallization or activity assays.
Crystallization.
All crystals of ERAP1 were grown by using the sitting drop vapor diffusion method. Crystals of the closed form were obtained by mixing 100 nl of protein solution (17 mg/ml) and 50 nl of a precipitant consisting of 1.4 M KCitrate; 0.1 M Cacodylate pH 5.7 and 0.17 mM n-Dodecyl-Beta-D-maltoside. The open form of ERAP1 was crystallized by mixing 2∶1 ratio of protein and buffer consisting of 0.1 M Tris pH 8.0 and 20% PEG3350. Before crystallization the protein was mixed with a peptide WRVYEKCALK (molar ratio 1∶10). A solution containing mother liquor, supplemented with 20% ethylene glycol was added to both crystal forms before they were flash frozen in liquid nitrogen. A mercury derivative of the closed crystal form was prepared by adding 1 μl of reservoir solution supplemented with 10 mM Thiomersal to a drop containing native crystals of space group P622. After 10 min, a crystal was transferred to a cryo solution containing reservoir solution supplemented with 25% ethylene glycol and the crystal was immediately flash frozen in liquid nitrogen.
Data Collection and Structure Solution.
Data on native and derivative crystals were collected on beamline I03 at the Diamond Light Source. All datasets were integrated with XDS (29) and scaled with SCALA (30). One mercury site was identified with the program HYSS (31) followed by phase refinement with SHARP (32) and solvent flattening with SOLOMON (33). Automated model building was performed with ARP/wARP (34), resulting in a more than 85% complete model. After several rounds of manual rebuilding in COOT (35) and subsequent cycles of refinement with REFMAC (36), the model of the closed form converged to a final Rfactor and Rfree of 15.5% and 21.5%, respectively. The structure of the open crystal form was determined by molecular replacement using the program PHASER (37). Individual domains of the previously solved closed state were used as search models. Alternating rounds of refinement with REFMAC and rebuilding with COOT gave a final Rfactor and Rfree of 22.9% and 28.3%, respectively. The program VOIDOO (38) was used to calculate cavity volumes and the program LSQMAN (39) was used to generate distance plots.
Peptide Trimming Assays.
100 nmol of the Chlamydia (protein accession no. C4PLH3) derived peptide 110–122 QITANRELIQQEL containing the HLA-B27-restricted epitope NRELIQQEL were incubated with 3.5 μg of wild-type ERAP1 or the AS protective variant K528R in 1ml of 50 mM Tris pH 7.8 containing 0.5 μg protease free bovine serum albumin (Sigma) in a time course experiment at 30 °C. The reaction was stopped by adding formic acid (1% final concentration) and the samples kept at -80 °C until analysis. Samples were desalted using C18 Zip-tip (Millipore) and analyzed by nano-LC-MS using a Chipcube coupled to an Agilent 6520 quadrupole time-of-flight (Q-Tof) tandem mass spectrometer (Agilent). Peptide separation was performed using a 10 min gradient from 3% acetonitrile, 0.1% formic acid in H2O to 45% acetonitrile, 0.1% formic acid in H2O at a flow rate of 600 nl/ min. Data was acquired in MS only mode. Quantitative data was obtained by the generation and integration of Extracted Ion Chromatograms (EIC) for the doubly charged ions of each peptide intermediate using the Masshunter Qualitative Analysis Software (Agilent). As a normalization, the areas of all EICs observed in each time point were added together, and the signal of each peptide intermediate was calculated as a percentage of the sum of all EICs.
Supplementary Material
Acknowledgments.
We thank the staff at Diamond Light Source for expert help at the beamline. The Structural Genomics Consortium is a registered charity (no. 1097737) funded by the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck and Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust. M.A.B. is funded by a National Health and Medical Research Council (Australia) Principal Research Fellowship. This study was also funded, in part, by the Arthritis Research UK (grants 19536, 18599, and 18797), by the Wellcome Trust (grant 076113), and by the National Institute for Health Research Oxford Comprehensive Biomedical Research Centre ankylosing spondylitis chronic disease cohort (theme code A91202). D.H. was in part funded by the National Ankylosing Spondylitis Society (UK), R.F., P.B., and B.M.K. are supported by an Action Medical Research Grant (charity no. 208701 and SC039284). The study was supported by the Oxford National Institute for Health Research Biomedical Research Unit.
Note.
During typesetting of this manuscript a bestatin-bound open structure of ERAP1 was disclosed (40), supporting the conclusions described in the current article.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The structures have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2YD0 and 3QNF).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1101262108/-/DCSupplemental.
References
- 1.Saric T, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–1176. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 2.Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci USA. 2005;102:17107–17112. doi: 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui X, et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest. 2002;110:515–526. doi: 10.1172/JCI13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui X, Rouhani FN, Hawari F, Levine SJ. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of TNF receptor type 1 shedding. J Immunol. 2003;171:6814–6819. doi: 10.4049/jimmunol.171.12.6814. [DOI] [PubMed] [Google Scholar]
- 5.Cui X, Rouhani FN, Hawari F, Levine SJ. An aminopeptidase, ARTS-1, is required for interleukin-6 receptor shedding. J Biol Chem. 2003;278:28677–28685. doi: 10.1074/jbc.M300456200. [DOI] [PubMed] [Google Scholar]
- 6.Tsujimoto M, Hattori A. The oxytocinase subfamily of M1 aminopeptidases. Biochim Biophys Acta. 2005;1751:9–18. doi: 10.1016/j.bbapap.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 8.Blanchard N, Shastri N. Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol. 2008;20:82–88. doi: 10.1016/j.coi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falk K, Rotzschke O. The final cut: how ERAP1 trims MHC ligands to size. Nat Immunol. 2002;3:1121–1122. doi: 10.1038/ni1202-1121. [DOI] [PubMed] [Google Scholar]
- 10.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 11.York IA, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 12.Kanaseki T, Blanchard N, Hammer GE, Gonzalez F, Shastri N. ERAAP synergizes with MHC class I molecules to make the final cut in the antigenic peptide precursors in the endoplasmic reticulum. Immunity. 2006;25:795–806. doi: 10.1016/j.immuni.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evnouchidou I, et al. The internal sequence of the peptide-substrate determines its N-terminus trimming by ERAP1. PLoS One. 2008;3:e3658. doi: 10.1371/journal.pone.0003658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hearn A, York IA, Rock KL. The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J Immunol. 2009;183:5526–5536. doi: 10.4049/jimmunol.0803663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MA. Genetics and the pathogenesis of ankylosing spondylitis. Curr Opin Rheumatol. 2009;21:318–323. doi: 10.1097/bor.0b013e32832b3795. [DOI] [PubMed] [Google Scholar]
- 16.Brown MA. Progress in spondylarthritis. Progress in studies of the genetics of ankylosing spondylitis. Arthritis Res Ther. 2009;11:254–259. doi: 10.1186/ar2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvey D, et al. Investigating the genetic association between ERAP1 and ankylosing spondylitis. Hum Mol Genet. 2009;18:4204–4212. doi: 10.1093/hmg/ddp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strange A, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kyrieleis OJ, Goettig P, Kiefersauer R, Huber R, Brandstetter H. Crystal structures of the tricorn interacting factor F3 from Thermoplasma acidophilum, a zinc aminopeptidase in three different conformations. J Mol Biol. 2005;349:787–800. doi: 10.1016/j.jmb.2005.03.070. [DOI] [PubMed] [Google Scholar]
- 20.Addlagatta A, Gay L, Matthews BW. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc Natl Acad Sci USA. 2006;103:13339–13344. doi: 10.1073/pnas.0606167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Addlagatta A, Gay L, Matthews BW. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry. 2008;47:5303–5311. doi: 10.1021/bi7022333. [DOI] [PubMed] [Google Scholar]
- 22.Thunnissen MM, Nordlund P, Haeggstrom JZ. Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat Struct Biol. 2001;8:131–135. doi: 10.1038/84117. [DOI] [PubMed] [Google Scholar]
- 23.Tholander F, et al. Structure-based dissection of the active site chemistry of leukotriene A4 hydrolase: implications for M1 aminopeptidases and inhibitor design. Chem Biol. 2008;15:920–929. doi: 10.1016/j.chembiol.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 24.Tsujimoto M, Goto Y, Maruyama M, Hattori A. Biochemical and enzymatic properties of the M1 family of aminopeptidases involved in the regulation of blood pressure. Heart Fail Rev. 2008;13:285–291. doi: 10.1007/s10741-007-9064-8. [DOI] [PubMed] [Google Scholar]
- 25.Goto Y, Tanji H, Hattori A, Tsujimoto M. Glutamine-181 is crucial in the enzymatic activity and substrate specificity of human endoplasmic-reticulum aminopeptidase-1. Biochem J. 2008;416:109–116. doi: 10.1042/BJ20080965. [DOI] [PubMed] [Google Scholar]
- 26.Helgstrand C, Hasan M, Uysal H, Haeggstrom JZ, Thunnissen MMGM. A leukotriene A(4) hydrolase-related aminopeptidase from yeast undergoes induced fit upon inhibitor binding. J Mol Biol. 2011;406:120–134. doi: 10.1016/j.jmb.2010.11.059. [DOI] [PubMed] [Google Scholar]
- 27.Appel H, et al. Use of HLA-B27 tetramers to identify low-frequency antigen-specific T cells in Chlamydia-triggered reactive arthritis. Arthritis Res Ther. 2004;6:R521–534. doi: 10.1186/ar1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haroon N, Inman RD. Endoplasmic reticulum aminopeptidases: Biology and pathogenic potential. Nat Rev Rheumatol. 2010;6(8):461–467. doi: 10.1038/nrrheum.2010.85. [DOI] [PubMed] [Google Scholar]
- 29.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 30.Evans P. Scaling and assessment of data quality. Acta Crystallogr D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 31.Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Crystallogr D. 2003;59:1966–1973. doi: 10.1107/s0907444903018043. [DOI] [PubMed] [Google Scholar]
- 32.La Fortelle Ed, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Method Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 33.Abrahams JP, Leslie AGW. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Crystallogr D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 34.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 39.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen T, et al. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat Struct Mol Biol. 2011 doi: 10.1038/nsmb.2021. 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.