

Abstract
Molecular-dynamics simulations were carried out to ascertain which of the potential multimeric forms of the transmembrane peptaibol channel, antiamoebin, is consistent with its measured conductance. Estimates of the conductance obtained through counting ions that cross the channel and by solving the Nernst-Planck equation yield consistent results, indicating that the motion of ions inside the channel can be satisfactorily described as diffusive. The calculated conductance of octameric channels is markedly higher than the conductance measured in single channel recordings, whereas the tetramer appears to be nonconducting. The conductance of the hexamer was estimated to be 115 ± 34 pS and 74 ± 20 pS, at 150 mV and 75 mV, respectively, in satisfactory agreement with the value of 90 pS measured at 75 mV. On this basis, we propose that the antiamoebin channel consists of six monomers. Its pore is large enough to accommodate K+ and Cl− with their first solvation shells intact. The free energy barrier encountered by K+ is only 2.2 kcal/mol whereas Cl− encounters a substantially higher barrier of nearly 5 kcal/mol. This difference makes the channel selective for cations. Ion crossing events are shown to be uncorrelated and follow Poisson statistics.
Introduction
Ion channels mediate and regulate transport of charged species across cell membranes. Not only do they play an essential role in the physiology of a cell but are also frequent drug targets. Despite their importance in biology and medicine, we know less about them than about nearly any other major class of proteins. Only in the last decade were high-resolution structures of a number of ion channels solved through x-ray crystallography (see, e.g., (1,2)). However, a considerable gap still remains in understanding the structure-function relationship, as information revealed by x-ray crystallography is static and often incomplete. Model building and molecular dynamics (MD) simulations combined with x-ray structures can, in principle, help close this gap by providing insight into the dynamics of functional states and processes associated with ion conductance.
To determine the relevance of MD simulations, they should be compared with experimental electrophysiological data, which directly measure the main function of ion channels—ionic conductance. At present, such comparisons are very difficult to perform for large, eukaryotic ion channels or their bacterial homologs, as they require long simulations, most likely extending to multi-microsecond timescales. Instead, we can test the approach and improve the computational tools using simple model channels. These channels not only inform us about how complex channels function, but also are of considerable interest in their own rights. Some are viral channels which are promising drug targets (3). Others, formed by nongenomic proteins from higher organisms, are themselves therapeutic, because they exhibit antimicrobial activity (4,5).
In this study, we focus on ion channels formed by antiamoebin 1 (AAM), a member of a nonribosomally synthesized family of fungal peptides called peptaibols (4,6). Peptaibols are short peptides that usually contain 15–20 residues and are rich in nonstandard amino acids, such as α-aminoisobutyric acid (Aib), D-isovaline (Iva), and hydroxyproline (Hyp). Many peptaibols form ion-conducting helical bundles that span cell membranes. Antiamoebin, first isolated from the fungal family Emericellopsis (7) and later found in other groups of fungi, is among the shortest peptaibols capable of forming channels, because it contains only 16 residues. The sequence of AAM is
| Ac-Phe-Aib-Aib-Aib-Iva-Gly-Leu-Aib-Aib- |
where “Phol” stands for phenylalinol. None of the residues is charged at channel-forming, physiological conditions and, except for Gln and Hyp, all side chains are relatively small and hydrophobic. The structure of a single AAM molecule, solved using both x-ray crystallography (8,9) and NMR (10), is largely helical.
The ion conductance properties of AAM have been studied in mixed palmitoyloleoylphosphatidylcholine/dioleoylphosphatidylcholine membranes (11). AAM channels exhibit only one conductance level, which indicates a single structure. This is in contrast to alamethicin, which is the best-studied peptaibol, both experimentally (12) and theoretically (13,14). It exhibits multiple conductance levels (15), which are thought to arise from a dynamic equilibrium between bundles containing different numbers of monomers (16). The microscopic conductance of AAM obtained from single channel recordings, adjusted to a 1 M concentration of KCl, is equal to 90 pS (11). It is believed that the channel exhibits some selectivity for K+ over Cl− (17). In addition to forming channels, AAM might also function as a carrier (9).
The number of polypeptide chains needed to form conducting AAM channels is not well established. Measurements of macroscopic conductance as a function of peptide concentration led to the suggestion that the channel might be tetrameric (11). Under somewhat different assumptions about the equilibrium constants that govern channel formation, hexamers and octamers are also possible. This led to a proposal that the pore of the tetrameric channel is too small to conduct ions and, instead, the channel is octameric (17). To resolve this issue, we built tetrameric, hexameric and octameric AAM channels, examined their short timescale stabilities in the membrane, calculated their conductances, and compared them with the conductance measured electrophysiologically. This approach, which directly links the structure of the channel with its function, provides the means of determining the number of monomers in the conducting helical bundle.
So far, channel conductances have been calculated mostly using methods that do not invoke full atomistic details of the system, but instead rely on solving the Nernst-Planck (NP) or Poisson-Nernst-Planck (PNP and 3d-PNP) equations (18,19) (see (20) for a recent review,) or on Brownian dynamics (19,21,22). Because important information about channel dynamics and energetics that influences conductance might be lost in these approaches, it is important to determine whether conductance can be accurately determined from all-atom simulations. In MD, channel conductance has been estimated directly by observing the ionic currents that develop in the presence of an applied electric field (23–26).
In this study, we calculate conductance using a combination of MD and NP methods. In the latter model it is assumed that ions move through the channel diffusively in response to the applied electric field and the underlying potential of mean force. Each method has its advantages and disadvantages. The direct method involves fewer approximations whereas the approach based on the Nernst-Planck equation can be applied to a broader range of conditions. Agreement between both methods indicates that the approximations underlying the diffusive model are satisfactory for simple channels.
Methods
Initial channel models
Initial structures of AAM bundles containing four, six, and eight monomers were constructed using the M-ZDOCK program (27). For subsequent MD simulations, we selected two octameric, one hexameric, and one tetrameric channel using the procedure described in the Supporting Material. One octameric model is a funnel-shaped bundle of parallel helices, as proposed by O'Reilly and Wallace (17). The lumen of the channel is hydrophilic due to the presence of the backbone carbonyl groups and the Gln-11 residues, which form a hydrogen-bonded ring pointing toward the center of the pore.
The other candidate channel represents a class of well-packed structures in which the Gln residues are oriented toward the backbones of neighboring helices, forming a network of interhelical hydrogen bonds stabilizing the bundle. The selected hexameric structure is similar to that of O'Reilly and Wallace, but is tighter and has a smaller pore because it is built of a smaller number of monomers. The selected tetrameric channel is a compact structure in which the side chains of Gln-11 are directed toward the neighboring helices. The minimum radius of the pore is 1.6 Å, which is sufficient for transporting dehydrated, but not hydrated, K+ ions. Structures in which the Gln residues are directed toward the center have pores that are too small to conduct ions.
MD simulations
Several systems containing bundles of AAM embedded in a palmitoyloleoylphosphatidylcholine (POPC) bilayer placed between two lamellae of water were simulated using the program NAMD (28). Periodic boundary conditions were applied in three spatial directions. Long-ranged electrostatic forces were calculated using the particle-mesh Ewald method with a grid of 128 × 128 × 128. The r-RESPA multiple time-stepping algorithm was used, in which the base time-step was 1 fs and the long-range forces were updated every four steps. Water molecules were treated as rigid bodies using the SETTLE algorithm. Unless otherwise noted, the calculations were carried out in an NPT ensemble. The pressure was maintained at 1 atm by the Langevin piston with a period of 100 fs and a decay time of 50 fs. The temperature was kept at 300 K using Langevin damping with a damping coefficient of 1 ps–1. Each system was initially equilibrated using a multistep procedure described in the Supporting Material. Then, 30-, 20-, and 45-ns equilibrium MD trajectories were obtained for the tetramer, hexamer, and octamer, respectively.
Potential energy functions
The CHARMM27 force field was used to describe the POPC phospholipids (29,30) and water molecules were represented by the TIP3P model. CHARMM22 was used to describe the standard amino acids in AAM (31). Nonbonded van der Waals parameters and partial charges for the methyl groups attached to the α-carbon in Aib and Iva were taken from alanine, and the ethyl group in Iva was based on the parameters for the β-carbon in valine and the methyl group in alanine. The parameters for the hydroxyl group in Hyp were taken from threonine. Finally, the C-terminal phenylalinol residue was built from phenylalanine with a -CH2OH blocking group on the α-carbon.
To test the potential parameters for nonstandard amino acids, a single AAM molecule was studied in hexane and methanol. The results were compared to the x-ray structure (9) and the NMR structure obtained in methanol solution (10). These simulations, described in the Supporting Material, indicate that the potential functions used to describe the peptide, including those for atypical amino acids, are suitable for modeling the AAM channel.
Free energy calculations
To compute the free energy profile (potential of mean force) encountered by K+ or Cl− as it traverses the pore, an order parameter was chosen as the z component (in the direction perpendicular to the membrane surface) of a vector joining a selected ion and the center-of-mass of the AAM bundle. The free energy profiles for both K+ and Cl− along the order parameter were computed using the adaptive biasing force method (32–34), as implemented in NAMD (35). The method has been shown to be very efficient in calculating free energy changes along different dynamical variables (35,36). Here, the transmembrane region was subdivided into several strata or windows, 5–8 Å wide. Then, trajectories at least 10-ns long were obtained with an ion constrained in each window, and the free energy profile across the pore was constructed by integrating the average force over all windows.
In addition, a 50-ns trajectory was computed in the absence of constraints. From this trajectory, equilibrium density profiles, ρeq(z), were obtained for each ion. On both sides of the channel, but not in the central region, the densities are sufficient to calculate the free energy profile,
with satisfactory accuracy. Here, kB is the Boltzmann constant and T is temperature. The total free energy profiles were obtained by combining the two methods for calculating A(z). Adaptive biasing force covered the range of z between −15 and 15 Å, whereas density profiles were considered reliable in the ranges of [−20,12] and [12,20] Å.
Conductance calculations
The single-channel conductance is defined as the ratio of the observed current, I, through the channel to the applied voltage (37), In MD simulations, the conductance can be computed directly by applying an external electric field to the system and observing the instantaneous current that develops (23,26),
| (1) |
where qi and zi(t) are the charge and position at time t of atom i, respectively, and Lz is the length of the simulation cell in the direction perpendicular to the membrane. The total current is obtained by integrating Eq. 1. The current can also be estimated by counting the number of ions that cross the channel during the simulation. As shown in the Supporting Material, these estimates yield the same conductance to within statistical errors.
Simulations in the presence of a constant, external electric field, were carried out in an NVT ensemble, as described by Aksimentiev and Schulten (23). The volume of the system was fixed because fluctuations in the box length lead to fluctuation in the applied transmembrane potential (23). Because the aqueous phase is conducting, the applied field corresponds to application of an external, transmembrane voltage. For the hexamer and the octamer, 150-ns and 15-ns trajectories were generated, respectively, at a potential of 150 mV. This potential is higher than 75 mV applied in the most reliable single-channel recording experiments (11) to observe a sufficient current for a meaningful estimate of the conductance, and for determining whether ion crossing events are uncorrelated. Slow relaxation of the hexameric channel was observed to occur over the first 50 ns of the trajectory, and this has been excluded from the analysis.
Channel conductance can be also estimated by assuming that ions diffuse through the channel in a one-dimensional potential of mean force and the applied field along z. If ion-ion correlations and the dependence of the diffusion coefficient, D, on z are ignored, which will be shown to be good approximations, the flow of ions can be calculated from the NP equation (37,38). In the steady-state approximation, net flow of ions of a given type through the channel, j, is
| (2) |
where ρ(z) is the linear number density of ions at position z in the channel (in units of number of ions/length) and V is the energy of the ion due to the applied voltage. Integrating this equation between za and zb with the exponential integrating factor yields
| (3) |
In macroscopic calculations, za and zb are set to the edges of the channel. Then it is assumed that
is expressed in terms of the bulk concentration and the area in the mouth of the channel (37,38). The equation, however, is valid for any choice of za and zb inside the channel. Calculating j from this equation requires knowledge of A(z) from separate simulations in the absence of voltage. Once A(z) is available, j can be calculated at different applied voltages. For two voltages across the membrane, ΔVμ and ΔVν, the ratio of the corresponding currents, jμ and jν is
| (4) |
if za and zb are at the edges of the channel.
Alternatively, Eq. 2 can be integrated through dividing both sides by ρ(z) and integrating. This yields
| (5) |
This equation is not practical for macroscopic simulations because the nonequilibrium density profile, ρ(z), needs to be known, but it is useful for testing the validity of the diffusion equation against MD. If za and zb are located at the edges of the channel, all required information is obtained from a single MD simulation with applied voltage. We pursued both methods in our analysis.
Results
Octameric and tetrameric channel models
Simulations of the octameric channels, initiated from the structure proposed by O'Reilly and Wallace (17), yield a stable bundle both in the absence and the presence of an applied electric field. However, the conductance of this model estimated from Eq. 1 is 910 pS at 1 M salt concentration. A similar estimate is obtained from the NP equation using the approach described above. Because these estimates are an order-of-magnitude higher than the measured conductance, it appears that the model does not correctly represent the AAM channel. The same is the case for the second octameric model considered here. Even though its initial structure had a smaller pore, and therefore was expected to exhibit lower conductance, the pore opened up and reached the same size as the pore in the first model during the first 10 ns of unrestrained simulation after equilibration. This yields a conductance that is also inconsistent with experiment.
The tetrameric channel was simulated for 30 ns in the absence of an electric field. To prevent the channel from closing and allow for its structural adjustments to the presence of the ion, a K+ ion was restrained in the middle of the pore for the first 9 ns. Once the restraints were removed, the channel remained stable and retained a continuous water strand across the pore, but the ion was expelled from the channel. No crossing events were observed during this simulation or during the simulation in the presence of a 0.15-V applied potential.
To increase the likelihood of generating an observable current, a higher potential of 0.5 V was applied. Assuming that the channel is Ohmic, a conductance of 90 pS should yield, on average, 14 crossing events in 20 ns under these conditions. In our simulations of this length, the channel remained stable, but no crossing events were observed. This null result suggests that the tetrameric structure is nonconducting or its conductance is markedly lower than 90 ps.
Structural stability and flexibility of the hexameric model
In the reminder of this section, we present results for the hexameric channel, shown in Fig. 1, which was simulated with and without electric field. In both types of simulations, the system remained stable. The density profiles of its major components—water, phospholipids, protein, and ions—as a function of the z coordinate perpendicular to the membrane did not show any signs of drift. These profiles obtained from the simulation without electric field are shown in Fig. 2. The density profiles for water and POPC overlap significantly due to penetration of water molecules into the phospholipid headgroup region. Similar profiles were obtained in simulations of a pure POPC-water system (39,40).
Figure 1.
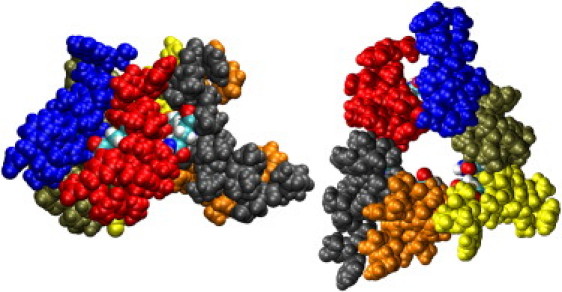
Side and top views of a model hexameric bundle of AAM. Each helix is colored separately, and the Gln 11 residues are shown in CPK colors.
Figure 2.
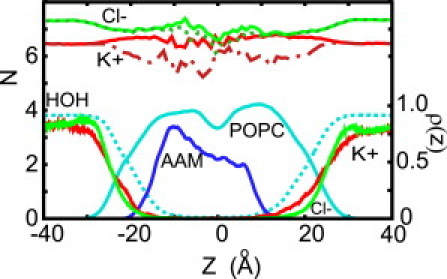
Density profiles of water (dotted cyan), lipids (cyan), AAM (blue), K+ (red), and Cl− (green) ions. Shown above the density profiles are the average hydration and solvation numbers for K+ (red and dash-dotted red) and Cl− (solid and dotted green) ions as a function of its position along the AAM6 channel. The hydration number is the number of oxygen atoms from water molecules that are located closer to the ion than the minimum between the first and second peaks in the ion-water oxygen radial distribution function. These distances are 3.4 Å and 3.75 Å for K+ and Cl−, respectively. Solvation number is defined in a similar fashion, but also includes oxygen and nitrogen atoms from the protein and oxygen atoms from the lipids that are within the same distances.
From the density profiles of the K+ and Cl− it follows that the ions exhibit some charge layering at the water-membrane interface, with K+ penetrating the membrane more readily than Cl−. This is primarily because negatively charged carbonyl and phosphate oxygen atoms, located closer to the hydrophobic core of the membrane than the positively charged NH3 groups, interact favorably with positive charges, but not with negative charges. Recent experiments on solid-supported phosphatidylcholine membranes showed a selectivity for the cations in KCl and NaCl, although this depends on the particular ion pair (41). A similar effect was noted in simulations of dipalmitoylphosphatidylcholine membranes with NaCl (42).
The length of the AAM bundle is only ∼23 Å (see Fig. 2), which is substantially less than the width of the hydrophobic core of the unperturbed membrane (∼30 Å). There is, therefore, hydrophobic mismatch between the peptide and the membrane. As predicted by the mattress model (43), this causes thinning of the membrane near the channel, as shown in Fig. S4 in the Supporting Material.
The channels, which are built of short helical monomers, appear to be quite flexible. This view is supported by the calculations of the pore radius along the channel axis, carried out with the aid of the HOLE program (44), shown in Fig. 3. The size of the pore undergoes substantial fluctuations, mostly due to inward and outward motions of the individual helices, with standard error varying between 0.5 and 1 Å. Its narrowest part is located in the middle, near the Gln-11 residues. This is a result of the overall shape of the channel and the bulkiness of Gln compared to the Aib and Iva residues that line other regions of the pore. At an applied voltage of 150 mV, the average minimum radius is initially equal to 4.8 Å. After 30 ns the channel narrows somewhat, to ∼3.5 Å, and the location of the minimum shifts slightly. In the absence of the electric field, the pore is quite similar. The Gln residues do not form a ring of hydrogen bonds that protrudes into the pore, as proposed by O'Reilly and Wallace. Instead, as detailed in the Supporting Material, they form hydrogen bonds with water inside the pore or, less frequently, with the neighboring peptide.
Figure 3.
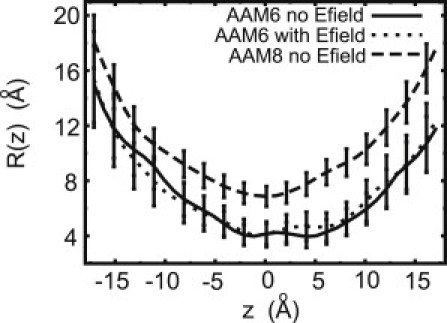
Average radius of the channel, along the axis of the pore, for the AAM hexamer with (solid) and without (dotted) applied field. The average pore radius for the AAM octamer model without applied field (dashed) is shown for comparison.
As shown in Fig. 2, the pore is sufficiently wide for ions to retain most of their first hydration shells inside the channel. Even in its narrowest region, only one water molecule is lost from these shells. For K+, this water molecule is replaced by the oxygen atoms from the side chains of Gln and Hyp residues with additional, smaller contributions from backbone oxygen atoms and the blocking alcohol of the phenylalinol. In the interfacial region, a slight decrease in the hydration of the K+ ion equal, on average, to 0.5 water molecule is compensated by the carbonyl and phosphate oxygen atoms from the lipids. As a result, the total solvation number for K+ remains fairly constant along the channel. The hydration number for Cl− also drops slightly in the membrane region. However, in contrast to K+, this drop is not compensated by polar groups in the protein lining the pore or in the lipids because these groups are also negatively charged. As a result, the solvation number decreases slightly, by ∼0.5.
Potential of mean force for ion transfer across the channel
The potential of mean force, A(z), experienced by K+ and Cl− in the channel is shown in Fig. 4. This quantity has long been recognized as important for understanding the mechanism of ion transport (45,46) and is a necessary ingredient of diffusion-based methods for estimating conductance. The free energy barrier associated with crossing the membrane through the channel, equal to 2.2 ± 0.5 kcal/mol and 5.0 ± 0.7 kcal/mol for K+ and Cl−, respectively, is markedly reduced compared to the barrier experienced by the ions in unassisted permeation (47). The difference in the barrier heights implies that the channel is selective for K+ over Cl−. No local free energy minima are present for either ion, indicating the absence of binding sites inside the channel. Analysis of the trajectories provided no evidence for correlations in the motions of ions of the same or opposite charge.
Figure 4.
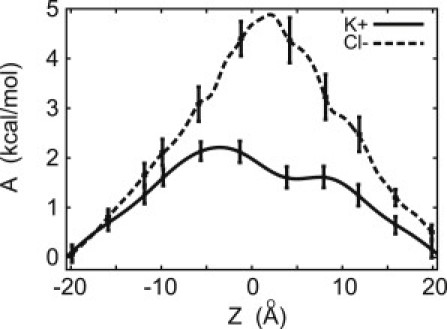
Free energy for transporting K+ (solid) and Cl− (dashed) ions through the transmembrane AAM6 pore.
Ion diffusion and transport properties
Diffusion inside wide channels is usually well described by Fick's law. However, if the channel radius is too small (∼1–2 Å) to allow permeants to pass each other inside the channel, their diffusion becomes single-file and Fick's law no longer applies (48)—as observed in computer simulations of ion transport through gramicidin (46), the KcsA channel (49), or small-sized carbon nanotubes (50). The pore in the hexameric AAM channel, which has a minimum radius of ∼2.5 Å, appears to be sufficiently wide for ions not to move in a single file. This assertion was tested by analyzing the time evolution of the probability distribution that an ion located at position z0 at time t = 0 is at position z at time t. This analysis, described in the Supporting Material, reveals that at timescales longer than 10 ps, ion diffusion is Fickian. The diffusion coefficient for K+ near the center of the channel is estimated at 0.5 × 10−5 cm2/s.
For Fickian diffusion, the diffusion coefficient, Dz(z), can also be estimated from the force autocorrelation function of an ion that is held stationary at position z in the channel (51). Inside the channel, the dependence of Dz(z) on z is weak, of the order of the statistical errors. Its estimated values for K+ and Cl− are 0.46 × 10−5 cm2/s and 0.70 × 10−5 cm2/s, respectively. For K+, this estimate is quite close to the value obtained from measuring ionic displacement from the center of the channel, as described above.
For comparison, the calculated diffusion coefficients in the bulk water region are ∼2.2 and 1.9 × 10−5 cm2/s for K+ and Cl−, respectively, and the corresponding measured values of D for the same ions are 1.96 and 2.03 × 10−5 cm2/s (37). Thus, the diffusion coefficient is reduced inside the hexameric AAM channel relative to its value in bulk water by approximately a factor of 3–5. A decrease in D for ions or water molecules was also observed in other channels, such as KcsA (49), several biological model channels (52), and gramicidin A (53). It might be caused by the increase in local friction due to the rough inside surface of a channel. Consistent with this view, D increases inside channels with smooth surfaces, such as carbon nanotubes (50).
Channel conductance
A total flux of 10 K+ ions was observed during the last 100-ns trajectory with an applied voltage of 150 mV and at 1 M KCl concentration, which yields a channel conductance of 109 pS. All crossing events occurred in the direction of the field. No Cl− ions crossed the pore during that time. This is expected, considering that the free energy barrier to channel crossing is substantially higher for Cl− than for K+. The ion trajectories, from which the crossing events are identified, are shown in Fig. 5 (top). The integrated current (see Eq. 1) is shown in Fig. 5 (bottom). The conductance estimated from the slope of a single straight line fitted to the integrated current (see Eq. 1), shown in Fig. 5 (bottom), is equal to 126 pS.
Figure 5.
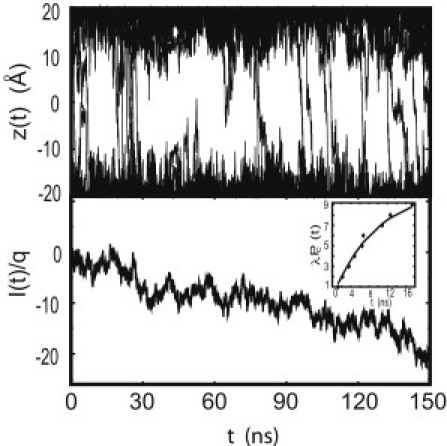
(Top) Trajectories of K+ ions in the pore region for a 150-ns simulation with an external potential of 0.15 V. In the first 30 ns there were four Cl− trajectories (not shown) and nine K+ (solid) trajectories that cross the pore. During the remaining 120-ns simulation, after the channel relaxed to a structure with a smaller pore, 10 K+ and no Cl− ions crossed the channel. (Bottom) integrated ionic current of K+as a function of time. (Inset) The number of waiting times shorter than t predicted from Poisson statistics for the total time interval of 100 ns (solid line) and encountered in MD simulations (solid diamonds).
Because ion-crossing events appear to be uncorrelated, they should be well described as a Poisson process. Then the probability distribution of waiting times (times between consecutive events) should decay exponentially with a characteristic constant, λ, which defines the number of events in the total time interval. As seen in Fig. 5 (bottom), the cumulative distribution, Pc(t), that shows waiting time between events shorter than t, follows the Poisson statistics very closely, indicating that ion transport through AAM can be described as a single, homogeneous Poisson process. The value of λ determined through best fit is 10.5 ions/100 ns. The corresponding statistical error is 3.24. This yields the conductance of 115 ± 34 pS. Good agreement between different methods for calculating conductance suggests consistency and convergence of the calculations.
Ionic current can be also calculated from the NP equation. Typically, this involves an area term, which is often identified with the cross-sectional area at the mouths of the pore (38). Information about the cross-sectional area of the rest of the pore is already included in the free energy profiles. Assigning the area term cannot be done unambiguously and, as a result, the accuracy of the calculated current might suffer considerably. To overcome these difficulties we used an alternative approach, in which only quantities calculated from the MD trajectory are used as input. These quantities are density profiles from equilibrium or nonequilibrium simulations. If the limits of integration in Eq. 3 or Eq. 5 are set to the edges of the channel, then one of those two simulations is sufficient. For other limits, they need to be combined. We explored both approaches. Equation 5 yields the current of ∼15 ions/100 ns and shows very good stability with respect to changes in the limits of integration (see the Supporting Material). In contrast, estimates of j based on Eq. 3 appear to be quite sensitive to boundary effects and the detailed shape of A(z). As a consequence, we were unsuccessful in obtaining currents that exhibited comparable stability with respect to the limits of integration.
Using Eq. 4, we estimated that the current is reduced by a factor of 3 when the applied voltage is reduced from 150 mV to 75 mV. On the basis of the calculated ratio, which, in contrast to the absolute current, is fairly insensitive to the details of A(z), we estimated that the conductance at 75 mV is 74 ± 20 pS, in good agreement with the experimental conductance of 90 pS.
Discussion
Relation between structure and conductance
The number of monomers forming the channel can be estimated from measurements of macroscopic conductance as a function of protein concentration. Using a simple chemical equilibrium model, in which n units aggregate to form a conducting channel (54), the value of n for AAM was determined to be between 3 and 4 (11). If the aggregating units are identified as monomers, this means that the channel is tetrameric (17), because trimers are too small to conduct ions. Alternatively, the formation of channels might be a two-step process, in which the initial aggregation of monomers into dimers is very strongly favored. This step is followed by self-assembly of dimers into conducting structures. If this mechanism is correct, then the AAM channel should be a hexamer or an octamer.
On this basis, O'Reilly and Wallace (17) proposed an octameric model, arguing that the pore in a tetramer would be too small to conduct. Other aggregation mechanisms, governed by equilibrium constants that yield a more complex relation between n and the number of monomers in the channel, are also possible. The interpretation of n is further complicated by possible coexistence of conducting and nonconducting aggregates. Thus, determining the number of monomers that form the channel, solely based on measuring macroscopic conductance, might be unreliable.
Octameric models yield conductances that are markedly higher than those measured experimentally. Tetrameric aggregates, in turn, are either too short to form transmembrane bundles, or have pores that are too small to conduct ions, or are too loosely bound to be stable. This conclusion is borne out by direct simulation of tetrameric bundles, which are not observed to conduct ions, even in the presence of a large, 500-mV transmembrane potential. This conclusion may not apply to all peptaibols. In alamethicin, tetrameric structures have been associated with the low conducting level. This interpretation is supported by the measured conductance of tethered alamethicin tetramers (55). However, some theoretical studies cast doubt on the role of tetramers in the conductance of alamethicin (13,14). Furthermore, even though the sequences of antiamoebin and alamethicin are related, their conductance properties are, in many respects, different (56). Therefore, indirect evidence for conductance of tetrameric structures of alamethicin may not be relevant to antiamoebin.
In contrast to tetrameric or octameric models, the hexameric structure yields satisfactory agreement between the calculated and measured microscopic conductance. At the applied voltage of 75 mV and 1 M concentration of KCl, the conductance calculated using the NP equation is equal to 74 pS, compared to 90 pS measured in single-channel recordings. Furthermore, the hexameric structure, as expected, is selective for K+. This is largely due to the favorable interactions of K+ with oxygen atoms of Gln and peptide backbone atoms and to the deeper penetration of K+ than Cl− into the membrane. On this basis, we propose that the AAM channel is built of six helices.
Comparison of different methods for calculating conductance
Direct simulation of the conductance involves fewer assumptions than stochastic models, such as the NP equation. For pores that are both large enough and stable enough to develop statistically significant currents at voltages of interest, this is the method of choice. Simulations of α-hemolysin, which has a much larger pore than AAM, were carried out at voltages between ±120 mV to ± 1200mV (23). In this system, good results for the current-voltage dependence were obtained in 10-ns simulations at each voltage. Simulations of OmpF (24) and OmpC channels (25) at an applied voltage of 1000 mV yielded good agreement with experimental conductances over a range of temperatures at lower salt concentrations (<5 M), but at higher concentrations the effects of temperature on conductance were underestimated.
For low-conducting channels, such as AAM, obtaining statistically significant results from direct simulations in the most interesting range of applied voltages would require MD trajectories of several microseconds. In shorter simulations, the applied voltage must be larger than those usually used in experiments, sometimes even nonphysiological. Mapping out the current-voltage curve, which is often available from electrophysiological measurements for non-Ohmic, low-conducting channels using direct simulations, becomes impractical. This limitation prevents stringent testing of theory against experiment.
In contrast to the direct approach, the NP equation can be used to calculate the conductance at any desired voltage. It relies on an assumption that motion of the ion in the channel is diffusive. For channels with sufficiently large pores, such as AAM, this appears to be the case. It is also assumed that the free energy profile of the ions at potentials of interest can be obtained simply by adding the free energy of a linear voltage ramp to the equilibrium free energy profile. This means that the applied potential has only a small effect on the structure of the protein and membrane. The comparison of the pore radii (see Fig. 3) of the AAM simulated at 150 mV and without applied voltage indicates that this assumption appears to be valid.
The limitations of continuum models have been discussed in the literature (37,57,58). In particular, the NP equation neglects ion correlations and the PNP or 3d-PNP equations are to be preferred (37,58), although 3d-PNP calculations tend to overestimate conductance, due to a lack of dynamic ion-ion correlations (19,46). In this article, the computed free energy profile contains all interactions that an ion encounters in the channel in the absence of an external electric field, including ion-ion interactions in a mean-field approximation. Because ions seem to cross the channel as uncorrelated events that follow Poisson statistics, it appears that any contributions that were not captured in this approximation do not play a significant role in conduction, and a more involved PNP approach is not necessary.
Even though the assumptions of the diffusion model are fulfilled, the agreement between the current calculated directly and estimated from the model is not as good as might be expected. The discrepancies might be due to uncertainties in ρ(z). However, the consistency in j calculated at different integration limits argues against it. A more likely explanation is that the applied electric field, which should disappear only in the conducting phase, might extend beyond the range of the relatively short AAM channel. Then the voltage drop along the channel would be smaller than assumed and would lead to overestimating currents calculated from the NP equation. If the field extends from −25 to 25 Å, which is close to the half-widths of most density profiles shown in Fig. 1, the current will be 12 ions/100 ns. The range of electric field can be, in principle, obtained from MD—but this would require markedly longer simulation times.
Our simulations have been carried out using standard, nonpolarizable models, although polarization effects have long been of concern (for recent reviews, see Illingworth and Domene (59) and Bucher and Rothlisberger (60)). For example, simulations of gramicidin with a polarizable force field yield a lower free energy barrier to ion permeation and estimated conductances that are in qualitative agreement with experimental result (61). Polarization effects become increasingly important, with the increase in the number of water molecules removed from the first hydration shell and balanced by interactions with polarized protein atoms (62). In AAM, the ions permeate the channel with their first solvation shells essentially intact; thus, effects of polarization on the computed free energy profiles are expected to be small.
Conclusions
We have investigated tetrameric, hexameric, and octameric models of the antiamoebin transmembrane channel in a water-POPC membrane system. The conductance of the hexameric model of AAM is estimated at 74 pS at 75 mV and 1 M KCl, which is in satisfactory agreement with the experimental value of 90 pS. The conductance observed in the octameric bundle is >10 times larger, and the tetrameric model does not conduct even at voltages of 500 mV. Consequently, we conclude that the conducting state of AAM is most likely hexameric.
Ions traverse the pore with their solvation shells mostly intact, which in turn means that the free energy barriers to ions crossing the channel are small. The pore is sufficiently large to allow for Fickian rather than single-file diffusion of ions. Other assumptions underlying the Nernst-Planck equation also appear to be fulfilled for simple channels. As a result, the estimates of ionic conductance obtained from the direct method that relies on counting ion crossing events and from the NP equation are in good agreement once the range of applied voltage is properly accounted for. This, in turn, allows for combining MD simulations at a single voltage and stochastic calculations to estimate conductance at any desired voltage without making explicit assumptions about the pore dimensions.
Using computer simulations to link structural information about ion channels with electrophysiological measurements, which provide the most relevant functional data, appears to be a useful strategy for investigating simple ion channels. It provides a sensitive test of whether our understanding how channels are built and how they conduct ions is satisfactory. As computational capabilities expand, this strategy might become very valuable for studying more-complex channels.
Acknowledgments
All simulations were performed at the NASA Advanced Supercomputing Division.
This work was supported by the National Aeronautics and Space Administration (NASA) Exobiology and NASA Astrobiology Programs. Partial support to B.A.W. was provided by a grant from the UK Biotechnology and Biological Research Council.
Footnotes
Pär Bjelkmar's present address is Department of Theoretical Physics, Royal Institute of Technology, SE-10691 Stockholm, Sweden.
Supporting Material
References
- 1.Walz T., Hirai T., Engel A. The three-dimensional structure of aquaporin-1. Nature. 1997;387:624–627. doi: 10.1038/42512. [DOI] [PubMed] [Google Scholar]
- 2.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 3.Fischer W.B., Sansom M.S.P. Viral ion channels: structure and function. Biochim. Biophys. Acta. 2002;1561:27–45. doi: 10.1016/s0304-4157(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 4.Chugh J.K., Wallace B.A. Peptaibols: models for ion channels. Biochem. Soc. Trans. 2001;29:565–570. doi: 10.1042/bst0290565. [DOI] [PubMed] [Google Scholar]
- 5.Duclohier H. Peptaibiotics and peptaibols: an alternative to classical antibiotics? Chem. Biodivers. 2007;4:1023–1026. doi: 10.1002/cbdv.200790094. [DOI] [PubMed] [Google Scholar]
- 6.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 7.Pandey R.C., Meng H., Rinehart K.L., Jr. Structure of antiamoebin I from high resolution field desorption and gas chromatographic mass spectrometry studies. J. Am. Chem. Soc. 1977;99:5203–5205. doi: 10.1021/ja00457a063. [DOI] [PubMed] [Google Scholar]
- 8.Karle I.L., Perozzo M.A., Balaram P. Crystal structure of the channel-forming polypeptide antiamoebin in a membrane-mimetic environment. Proc. Natl. Acad. Sci. USA. 1998;95:5501–5504. doi: 10.1073/pnas.95.10.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snook C.F., Woolley G.A., Wallace B.A. The structure and function of antiamoebin I, a proline-rich membrane-active polypeptide. Structure. 1998;6:783–792. doi: 10.1016/s0969-2126(98)00079-3. [DOI] [PubMed] [Google Scholar]
- 10.Galbraith T.P., Harris R., Wallace B.A. Solution NMR studies of antiamoebin, a membrane channel-forming polypeptide. Biophys. J. 2003;84:185–194. doi: 10.1016/S0006-3495(03)74841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duclohier H., Snook C.F., Wallace B.A. Antiamoebin can function as a carrier or as a pore-forming peptaibol. Biochim. Biophys. Acta. 1998;1415:255–260. doi: 10.1016/s0005-2736(98)00184-9. [DOI] [PubMed] [Google Scholar]
- 12.Leitgeb B., Szekeres A., Kredics L. The history of alamethicin: a review of the most extensively studied peptaibol. Chem. Biodivers. 2007;4:1027–1051. doi: 10.1002/cbdv.200790095. [DOI] [PubMed] [Google Scholar]
- 13.Breed J., Biggin P.C., Sansom M.S. Alamethicin channels—modeling via restrained molecular dynamics simulations. Biochim. Biophys. Acta. 1997;1325:235–249. doi: 10.1016/s0005-2736(96)00262-3. [DOI] [PubMed] [Google Scholar]
- 14.Tieleman D.P., Hess B., Sansom M.S. Analysis and evaluation of channel models: simulations of alamethicin. Biophys. J. 2002;83:2393–2407. doi: 10.1016/s0006-3495(02)75253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon L.G., Haydon D.A. The unit conductance channel of alamethicin. Biochim. Biophys. Acta. 1972;255:1014–1018. doi: 10.1016/0005-2736(72)90415-4. [DOI] [PubMed] [Google Scholar]
- 16.Sansom M.S. The biophysics of peptide models of ion channels. Prog. Biophys. Mol. Biol. 1991;55:139–235. doi: 10.1016/0079-6107(91)90004-c. [DOI] [PubMed] [Google Scholar]
- 17.O'Reilly A.O., Wallace B.A. The peptaibol antiamoebin as a model ion channel: similarities to bacterial potassium channels. J. Pept. Sci. 2003;9:769–775. doi: 10.1002/psc.514. [DOI] [PubMed] [Google Scholar]
- 18.Kurnikova M.G., Coalson R.D., Nitzan A. A lattice relaxation algorithm for three-dimensional Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys. J. 1999;76:642–656. doi: 10.1016/S0006-3495(99)77232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noskov S.Y., Im W., Roux B. Ion permeation through the α-hemolysin channel: theoretical studies based on Brownian dynamics and Poisson-Nernst-Plank electrodiffusion theory. Biophys. J. 2004;87:2299–2309. doi: 10.1529/biophysj.104.044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coalson R.D., Kurnikova M.G. Poisson-Nernst-Planck theory approach to the calculation of current through biological ion channels. IEEE Trans. Nanobiosci. 2005;4:81–93. doi: 10.1109/tnb.2004.842495. [DOI] [PubMed] [Google Scholar]
- 21.Chung S.H., Kuyucak S. Ion channels: recent progress and prospects. Eur. Biophys. J. 2002;31:283–293. doi: 10.1007/s00249-002-0216-4. [DOI] [PubMed] [Google Scholar]
- 22.Cheng M.H., Cascio M., Coalson R.D. Theoretical studies of the M2 transmembrane segment of the glycine receptor: models of the open pore structure and current-voltage characteristics. Biophys. J. 2005;89:1669–1680. doi: 10.1529/biophysj.105.060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aksimentiev A., Schulten K. Imaging α-hemolysin with molecular dynamics: ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005;88:3745–3761. doi: 10.1529/biophysj.104.058727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pezeshki S., Chimerel C., Kleinekathöfer U. Understanding ion conductance on a molecular level: an all-atom modeling of the bacterial porin OmpF. Biophys. J. 2009;97:1898–1906. doi: 10.1016/j.bpj.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biró I., Pezeshki S., Kleinekathöfer U. Comparing the temperature-dependent conductance of the two structurally similar E. coli porins OmpC and OmpF. Biophys. J. 2010;98:1830–1839. doi: 10.1016/j.bpj.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faraudo J., Calero C., Aguilella-Arzo M. Ionic partition and transport in multi-ionic channels: a molecular dynamics simulation study of the OmpF bacterial porin. Biophys. J. 2010;99:2107–2115. doi: 10.1016/j.bpj.2010.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce B., Tong W., Weng Z. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics. 2005;21:1472–1478. doi: 10.1093/bioinformatics/bti229. [DOI] [PubMed] [Google Scholar]
- 28.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 30.Feller S.E., Yin D., MacKerell A.D., Jr. Molecular dynamics simulation of unsaturated lipid bilayers at low hydration: parameterization and comparison with diffraction studies. Biophys. J. 1997;73:2269–2279. doi: 10.1016/S0006-3495(97)78259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacKerell A.D., Jr., Brooks B., Karplus M. CHARMM: the energy function and its parameterization with an overview of the program. In: Schleyer P.v. R., Allinger N.L., Clark T., Gasteiger J., Kollman P., Schaefer H.F., III, Schreiner P.R., editors. The Encyclopedia of Computational Chemistry. Vol. 1. John Wiley and Sons; New York: 1998. pp. 271–277. [Google Scholar]
- 32.Darve E., Pohorille A. Calculating free energies using average force. J. Chem. Phys. 2001;115:9169–9183. [Google Scholar]
- 33.Darve E., Wilson M.A., Pohorille A. Calculating free energies using a scaled-force molecular dynamics algorithm. Mol. Simul. 2002;28:113–144. [Google Scholar]
- 34.Darve E., Rodríguez-Gómez D., Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- 35.Hénin J., Chipot C. Overcoming free energy barriers using unconstrained molecular dynamics simulations. J. Chem. Phys. 2004;121:2904–2914. doi: 10.1063/1.1773132. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Gomez D., Darve E., Pohorille A. Assessing the efficiency of free energy calculation methods. J. Chem. Phys. 2004;120:3563–3578. doi: 10.1063/1.1642607. [DOI] [PubMed] [Google Scholar]
- 37.Hille B. 3rd Ed. Sinauer Associates; Sunderland, MA: 2001. Ion Channels of Excitable Membranes. [Google Scholar]
- 38.Levitt D.G. Interpretation of biological ion channel flux data—reaction-rate versus continuum theory. Annu. Rev. Biophys. Biophys. Chem. 1986;15:29–57. doi: 10.1146/annurev.bb.15.060186.000333. [DOI] [PubMed] [Google Scholar]
- 39.Heller H., Schaefer M., Schulten K. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid-crystal phases. J. Phys. Chem. 1993;97:8343–8360. [Google Scholar]
- 40.Chiu S.W., Subramaniam S., Jakobsson E. Simulation study of a gramicidin/lipid bilayer system in excess water and lipid. I. Structure of the molecular complex. Biophys. J. 1999;76:1929–1938. doi: 10.1016/S0006-3495(99)77352-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Celma J.J., Hatahet L., Fendler K. Specific anion and cation binding to lipid membranes investigated on a solid supported membrane. Langmuir. 2007;23:10074–10080. doi: 10.1021/la701188f. [DOI] [PubMed] [Google Scholar]
- 42.Pandit S.A., Bostick D., Berkowitz M.L. Molecular dynamics simulation of a dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 2003;84:3743–3750. doi: 10.1016/S0006-3495(03)75102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouritsen O.G., Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys. J. 1984;46:141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smart O.S., Goodfellow J.M., Wallace B.A. The pore dimensions of gramicidin A. Biophys. J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuckerman D.M., Woolf T.B. Efficient dynamic importance sampling of rare events in one dimension. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2001;63:016702. doi: 10.1103/PhysRevE.63.016702. [DOI] [PubMed] [Google Scholar]
- 46.Roux B., Allen T., Im W. Theoretical and computational models of biological ion channels. Q. Rev. Biophys. 2004;37:15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 47.Wilson M.A., Pohorille A. Mechanism of unassisted ion transport across membrane bilayers. J. Am. Chem. Soc. 1996;118:6580–6587. doi: 10.1021/ja9540381. [DOI] [PubMed] [Google Scholar]
- 48.Levitt D.G. Dynamics of a single-file pore: non-Fickian behavior. Phys. Rev. A. 1973;8:3050–3054. [Google Scholar]
- 49.Bernèche S., Roux B. A microscopic view of ion conduction through the K+ channel. Proc. Natl. Acad. Sci. USA. 2003;100:8644–8648. doi: 10.1073/pnas.1431750100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hummer G., Rasaiah J.C., Noworyta J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 51.Zwanzig R.W. Elementary derivation of time-correlation formulas for transport coefficients. J. Chem. Phys. 1964;40:2527–2533. [Google Scholar]
- 52.Smith G.R., Sansom M.S.P. Effective diffusion coefficients of K+ and Cl− ions in ion channel models. Biophys. Chem. 1999;79:129–151. doi: 10.1016/s0301-4622(99)00052-6. [DOI] [PubMed] [Google Scholar]
- 53.Allen T.W., Andersen O.S., Roux B. Molecular dynamics—potential of mean force calculations as a tool for understanding ion permeation and selectivity in narrow channels. Biophys. Chem. 2006;124:251–267. doi: 10.1016/j.bpc.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 54.Hall J.E., Vodyanoy I., Marshall G.R. Alamethicin. A rich model for channel behavior. Biophys. J. 1984;45:233–247. doi: 10.1016/S0006-3495(84)84151-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woolley G.A. Channel-forming activity of alamethicin: effects of covalent tethering. Chem. Biodivers. 2007;4:1323–1337. doi: 10.1002/cbdv.200790113. [DOI] [PubMed] [Google Scholar]
- 56.Duclohier H. Helical kink and channel behavior: a comparative study with the peptaibols alamethicin, trichotoxin and antiamoebin. Eur. Biophys. J. 2004;33:169–174. doi: 10.1007/s00249-003-0383-y. [DOI] [PubMed] [Google Scholar]
- 57.Cooper K., Jakobsson E., Wolynes P. The theory of ion transport through membrane channels. Prog. Biophys. Mol. Biol. 1985;46:51–96. doi: 10.1016/0079-6107(85)90012-4. [DOI] [PubMed] [Google Scholar]
- 58.Eisenberg R.S. Computing the field in proteins and channels. J. Membr. Biol. 1996;150:1–25. doi: 10.1007/s002329900026. [DOI] [PubMed] [Google Scholar]
- 59.Illingworth C.J., Domene C. Many-body effects and simulations of potassium channels. Proc. R. Soc. A. 2009;465:1701–1716. [Google Scholar]
- 60.Bucher D., Rothlisberger U. Molecular simulations of ion channels: a quantum chemist's perspective. J. Gen. Physiol. 2010;135:549–554. doi: 10.1085/jgp.201010404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patel S., Davis J.E., Bauer B.A. Exploring ion permeation energetics in gramicidin A using polarizable charge equilibration force fields. J. Am. Chem. Soc. 2009;131:13890–13891. doi: 10.1021/ja902903m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bucher D., Kuyucak S. Importance of water polarization for ion permeation in narrow pores. Chem. Phys. Lett. 2009;477:207–210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.