

Abstract
We demonstrate electron microscopy of fully hydrated eukaryotic cells with nanometer resolution. Living Schizosaccaromyces pombe cells were loaded in a microfluidic chamber and imaged in liquid with scanning transmission electron microscopy (STEM). The native intracellular (ultra)structures of wild-type cells and three different mutants were studied without prior labeling, fixation, or staining. The STEM images revealed various intracellular components that were identified on the basis of their shape, size, location, and mass density. The maximal achieved spatial resolution in this initial study was 32 ± 8 nm, an order of magnitude better than achievable with light microscopy on pristine cells. Light-microscopy images of the same samples were correlated with the corresponding electron-microscopy images. Achieving synergy between the capabilities of light and electron microscopy, we anticipate that liquid STEM will be broadly applied to explore the ultrastructure of live cells.
Introduction
Electron microscopy (EM) has been a key provider of our knowledge about subcellular and molecular structures in cells (1). Without EM it would not have been possible to integrate biochemical and atomic-scale structural information, obtained, for instance, from x-ray crystallography and nuclear magnetic resonance studies, into a realistic cellular framework (2). Scientists have hoped since the early days of electron microscopy to achieve better resolution than the diffraction-limited resolution of light microscopy for imaging live eukaryotic cells, to gain insights into the native intracellular ultrastructure (3). Despite various attempts, the spatial resolution obtained with EM on pristine cellular samples in aqueous solutions was not better than that achievable with light microscopy (4,5). Nanometer resolution is achieved with cryo-EM (6,7) and x-ray microscopy (8,9), but both require frozen samples to preserve the cellular ultrastructure. Consequently, the imaged cells are not in their native liquid environment, nor in a living state. Nanoscale scanning probe microscopy is limited to the imaging of cellular surfaces (10,11). Superresolution light microscopy (3,12) reaches a subdiffraction resolution of <50 nm in live cells, but only on fluorescent labels attached to specific sets of proteins, and not on the native cellular ultrastructure. It is thus not possible at the present time to study the ultrastructure of pristine eukaryotic cells.
Here, we demonstrate electron microscopy of fully hydrated eukaryotic cells with up to 32-nm resolution, an order of magnitude better than the resolution of conventional light microscopy. Schizosaccaromyces pombe cells, widely used as a model organism in molecular and cell biology (13), were loaded in a microfluidic chamber, kept alive, and then imaged in liquid with scanning transmission electron microscopy (STEM) (14). The cells were imaged in their pristine state, without genetic modification, to include fluorescent labels, staining, sectioning, etc. The native intracellular ultrastructure of wild-type cells and three different mutants was studied in vivo.
Materials and Methods
Yeast cell cultures
Liquid cultures of S. pombe cells, wild-type 972, spn3Δ mutant, and temperature sensitive orb6-25, and cdc25-22 cdc15(27A) mutants, were grown for 24 h in 25 ml liquid consisting of YES broth media, 0.3% yeast extract, 0.3% malt extract, 0.5% peptone, and 1% D-glucose, with 50 mg/L each of adenine, histidine, leucine, uracil, and lysine (Sunrise Science Products, San Diego, CA) in a 25°C incubator with shaking at 250 rpm. The optical density (OD) was determined with a spectrometer at 595 nm (Evolution 60, Thermo Scientific, Waltham, MA) and the wild-type and spn3Δ mutant cells were harvested when the OD value reached 0.3, indicating that the cultures were in the log phase of their growth curve. The orb6-25 mutant cultures were further grown at 36°C for additional 3 h. From each culture, 10 ml was harvested by centrifugation (10 min/2000 rpm). The cells were washed with 10 ml of sterile 10-mM Na-HEPES supplemented with 2% D-glucose (both from Sigma Aldrich, St. Louis, MO) at pH 7.2 (NaHEPES). Pellets were resuspended in 1 or 2 ml of NaHEPES. A 20 μM FUN-1 (Invitrogen, Carlsbad, CA) stock solution in NaHEPES was prepared and added 1:1 to the yeast-cell suspension, yielding 10 μM FUN-1 final concentration. The cultures were incubated in the dark for at least 60 min before fluorescence microscopy was performed to check for the red fluorescent staining of vacuoles, indicating viability of the cells, or for bright and more uniform yellow-green fluorescence, indicating a dead or dying status (15).
Preparation of the microfluidic chamber with yeast cells
The liquid STEM system consisted of a microfluidic chamber assembled from two silicon microchips with electron-transparent windows and a liquid flow specimen holder (Protochips, Raleigh, NC) (14,16). The electron-transparent windows spanned an area of 50 × 400 μm and were made of 50-nm-thick silicon nitride. The microchips were plasma-cleaned to render the surfaces hydrophilic, then coated with poly-L-lysine (Sigma-Aldrich) to enhance cell adherence and to maintain the surface hydrophilic. Gold nanoparticles of sizes 5, 10, and 30 nm were applied to the upward-facing window, serving as a guide for focusing of the STEM (except for Figs. 5 A and 6 A). The microfluidic chamber was loaded with live S. pombe cells by placing a droplet of a suspension of cells in buffer solution on a microchip forming the lower half of the chamber. The microfluidic chamber was then closed with a second microchip. The loading procedure was completed within 1 min, and light-microscopy and STEM images were recorded within a few minutes.
Figure 5.
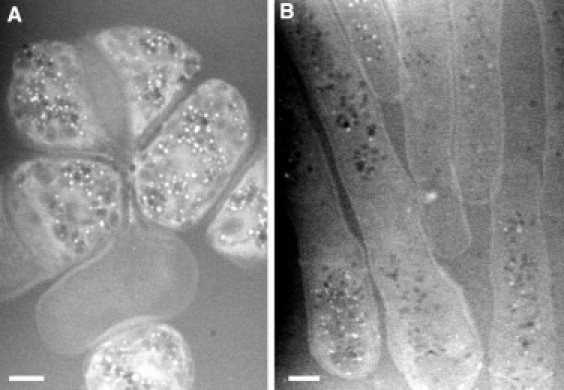
Liquid STEM of S. pombe mutants. (A) Image of orb6–25 mutant cells. (B) Image showing several cells of the temperature-sensitive mutant cdc25–22 cdc15(27A). The cells arrest at the G2 phase before entering mitosis and are not able to build septa. This leads to the development of an abnormally elongated phenotype.
Figure 6.
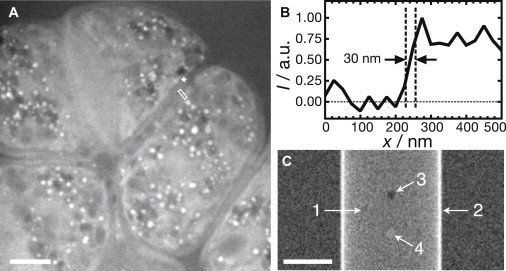
Analysis of the spatial resolution of liquid STEM of fully hydrated yeast cells. (A) Selected region of the image recorded of an orb-25 mutant shown in Fig. 5B. (B) Line scan representing the average intensity, I, versus the position, x, of a 5-pixel-wide line drawn over a sharp edge indicated by an asterisk in A. The 25–75% rising edge width is 30 nm. The signal was normalized to unity at the maximal intensity, and to zero at the intensity of the lower side of the edge. (C) Simulated liquid STEM image of a model yeast cell in a water layer. The image is the top view of a simple cell model (arrow 1) consisting of a cylinder (arrow 2 points to the cylinder wall), with one lipid droplet (arrow 3), and one protein vesicle (arrow 4). Scale bars, 2 μm.
Light microscopy
After the specimen holder was loaded with live yeast cells, it was placed on a mechanical translation stage with three directions of movement. The tip of the specimen holder containing the microchips was positioned in a water droplet above a 60×, 1.0-numerical-aperture water-immersion lens on an inverted microscope (TS100, Nikon, Tokyo, Japan). Images were recorded at room temperature using a 3-megapixel charge-coupled device camera (Micropublisher 3.3 RTV, QImaging, Surrey, British Columbia, Canada), and Qcapture software, and stored in 8-bit tiff format. The positions of the cells with respect to the four corners of their silicon nitride windows were used to correlate the STEM images with the light-microscopy images. Fluorescence imaging showed that the S. pombe yeast cells could be kept alive for extended periods of up to hours in the microfluidic chamber (data not shown). The images were adjusted for optimum brightness and contrast, cropped, and color-enhanced using Image J software (National Institutes of Health, Bethesda, MD).
Liquid STEM imaging
The STEM (CM200 TEM/STEM, Philips/FEI, Hillsboro, OR) was set to 200 kV, with a beam semiangle α of 5.6 mrad, a probe current of 0.22 nA, and an annular dark field (ADF) detector semiangle of 70 mrad (Fischione Instruments, Export, PA). STEM images of 1024 × 1024 pixels were recorded at room temperature using ES Vision software (Philips/FEI) with a pixel dwell time of 10 μs and a pixel size of 25 nm (magnification 4800×), and stored as 16-bit tiff files. The electron probe diameter containing 50% of the current was calculated to be 0.9 nm, resulting from 0.85 nm under ideal circumstances at the used beam semiangle (17) plus an estimated contribution of ∼0.3 nm due to instabilities and imperfections of the alignment, added quadratically. An imaging session started by localizing the edge of a window with the electron microscope set to fast scanning in searching mode, such that the positions of the yeast cells in the STEM images could be correlated with their positions in the light-microscopy images. The microscope was then focused using the contrast obtained on the gold nanoparticles. The stage position was changed to the direction of a yeast cell of interest as located from its fluorescence image. By repeating stage movements and refocusing, the cell of interest was approached. As soon as the cell of interest appeared in the field of view, a STEM image was recorded. To enhance the visibility of the cellular structures, the images were filtered with a convolution filter with a kernel of (1,1,1,1,3,1,1,1,1) in ImageJ (NIH). The gamma level was set to 0.75, and the contrast curve, contrast level, and brightness level were adjusted for maximal visibility of the biological structures (Adobe Photoshop, Adobe, San Jose, CA). The red channel of the fluorescence image (see Fig. 3 B) was overlaid after adjustment for the difference in magnification and for image rotation (Adobe Photoshop) (see Fig. 3 C).
Figure 3.
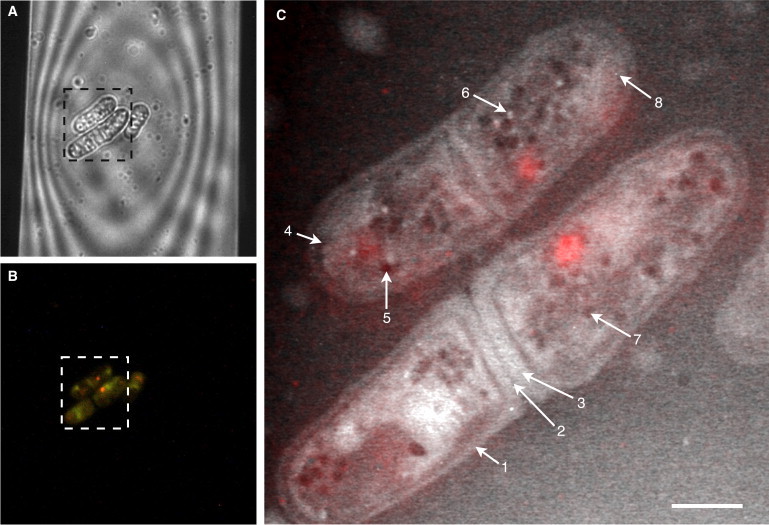
Light microscopy and liquid STEM of fully hydrated wild-type S. pombe yeast cells, which were alive at the onset of the recording of the first STEM image. (A) Phase-contrast image showing S. pombe cells within a portion of the viewing window of the microfluidic chamber. (B) In the corresponding fluorescence image, it can be seen that all cells accumulated FUN-1 dye and emitted a punctuated red fluorescence, the typical signal of living yeast cells. (C) Liquid STEM image recorded in the fully hydrated state of the same pristine yeast cells as shown in A and B. Numbered arrows indicate examples of allocated organelles: the cell wall (1), the primary septum (2), the secondary septum (3), a cell membrane invagination (4), a lipid droplet (5), a peroxisome (6), an unclassified vesicle (7), and a gold nanoparticle (8). The color is an overlay of the red channel of the fluorescence image (B). Scale bar, 2 μm.
Measuring the liquid thickness
The liquid thickness was measured using STEM by comparing the fraction N/N0 of the incoming electrons scattered onto the ADF detector. The thickness of the liquid T follows from this fraction as (14,18) T = −l(β)ln(1 − N/N0), with l(β) the mean-free-path length for elastic scattering into detector opening semiangle β or larger. Water has lwater = 10.5 μm for β = 70 mrad. We determined a liquid thickness of 6 ± 2 μm, which is consistent with the diameter of the yeast cells (see Fig. 3 C). The thickness was 3 ± 1 μm at the corner of the window, i.e., the windows bulged outward at the locations of the yeast cells. The fringes in the phase-contrast microscopy image (see Fig. 3 A) confirm bending of the silicon nitride window. The liquid thicknesses were 4 ± 1 μm, 4 ± 1 μm, 3 ± 1 μm, and 3 ± 1 μm, respectively (see Figs. 4, C and D, and 5, A and B). In the latter images, the yeast cells had probably flattened in the microfluidic chamber.
Figure 4.
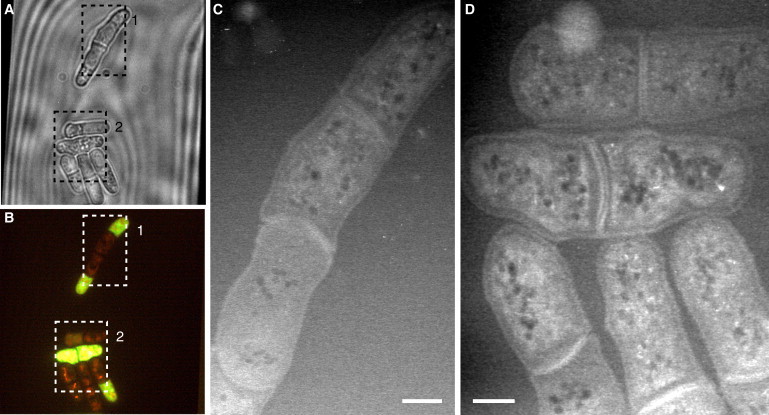
Light microscopy and liquid STEM of two Spn3Δ mutants of S. pombe. (A and B) Phase-contrast (A) and fluorescence (B) images of mutant yeast cells. The bright horizontally oriented cell represents a dead cell, whereas the others are alive. (C) Liquid STEM image of an elongated cell with multiseptal phenotype shown in dashed rectangle 1 in A and B. (D) Image of a group of cells showing the wild-type phenotype, from rectangle 2 in A and B.
Measuring object dimensions
The cell wall thickness observed (see Fig. 3 C) was measured from line scans made with a width of 6 pixels and a direction perpendicular to the bright line outlining the yeast cells, and by calculating the full width at half-maximum of the intensity peak of the line scan. Measurements were taken at five positions to produce an average value of 0.19 ± 0.04 μm. The diameters of six vesicles with dark contrast (lipid vesicles) were determined from their full width at half-minimum values, for an average value of 0.35 ± 0.08 μm. The average diameter of seven bright vesicles was 0.16 ± 0.02 μm.
Results
Correlative light microscopy and STEM of live S. pombe cells
S. pombe cells are cylindrical, with a diameter of ∼4 μm and a length of ∼6–15 μm. They grow by elongation of their ends and divide by medial septation, followed by cleavage of the primary septum. The yeast cells were placed in their fully hydrated, normal physiological state at ambient temperature in a saline-filled microfluidic chamber (16) with ultrathin windows for STEM of liquid specimens (Fig. 1). The windows separated the liquid from the vacuum of the EM, and were transparent to the photons and electrons of the energies used here. The microfluidic chamber was contained in an EM specimen holder for liquid specimens. The loading procedure was completed within 1 min. To verify that the cells in the microfluidic device were indeed living before STEM, the cells were incubated with a yeast-specific fluorescent live-dead indicator. For light-microscopy examination of the yeast cells, the EM specimen holder was positioned on a water immersion lens (Fig. 2). The phase-contrast image of Fig. 3 A depicts three S. pombe cells, two of which had just divided. The fluorescence image of Fig. 3 B shows bright red spots within the cells, indicating that the dye was transported into vacuoles in living cells, a process that is only possible in living cells. There was no sample preparation apart from the incubation with the dye after the yeast cells had been washed and transferred into the imaging buffer at the time the culture had reached the log phase of its growth curve.
Figure 1.
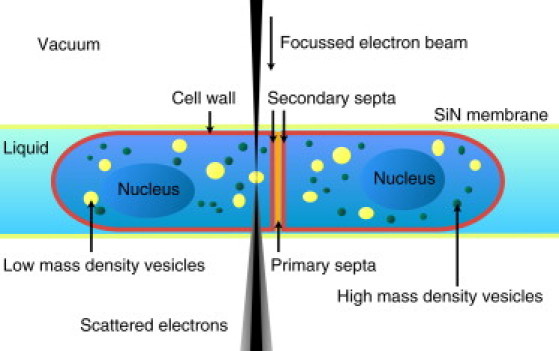
Schematic of a dividing S. pombe cell surrounded by liquid and contained between two silicon-nitride windows, transparent for photons and electrons. In STEM, the electron beam scans a defined area of the specimen, and the scattered transmitted electrons are used for detection. The contrast in the STEM images depends on the mass density and the atomic composition of the biological materials.
Figure 2.

Tip of a STEM specimen holder for liquid specimens placed on a 60×, 1.0 NA water immersion lens of a light microscope.
Within a few minutes after the recording of the light-microscopy images, the specimen holder was transferred to the vacuum chamber of the electron microscope. Here, the same yeast cells were localized and imaged while still in their liquid environment. The cellular structures as imaged with light microscopy were correlated with their EM counterparts via their previously determined coordinates on the SiN window. The STEM image of Fig. 3 C shows the two dividing cells and the edge of a third cell seen in the light-microscopy image. Fig. 3 C reveals intracellular components with details down to the ultrastructural level, such as the cell wall, the primary and secondary septa, and different types of intracellular vesicles. The upper cell appears to be in the process of division, whereas the lower cells were already in the process of separation after cell division. The red color indicates the locations of the vacuoles, albeit with a limited precision on account of the limited spatial resolution of the fluorescence image. The background appears darker in the left upper corner with respect to the right lower corner, due to a variation of the liquid thickness over the field of view. Gold nanoparticles applied to the upper window for focusing purposes are also visible. Fluorescence microscopy of similar samples showed that the yeast cells were not viable after STEM imaging. Even though the cells were killed by exposure to the electron beam in the STEM, the ameliorating fact is that the cells were living at the onset of the recording of the first micrograph. The STEM image of Fig. 3 C thus represents the ultrastructure of pristine S. pombe cells in liquid.
Assignment of visible structures to known organelles
We have assigned the visible structures in the STEM images to known yeast organelles using information about organelle morphology, size, and mass density. The first and most prominent feature of all yeast cells is their outer 0.1- to 0.2-μm-thick cell wall, composed mainly of polysaccharides (19). The cell wall emerged brighter in the image than the surrounding buffer (Fig. 3 C, arrow 1). The contrast obtained with STEM depends on the atomic number(s) and the mass density of the material in the path of the electron beam (20). The brighter signal indicates a higher mass density than the aqueous medium surrounding the cells, consistent with the higher mass density of 1.3 g/cm3 of the cell wall (21). The measured thickness of the bright line was 0.19 ± 0.04 μm, in agreement with published values for the thickness (19), noting that the cell wall will appear broader in the image, since it represents a projection through the three-dimensional shape of the cell wall. The cellular regions enclosed by the cell wall appear brighter than the surrounding liquid, as explained by their content of protein, lipids, and DNA, all with a higher mass density than the surrounding imaging buffer. As expected from the harvesting at their log phase growth, many cells in our samples were in the process of division and had a septum composed of a central primary septum (Fig. 3 C, arrow 2) flanked on each side by secondary septa (Fig. 3 C, arrow 3). Some cells exhibited fingerlike, ∼0.2-μm long structures close to the cell wall (Fig. 3 C, arrow 4). We suggest that these structures are invaginations in the cell membranes (22).
A distinct group of intracellular structures in yeast have spheroid forms and are classified as vacuoles or vesicles. These can be sorted on the basis of their size and their mass density (23). Two groups are particularly prominent in the STEM images. The first is composed of dark, round shapes (Fig. 3 C, arrow 5), which we assume represent lipid droplets known to exist in S. pombe cells (24). Our measured diameter of 0.35 ± 0.08 μm agrees with the reported lipid droplet size (24,25) of 0.32 ± 0.10 μm. The mass density of lipids is ∼0.9 g/cm3 and thus lower than the density of water; this explains the darker appearance of these spheres in the STEM image. A second class of vesicles is brighter than the cytoplasm and has an average diameter of 0.16 ± 0.02 μm (Fig. 3 C, arrow 6). These are presumably the cores of peroxisomes, which may include a dense crystalloid core consisting of urate oxidase molecules (26). Several other spheroid structures with intermediate gray tones (see, for instance, Fig. 3 C, arrow 7) can be discerned. Those structures could be lipid vesicles in regions of higher protein density. Some of the gold nanoparticles on the silicon nitride window that were used as aids for focusing the STEM are also visible (arrow 8). This analysis of the STEM image of Fig. 3 C shows that the obtained information is consistent with existing knowledge of the (ultra)structure of S. pombe.
Screening of mutant cells with liquid STEM
The short sample preparation and imaging time is beneficial for the screening of series of samples, for example, from cells with different mutations. To test STEM for its applicability to screening, we imaged cells of three mutants of S. pombe yeast. Fig. 4 A and B display light-microscopy images of the septin mutant spn3Δ. Cells of this mutant are delayed in separation and often grow in chains of typically two to four cell compartments, as seen in the liquid STEM image of Fig. 4 C. Several spn3Δ cells that have not developed a multiseptal phenotype are depicted in Fig. 4 D. The fluorescence image of Fig. 4 B shows both, live cells with punctuated red fluorescence, and a dead cell, the horizontally oriented yeast cell with a bright yellow-green fluorescence.
Cells of another mutant, orb-25, are shown in Fig. 5 A. These cells have a disturbed cell polarity displaying isotropic cell growth and a shortened length. In addition, most of these mutants accumulated an abundance of vacuoles compared to the wild-type. A few cells, like the one in the lower half of Fig. 5 A, appeared empty of organelles. A temperature-sensitive cdc25–22 cdc15(27A) mutant was also imaged (Fig. 5 B). These studies required only a few hours per mutant, a time that is similar to that needed for light microscopy, and much shorter (hours versus weeks) than what is possible with existing electron microscopy approaches (7).
Spatial resolution and imaging contrast of STEM of hydrated yeast cells
The maximal spatial resolution in the STEM images of the yeast cells in liquid was determined from Fig. 6 B (orb6–25 mutant), which corresponded to the smallest liquid thickness in the experimental series. Fig. 6 A shows a selected region of this image. As a measure of the resolution, we have used the 25–75% rising edge width (27) r25–75, applicable because the electron probe size was smaller than the pixel size for the upper ∼2 μm of the sample (electron beam-sample interactions broadened the probe to values larger than the pixel size of 25 nm for deeper layers (18)). The average intensity of the 5-pixel-wide line at marker 1 versus position is shown in Fig. 6 B. Five of the sharpest edges on elongated structural components in the image resulted in an average r25–75 = 32 ± 8 nm, approaching the pixel size of 25 nm. This value of r25–75 represents the maximal achieved resolution in this study on yeast ultrastructure.
The contrast of liquid STEM is determined by the signal/noise ratio observed between pixels recorded at the location of a certain material versus the background signal from the surrounding liquid. This contrast depends on the thickness of the liquid and on the electron density of the object on which the contrast is obtained (18). The resolution is given by the minimum size of the object, or the sharpest edge of the object that can be observed above the noise. The larger the difference is between the electron density of a biological material and the electron density of water, the larger the contrast, and hence the higher the resolution. Furthermore, the thinner the liquid, the higher is the resolution. The image of Fig. 3 C has a maximal resolution of 50 ± 10 nm on biological ultrastructure, as measured for a line scan over the edge of the dark-appearing vesicle (5). Indeed, this sample was thicker than the sample used for Fig. 6. The resolution on the gold nanoparticles is much higher (see Fig. 3 C).
There is no simple equation to calculate the resolution, because it depends on the geometry and the composition of the sample. To understand the achieved resolution and contrast, we have simulated STEM of a model sample by Monte Carlo methods (28,29). The sample consisted of a water layer 4 μm thick enclosed between two silicon nitride layers each 50 nm thick, with an outer cylinder radius of 2 μm and a wall thickness of 100 nm, as a simplified model of a yeast cell. As approximation of the cell wall material (19), we used glucose with a chemical composition of H10C6O5 and a mass density of 1.5 g/cm3. The cylinder was filled with water. The cell model further contained a sphere with a diameter of 400 nm placed at a depth of 500 nm in the cell (as seen from the top), with a chemical composition of H98C55O6 and a mass density of 0.92 g/cm3, as a model for lipid droplets (30). A vesicle was included as a sphere with a diameter of 400 nm at a depth of 500 nm with the chemical composition and mass density of an average protein of H5C3NO and a mass density (31) of 1.3 g/cm3. STEM images were simulated with the same parameters (but with a smaller image size) as used in the experiments, and filtered. The simulated image of Fig. 6 C reproduces the basic features of the experimental figures with a comparable contrast level. The cell (arrow 1) is brighter than the surrounding liquid, the wall of the cylinder is visible as a bright line (arrow 2), the lipid droplet is visible as a dark shape (arrow 3), and the vesicle filled with protein can be recognized as a feature with a brighter contrast than its surroundings (arrow 4). Since this simulation reproduces the basic features of the experimental STEM images, we can use such simulations to predict the contrast and resolution for future studies, or to analyze the composition of the materials in the sample.
Discussion
The maximal spatial resolution of STEM achievable on fully hydrated, pristine yeast cells in this initial study was 32 ± 8 nm, which is an order of magnitude better than the ∼200-nm resolution of diffraction-limited conventional light microscopy (32), and around one-sixth of the ∼5-nm electron-dose-limited resolution obtainable with TEM in cryosections of eukaryotic cells (7). Light microscopy is a standard tool for cell biology, but its use is limited for many cell constituents that are smaller than the wavelength of light. Super-resolution microscopy permits visualization of smaller objects but requires fluorescent labeling of specific proteins (3). Conventional EM relies on an extensive preparation of the cells through fixation, staining, and sectioning into thin sections (33), and is prone to artifacts. In modern cryo-EM (6,7) cells are rapidly frozen at high pressure to convert the cellular water into amorphous ice (34,35), sometimes with the help of high concentrations of glass-inducing solutes, thereby avoiding intracellular damage by ice crystals. Thin, peripheral regions of whole cells can be studied, but when the region of interest lies in a part of the cell that exceeds ∼0.3 μm of thickness, the cell has to be cryosectioned. The cells are thus not intact, nor alive. Furthermore, sample preparation, imaging, and analysis are highly time-consuming. The intermediate resolution achieved with STEM on hydrated cells, in combination with a sample preparation similar to that for light microscopy, or rather, the absence of EM sample preparation, is potentially of great value for the study of cellular ultrastructure and function.
An important questions associated with each biological EM technique is the effect of radiation damage. The hydrated yeast samples were exposed to 1.4 × 104 electrons/scan pixel of a size of 25 nm, which translates into an average electron dose of 22 e−/nm2. This electron dose is well below the dose limit for EM of wet biological specimens (36), and two orders of magnitude below the electron dose of 2 × 103 e−/nm2 or greater used in cryo-EM (7). A particular advantage of imaging at an intermediate resolution between those of light microscopy and cryo-EM is that the radiation dose typically scales with the square of the resolution (20). The local electron dose directly in the focal plane has likely been higher, maximally 1 × 104 e−/nm2 within the diameter of the electron probe containing 50% of the current of 0.9 nm assuming optimal focus at the beam entrance window. However, only a minor portion of a yeast cell was exposed to the higher dose, because electron beam scanning occurred with lines separated by the pixel size of 25 nm, and beam broadening rapidly decreased the intensity for deeper layers (18). In future experiments, the electron probe size could be changed to a value equal to the pixel size, which can be done while maintaining the same image contrast and resolution. The electron dose of 22 e−/nm2 can thus be considered as the dose required to obtain the images shown here.
We do not know at this point whether the liquid STEM approach can be used for tomography. The liquid specimen holder can be tilted and the geometry of the tapered windows in the microchips allows for tilt angles up to ∼35°, which could be used for tomography with a limited axial resolution. However, the recording of a tilt series on pristine cells is problematic, because the cells are not alive after the recording of one STEM image. One future possibility could be to record a series of ∼10 images at a low dose, using a special type of specimen stage that allows for rapid tilting with low drift.
These results represent what to our knowledge is a new approach in nanoscale microscopy. Pristine cells can now be examined with a resolution of a few tens of nanometers. We expect that this approach will be useful for research in fields such as cell biology to study questions requiring a spatial resolution better than that achievable with light microscopy, but not yet involving a spatial resolution as high as ∼5 nm. This methodology can easily be combined in correlative approaches with other microscopy techniques, to study the location and the function of single proteins within the cellular framework (2). We were able to obtain images from both light microscopy and STEM on the same cells in the microfluidic chamber. The temporal correlation between the images was in the range of a few minutes, but could be reduced to less than a second by the integration of a light microscope into the electron microscope (37). In our study, the identification of intracellular organelles was based on structural information (size, shape, and location), and on differences in mass density. Further organelle classification is possible by using specific fluorescent markers for proteins of interest (38). STEM could also be combined with superresolution imaging of fluorescent labels to correlate protein locations with <50 nm precision with ultrastructural information of intact cells (3). Nanoparticles, like colloidal gold or quantum dots, serving as protein tags could be used for the imaging of specifically labeled surface proteins with a resolution (14,39) of ∼3 nm. The capability of imaging pristine cells could be combined with the technique to image with a short pulse of electrons, the so-called four-dimensional EM (40). This would open the possibility to capture native cellular configurations before radiation-induced effects would have time to propagate through the structure. A burst of short pulses could potentially be used to examine processes of short duration occurring in liquid. We anticipate that liquid STEM will be broadly applied to explore pristine cells that are living at the onset of imaging, bridging the capabilities of light microscopy and cryo-EM.
Acknowledgments
We thank B. Binder, H. Demers, T. E. McKnight, D. W. Piston, R. Roberts-Galbraith, and Protochips, Inc. STEM images were recorded at the SHaRE User Facility, sponsored by the Division of Scientific User Facilities, Office of Basic Energy Sciences, U.S. Department of Energy. K.L.G is an Investigator of the Howard Hughes Medical Institute.
This research was supported by Vanderbilt University Medical Center and by National Institutes of Health grants R01RR018470 (to P. Mazur) and 1R43EB008589 (to S. Mick).
References
- 1.Stahlberg H., Walz T. Molecular electron microscopy: state of the art and current challenges. ACS Chem. Biol. 2008;3:268–281. doi: 10.1021/cb800037d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sali A., Glaeser R., Baumeister W. From words to literature in structural proteomics. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- 3.Lippincott-Schwartz J., Manley S. Putting super-resolution fluorescence microscopy to work. Nat. Methods. 2009;6:21–23. doi: 10.1038/nmeth.f.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parsons D.F. Structure of wet specimens in electron microscopy. Improved environmental chambers make it possible to examine wet specimens easily. Science. 1974;186:407–414. doi: 10.1126/science.186.4162.407. [DOI] [PubMed] [Google Scholar]
- 5.Thiberge S., Nechushtan A., Moses E. Scanning electron microscopy of cells and tissues under fully hydrated conditions. Proc. Natl. Acad. Sci. USA. 2004;101:3346–3351. doi: 10.1073/pnas.0400088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lucić V., Leis A., Baumeister W. Cryo-electron tomography of cells: connecting structure and function. Histochem. Cell Biol. 2008;130:185–196. doi: 10.1007/s00418-008-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierson J., Sani M., Peters P.J. Toward visualization of nanomachines in their native cellular environment. Histochem. Cell Biol. 2009;132:253–262. doi: 10.1007/s00418-009-0622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao W., Harteneck B.D., Attwood D.T. Soft x-ray microscopy at a spatial resolution better than 15 nm. Nature. 2005;435:1210–1213. doi: 10.1038/nature03719. [DOI] [PubMed] [Google Scholar]
- 9.Larabell C.A., Nugent K.A. Imaging cellular architecture with x-rays. Curr. Opin. Struct. Biol. 2010;20:623–631. doi: 10.1016/j.sbi.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betzig E., Trautman J.K., Kostelak R.L. Breaking the diffraction barrier: optical microscopy on a nanometric scale. Science. 1991;251:1468–1470. doi: 10.1126/science.251.5000.1468. [DOI] [PubMed] [Google Scholar]
- 11.Müller D.J., Helenius J., Dufrêne Y.F. Force probing surfaces of living cells to molecular resolution. Nat. Chem. Biol. 2009;5:383–390. doi: 10.1038/nchembio.181. [DOI] [PubMed] [Google Scholar]
- 12.Hell S.W. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 13.Ohi R., Gould K.L. Regulating the onset of mitosis. Curr. Opin. Cell Biol. 1999;11:267–273. doi: 10.1016/s0955-0674(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 14.de Jonge N., Peckys D.B., Piston D.W. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. USA. 2009;106:2159–2164. doi: 10.1073/pnas.0809567106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millard P.J., Roth B.L., Haugland R.P. Development of the FUN-1 family of fluorescent probes for vacuole labeling and viability testing of yeasts. Appl. Environ. Microbiol. 1997;63:2897–2905. doi: 10.1128/aem.63.7.2897-2905.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ring E.A., de Jonge N. Microfluidic system for transmission electron microscopy. Microsc. Microanal. 2010;16:622–629. doi: 10.1017/S1431927610093669. [DOI] [PubMed] [Google Scholar]
- 17.Barth J.E., Kruit P. Addition of different contributions to the charged particle probe size. Optik (Stuttg.) 1996;101:101–109. [Google Scholar]
- 18.de Jonge N., Poirier-Demers N., Drouin D. Nanometer-resolution electron microscopy through micrometers-thick water layers. Ultramicroscopy. 2010;110:1114–1119. doi: 10.1016/j.ultramic.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osumi M. The ultrastructure of yeast: cell wall structure and formation. Micron. 1998;29:207–233. doi: 10.1016/s0968-4328(97)00072-3. [DOI] [PubMed] [Google Scholar]
- 20.Reimer L., Kohl H. Springer; New York: 2008. Transmission Electron Microscopy: Physics of Image Formation. [Google Scholar]
- 21.Mazur P. Studies on rapidly frozen suspensions of yeast cells by differential thermal analysis and conductometry. Biophys. J. 1963;3:323–353. doi: 10.1016/s0006-3495(63)86824-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulholland J., Preuss D., Botstein D. Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane. J. Cell Biol. 1994;125:381–391. doi: 10.1083/jcb.125.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinser E., Daum G. Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae. Yeast. 1995;11:493–536. doi: 10.1002/yea.320110602. [DOI] [PubMed] [Google Scholar]
- 24.Grimard V., Massier J., Thiele C. siRNA screening reveals JNK2 as an evolutionary conserved regulator of triglyceride homeostasis. J. Lipid Res. 2008;49:2427–2440. doi: 10.1194/jlr.M800168-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Codlin S., Mole S.E. S. pombe btn1, the orthologue of the Batten disease gene CLN3, is required for vacuole protein sorting of Cpy1p and Golgi exit of Vps10p. J. Cell Sci. 2009;122:1163–1173. doi: 10.1242/jcs.038323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi H., Suga T., Niinobe S. Studies on peroxisomes. I. Intraparticulate localization of peroxisomal enzymes in rat liver. Biochim. Biophys. Acta. 1971;252:58–68. doi: 10.1016/0304-4165(71)90092-4. [DOI] [PubMed] [Google Scholar]
- 27.Reimer L. Springer; Berlin: 1998. Scanning Electron Microscopy: Physics of Image Formation and Microanalysis. [Google Scholar]
- 28.Drouin D., Couture A.R., Gauvin R. CASINO V2.42: a fast and easy-to-use modeling tool for scanning electron microscopy and microanalysis users. Scanning. 2007;29:92–101. doi: 10.1002/sca.20000. [DOI] [PubMed] [Google Scholar]
- 29.Demers H., Poirier-Demers N., de Jonge N. Simulating STEM imaging of nanoparticles in micrometers-thick substrates. Microsc. Microanal. 2010;16:795–804. doi: 10.1017/S1431927610094080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fidanza F., Keys A., Anderson J.T. Density of body fat in man and other mammals. J. Appl. Physiol. 1953;6:252–256. doi: 10.1152/jappl.1953.6.4.252. [DOI] [PubMed] [Google Scholar]
- 31.Fischer H., Polikarpov I., Craievich A.F. Average protein density is a molecular-weight-dependent function. Protein Sci. 2004;13:2825–2828. doi: 10.1110/ps.04688204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawley J.B. Springer; New York: 1995. Handbook of Biological Confocal Microscopy. [Google Scholar]
- 33.Bozzola J.J., Russell L.D. Jones and Bartlett; Boston: 1992. Electron Microscopy. [Google Scholar]
- 34.Webster P., Schwarz H., Griffiths G. Preparation of cells and tissues for immuno EM. Methods Cell Biol. 2008;88:45–58. doi: 10.1016/S0091-679X(08)00403-2. [DOI] [PubMed] [Google Scholar]
- 35.McDonald K.L. A review of high-pressure freezing preparation techniques for correlative light and electron microscopy of the same cells and tissues. J. Microsc. 2009;235:273–281. doi: 10.1111/j.1365-2818.2009.03218.x. [DOI] [PubMed] [Google Scholar]
- 36.Hui S.W., Parsons D.F. Electron diffraction of wet biological membranes. Science. 1974;184:77–78. doi: 10.1126/science.184.4132.77. [DOI] [PubMed] [Google Scholar]
- 37.Agronskaia A.V., Valentijn J.A., Gerritsen H.C. Integrated fluorescence and transmission electron microscopy. J. Struct. Biol. 2008;164:183–189. doi: 10.1016/j.jsb.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Kohlwein S.D. The beauty of the yeast: live cell microscopy at the limits of optical resolution. Microsc. Res. Tech. 2000;51:511–529. doi: 10.1002/1097-0029(20001215)51:6<511::AID-JEMT3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 39.Dukes M.J., Peckys D.B., de Jonge N. Correlative fluorescence microscopy and scanning transmission electron microscopy of quantum-dot-labeled proteins in whole cells in liquid. ACS Nano. 2010;4:4110–4116. doi: 10.1021/nn1010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flannigan D.J., Barwick B., Zewail A.H. Biological imaging with 4D ultrafast electron microscopy. Proc. Natl. Acad. Sci. USA. 2010;107:9933–9937. doi: 10.1073/pnas.1005653107. [DOI] [PMC free article] [PubMed] [Google Scholar]