

Abstract
Fluorescence spectroscopy is an indispensible tool for studying the structure and conformational dynamics of protein molecules both in isolation and in their cellular context. The ideal probes for monitoring intramolecular protein motions are small, cysteine-reactive fluorophores. However, it can be difficult to obtain specific labeling of a desired cysteine in proteins with multiple cysteines, in a mixture of proteins, or in a protein's native environment, in which many cysteine-containing proteins are present. To obtain specific labeling, we developed a method we call cysteine metal protection and labeling (CyMPL). With this method, a desired cysteine can be reversibly protected by binding group 12 metal ions (e.g., Cd2+ and Zn2+) while background cysteines are blocked with nonfluorescent covalent modifiers. We increased the metal affinity for specific cysteines by incorporating them into minimal binding sites in existing secondary structural motifs (i.e., α-helix or β-strand). After the metal ions were removed, the deprotected cysteines were then available to specifically react with a fluorophore.
Introduction
Fluorescence spectroscopy is commonly employed to study conformational changes in purified proteins and proteins in their native, cellular context (1,2). To faithfully and quantitatively report atomic-scale motions, one must use small fluorescent probes with short linkers that only minimally perturb the structure of the protein under study (3). Furthermore, it is critical to ensure that the protein molecule of interest is specifically labeled and that the overall fluorescence background is not too large.
Genetically encoded tags based on green fluorescent protein (GFP) are highly specific (4). However, GFP is large (∼27 kDa), often as large as the protein of interest, which makes it less useful for studying small movements (5). Specific labeling has also been accomplished with the use of genetically encoded His tags on proteins that bind metal-chelating fluorophores (6). Although these tags are much smaller than FP labels, both the tags and the fluorophores used to label them are relatively long and flexible, making them unfaithful reporters of the protein backbone positions to which they are attached. Furthermore, they are limited in terms of the location on the primary amino acid sequence into which they can be inserted. Tetracysteine motifs (cys-cys-xaa-xaa-cys-cys, where xaa is any amino acid) that react with arsenated fluorophores also provide specificity, but they require at least six residues in the proper orientation and are likely to disrupt the local structure of the protein into which they have been inserted (7).
The most faithful fluorescent reporters of protein backbone positions and conformational changes are small, cysteine-reactive fluorophores (3,5). These dyes covalently attach to cysteine residues that are found natively in proteins or introduced by site-directed mutagenesis. The smallest dyes are similar in size to tryptophan and have very short linkers, which enables them to attach to proteins without causing any significant structural perturbation. However, these fluorophores are not well suited for studies involving mixtures of proteins or proteins in their native, cellular context, where many other cysteine-containing proteins are available to react with a given fluorophore, creating a high-fluorescence background. Even purified proteins may have multiple reactive cysteines that can obscure the fluorescence signal originating from a labeled cysteine of interest.
Various strategies have been developed to circumvent this problem. One approach identifies surface-accessible cysteines on a protein of known structure, measures the kinetics of cysteine modification at each residue, and uses the difference in modification rates to specifically label residues (8). However, this method requires all accessible cysteines to have substantially different reaction rates from the cysteine of interest. Rather than relying on such good fortune, an ideal labeling method would allow the experimenter to directly manipulate the reaction kinetics of a desired cysteine relative to background cysteines to provide specificity. One technique uses proteins with multiple conformations, which may contain cysteine residues that are accessible only in a subset of states. These proteins can be maintained in an inaccessible state, which prevents the reaction of the desired cysteine while background cysteines are blocked with a nonfluorescent modifier such as N-ethylmaleimide (NEM) (9,10). After blocking is completed, the proteins can be shifted to a state in which the cysteine is accessible and labeled with fluorophore. This method requires a known residue with state dependence and allows little control over which part of a given protein can be labeled. Another strategy to control the reaction kinetics of specific cysteines involves their incorporation into reversible disulfide bonds (11). This method is highly specific when applied to purified proteins, but in a native environment many cellular proteins have disulfide bonds or cysteines that can form disulfides under the conditions used to oxidize the cysteines intended for labeling. These cysteines would therefore still contribute to a high-fluorescence background after reduction. Zinc finger domains, which contain cysteines that can be protected by metal ion binding, can be incorporated into proteins and specifically labeled (11). However, this requires the insertion of a structured domain of 18 amino acids, which will likely perturb the protein intended for study. Kuiper and colleagues (12) developed a labeling method whereby pairs of cysteines in close proximity to one another are protected with phenylarsine oxide while other cysteines are blocked. The phenylarsine oxide is then reduced off with dithiothreitol, and the cysteine pairs are subsequently available for modification. However, this method is limited in that it can only protect pairs of cysteines, both of which are then free to react with fluorophore. Furthermore, it requires harsh reducing agents to remove the protecting group, which may also expose cysteines in native disulfide bonds to reaction with fluorophores.
We have devised a novel (to our knowledge) method for the reversible protection of cysteine residues using group 12 metal ion binding to minimal sites incorporated into existing secondary structural elements. We call this method cysteine metal protection and labeling (CyMPL). CyMPL requires only a slight perturbation of the protein of interest, which makes it quite suitable for labeling a protein for subsequent mapping of structures or conformational changes. This technique can be applied to any region of known secondary structure in virtually any protein, and can be employed to specifically label a desired protein in a mixture, specifically label a given cysteine in a protein with multiple cysteines, or reduce the background fluorescence labeling in a cellular environment.
Materials and Methods
Molecular biology
The gene for the cysteineless HCN2I fragment (encoding residues 443–640 of the mouse HCN2 ion channel) was synthesized (Blue Heron Biotechnology, Bothell, WA) and cloned into the pMALc2T vector (New England Biolabs, Ipswich, MA), which contains maltose binding protein (MBP) separated from HCN2 by a thrombin cleavable linker (5,13). Mutant MBP/HCN2 fusion proteins were prepared by polymerase chain reaction. The GFP/Syntaxin1A/MBP K25C,K26C fusion (GFP-Syx-MBP) was created and cloned into the GEMHE vector via standard polymerase chain reaction techniques. mRNA was transcribed with the use of mMessage mMachine (Ambion, Austin, TX) and the T7 polymerase.
Peptides
Peptides were modeled after those published by Marqusee et al. (14) and obtained from Sigma Genosys (Sigma-Aldrich, St. Louis, MO) and American Peptide (Sunnyvale, CA). The basic sequence was AAAAKAACAKAAAAKA, with the N-terminus acetylated and the C-terminus amidated. Histidines and cysteines were substituted at position 7 (i+1), position 11 (i+3), and position 12 (i+4) as indicated in the text. The peptides arrived lyophilized at >95% purity and were dissolved at a stock concentration of 400 μM and stored at −80°C.
Protein purification
HCN2/MBP constructs were expressed in BL-21 (DE3) bacteria. Cultures (2 L) were grown to OD600 between 0.6 and 0.9 at 37°C, induced with 1 mM isopropyl-1-thio-β-d-galactopyranoside, and shaken overnight at 18°C. Bacteria were harvested by centrifugation at 4420 × g for 15 min at 4°C and lysed into 150 mL of buffer containing 150 mM KCl, 10 μM adenosine-3′,5′-cyclic monophosphate, 10% glycerol, and 30 mM HEPES, pH 7.2, using an Emulsiflex-C5 (Avestin, Ottawa, Ontario, Canada). Before the solution was homogenized, 1 mM phenylmethylsulfonyl fluoride, 2.5 mg/mL DNase, and one complete protease inhibitor tab (Roche, Indianapolis, IN) were added to the slurry. Cellular debris and insoluble proteins were removed by centrifugation at 186,000 × g for 45 min at 4°C. The fusion protein was purified on an amylose column (New England Biolabs) and eluted into the above buffer plus 50 mM maltose. Some proteins were cleaved with thrombin (100 U in 20 mL + 5 mM CaCl2) for 4 h at room temperature (Calbiochem, La Jolla, CA) and either used for subsequent experiments or purified by ion exchange chromatography using a HiTrap SP FF (GE Healthcare, Piscataway, NJ) column to purify HCN2 or HiTrap Q FF (GE Healthcare) column for MBP, eluting on a KCl gradient from 10 mM to 1 M.
Circular dichroism
For circular dichroism measurements, the peptide ACAAKAAAAKAAWAKA, which could be readily quantified by A280 nm, was diluted to a final concentration of 96 μM in 65 mM Na2SO4, 5 mM NaH2PO4, 5 mM Na2HPO4, pH 7.2. Data were acquired with the use of an Aviv 62A DS spectropolarimeter (Aviv Associates, Lakewood, NJ) at 25°C in a 1-mm path-length cuvette. Data were quantified according to the method of Greenfield and Fasman (15). The spectra were similar for all the peptides used in the experiments, but percent helicity was only quantified with the tryptophan-containing peptide because it was the only peptide for which the concentration was accurately determined by ultraviolet absorbance.
Time course experiments
Cys-his peptides and glutathione were diluted to 20 μM in buffer containing 130 mM NaCl, 30 mM HEPES, pH 7.2. Bimane-C3 maleimide was diluted to a final concentration of 2 μM in the same buffer. Then 125 μL of bimane were mixed with 125 μL of peptide by pipetting the mixture up and down for a final concentration of 10 μM peptide and 1 μM bimane. The increase in bimane fluorescence was monitored with a Spex Fluorolog-3 spectrofluorometer (HORIBA Jobin Yvon, Edison, NJ) with excitation at 390 nm and emission at 480 nm, each with a 5-nm slit width, sampling at 1 Hz. Data were acquired with the use of FluorEssence software. The reaction of cys-cys peptides was followed in the same way with a final concentration of 1 μM peptide and 1 μM bimane. Proteins were diluted to 1– 10 μM for time-course experiments. For the experiment shown in Fig. S2 of the Supporting Material, modification time courses were acquired with the above buffers plus 50% TFE or 4 M guanidinium chloride (GuHCl) as indicated.
Protein-labeling experiments
For selective labeling experiments, HCN2cys-free S563C,K565H (HCN-cys,his) and MBP-HCN2cys-free K570C (MBP-HCN-cys) were combined at 1–2 μM each in buffer containing 130 mM NaCl and 30 mM HEPES, pH 7.2. To selectively label MBP-HCN-cys, the mixture was reacted for 10 min using 1 μM fluorescein-5-maleimide (Flc) in the presence of 0–10 mM CdCl2. The products were run on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and imaged on a Fluorchem gel imaging system (Cell Biosciences, Santa Clara, CA) with excitation set to 365 nm and standard ethidium bromide filters used for emission. For selective labeling of HCN-cys,his, the protein mixture was reacted for 10 min with 100 μM NEM in the presence of 0–10 mM CdCl2. After 10 min, Cd2+ was chelated with 25 mM EDTA and the remaining protein was reacted for 10 min with 100 μM Flc. The products were visualized as above. For experiments with HCN2cys-free K570C and MBP K25C,K26C, the proteins were purified as a fusion, digested with thrombin, and diluted to ∼1 μM. Reactions were carried out in the same fashion as for HCN-cys,his and MBP-HCN-cys.
Oocyte-labeling experiments
Stage IV and V oocytes were removed from female Xenopus laevis frogs under tricaine anesthesia as previously described (16). Defolliculated oocytes were injected with 50 nL of water or mRNA encoding EGFP-Syntaxin1A-MBP K25C,K26C. For labeling, oocytes were shaken in 24-well culture dishes for 30 min in OR2 (82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.6), OR2 plus 100 μM NEM, or OR2 with 100 μM NEM and 0.1–1 mM CdCl2. All solutions contained 100 μg/mL bovine serum albumin to prevent the oocytes from sticking to the plastic culture dishes. The oocytes were subsequently washed and incubated for 30 min in OR2 containing 5 mM EDTA and 10 μM AlexaFluor 546 maleimide. The oocytes were washed again and imaged with a Leica SL (Leica, Bannockburn, IL) confocal microscope with laser excitation at 488 nm (GFP) and 543 nm (Alexa).
Data analysis
We analyzed the images using MetaMorph (Molecular Devices, Sunnyvale, CA), and analyzed the image intensities and fluorescence data using Origin 8 (OriginLab, Northampton, MA). Time-course data were fit with single exponential decays to determine the rate constants for reactions run under pseudo-first-order conditions or by linear regression to the first ∼10% of the reaction. Concentration-response data were normalized to the reaction rate at 0 Cd2+ and fit with the following expression for single-cysteine peptides and proteins, or cysteine-histidine peptides and proteins:
| (1) |
For cys-cys peptides and proteins, we used the following expression:
| (2) |
The second term in the equation for cys-cys peptides/proteins accounts for the small but significant contamination by proteins and peptides in which one cysteine in a pair was oxidized or otherwise prevented from reacting and binding Cd2+. Kd1 is the Cd2+ affinity of the cys-cys peptide; Kd2 refers to the affinity of a single cysteine for Cd2+ and was fixed to 158 μM, the measured value; and f refers to the fraction of protein or peptide with only one cysteine. Box plots show the median (horizontal line), the 25th and 75th percentile (box), and the minimum and maximum observations (whiskers). Numbers reported in the text and Table 1 are given as the mean ± SE.
Table 1.
Metal affinities and binding energies
| Kd (μM) | ΔG (kcal mol−1) | n | ||
|---|---|---|---|---|
| Cys Only | 158 ± 8 | −5.10 ± 0.032 | 5 | |
| Glutathione | 112 ± 4 | −5.30 ± 0.022 | 5 | |
| HCN2 S563C | 157 ± 42 | −5.17 ± 0.143 | 5 | |
| HCN2 K570C | 158 ± 28 | −5.14 ± 0.110 | 5 | |
| Cys/His i+1 | 27.8 ± 3.2 | −6.13 ± 0.066 | 5 | |
| Cys/His i+3 | 13.5 ± 0.8 | −6.54 ± 0.038 | 5 | |
| Cys/His i+4 | 16.2 ± 1.8 | −6.44 ± 0.065 | 5 | |
| Cys/His i+3,4 | 4.69 ± 0.58 | −7.17 ± 0.087 | 5 | |
| Cys/Cys i+1 | 1.79 ± 0.67 | −8.03 ± 0.361 | 5 | |
| Cys/Cys i+3 | 1.77 ± 0.52 | −7.89 ± 0.219 | 6 | |
| Cys/Cys i+4 | 0.94 ± 0.24 | −8.18 ± 0.174 | 5 | |
| HCN2 S563C/K565H | 6.04 ± 2.32 | −7.27 ± 0.267 | 6 | |
| MBP E22C,K25C | 3.68 ± 1.29 | −7.47 ± 0.205 | 6 | |
| MBP K25C,K26C | 3.06 ± 1.08 | −7.53 ± 0.195 | 5 | |
| GuHCl | Cys Only | 59.0 ± 3.5 | −5.68 ± 0.034 | 5 |
| Glutathione | 215 ± 11 | −4.92 ± 0.031 | 5 | |
| Cys/His i+4 | 30.3 ± 3.2 | −6.08 ± 0.067 | 6 | |
| Cys/His i+3,4 | 25.5 ± 6.1 | −6.23 ± 0.135 | 5 | |
| TFE | Cys Only | 102 ± 9 | −5.36 ± 0.055 | 5 |
| Cys/His i+4 | 24.5 ± 6.1 | −6.25 ± 0.139 | 5 | |
| Cys/His i+3,4 | 6.72 ± 1.49 | −7.00 ± 0.111 | 6 | |
| Zn2+ | Cys Only | 1,090 ± 290 | −4.07 ± 0.171 | 5 |
| Cys/His i+4 | 28.1 ± 3.3 | −6.12 ± 0.071 | 5 |
Values for Kd and ΔG of Cd2+ binding are given for peptides and proteins in aqueous buffer, 4 M GuHCl (GuHC1), and 50% TFE (Zn2+). Values for Kd and ΔG of Zn2+ binding are also indicated (top row). All numbers are expressed as mean ± SE; n refers to the number of experiments.
It should be noted that the affinities measured for the cys-cys peptides (in the low micromolar range) were on the same order of magnitude as the 1 μM peptide concentration used in the experiment. Ideally, a peptide concentration lower than the Kd should be used to provide accurate measurements of affinity. However, we did not use lower concentrations of peptide, because they did not produce reliably measurable time courses. The resulting concentration-response curves for Cd2+ binding to cys-cys pairs were steep and the apparent Kd was difficult to determine. The actual affinities are probably higher than those reported.
Chemicals and reagents
Chemicals were obtained from Sigma (St. Louis, MO). All fluorophores were purchased from Molecular Probes (Carlsbad, CA).
Results
Specific cysteine-labeling scheme
In addition to its distinctive chemical reactivity among amino acids, cysteine readily binds the soft metal Cd2+ (17). Cd2+ binding can slow the rate of reaction of a cysteine with thiol-modifying reagents. We hypothesized that if we could selectively enrich Cd2+ binding to a cysteine of interest while leaving the background cysteines unbound, we could then use reversible Cd2+ binding as a tool to protect specific cysteines from modification while blocking the background cysteines. We could then specifically label the desired cysteine after removing the Cd2+. With this in mind, we designed a scheme whereby the Cd2+ affinity of a given cysteine is increased by the addition of one or two metal binding amino acids in the vicinity that will act with cysteine as a bidentate (or tridentate) ligand to coordinate Cd2+ with relatively high affinity. In Fig. 1 A, a histidine residue is introduced into a protein near a desired cysteine. Upon addition of a low concentration of Cd2+ (i), these residues will coordinate the metal ion but individual background cysteines will not. Unbound cysteines can then react with a thiol-modifying reagent (e.g., NEM (ii)) while at the same time the metal-bound cysteine will be protected. After metal chelation (e.g., with EDTA (iii)), the cysteine of interest is again available to react with the applied fluorophore (iv) while the background cysteines are covalently blocked.
Figure 1.

Design of the cysteine protection method. (A) Schematic diagram depicting the specific cysteine protection method, CyMPL. The Cd2+ binding affinity of a desired cysteine is increased by placing an additional metal-binding amino acid nearby. Cd2+ is used to protect the desired cysteine while background cysteines are reacted with a nonfluorescent modifying reagent. Upon removal of Cd2+, the specific cysteine is available to react with fluorophore. (B) Reaction scheme for cysteine reacting with bimane C3-maleimide and competitive binding by Cd2+. (C) Time course of fluorescence increase upon reaction of the Cys Only peptide with bimane C3-maleimide at different [CdCl2]. (D) Concentration-response curve showing the rate (normalized to 0 Cd2+) of the reaction of the Cys Only peptide with bimane C3-maleimide as a function of [CdCl2]total. Data were fit with a single-site binding curve (see Materials and Methods). (E) Box plot showing the apparent Kd and ΔG for Cd2+ binding to Cys Only peptide, GSH, and the cysteine-free C-terminal region of the HCN2 ion channel with a single cysteine inserted at position 563 (HCN2 S563C) or position 570 (HCN2 K570C).
Design of metal-binding sites
To optimize the metal-binding sites for cysteine protection, we created a series of peptides based on the design of Marqusee et al. (14), which are substantially helical in aqueous solution. Into these peptides we introduced a cysteine alone or in combination with additional chelating residues. We measured the rate of peptide reaction with the thiol-modifying reagent bimane C3-maleimide (bimane), which showed a large fluorescence increase upon reaction (Fig. 1 B). Under pseudo-first-order conditions, the reaction time course can be fit with a single exponential decay to determine an apparent rate constant for the reaction, kapp.
Cd2+ slowed the reaction rate of a single cysteine with bimane in a concentration-dependent manner (Fig. 1, B and C). By plotting the normalized (to zero added Cd2+) rate constants of the reaction as a function of the total [Cd2+] and fitting with the appropriate expression, we were able to determine the Cd2+ affinity (Fig. 1 D; see Materials and Methods). Fig. 1 E shows the apparent Cd2+ affinity and binding free energy for single cysteines in a helical peptide (Cys Only peptide), in glutathione (GSH), and in two purified proteins with single cysteines (the cysteine-free cytoplasmic domain of the mouse HCN2 ion channel with single cysteines introduced at positions 563 or 570) (5). In all cases, the Cd2+ protection of cysteine occurred over nearly identical concentration ranges, regardless of the protein or peptide context, with affinities similar to those previously reported for Cd2+ binding to GSH (18). It should be noted that a significant amount of Cd2+ is expected to bind to Cl− in the buffer and potentially to the quartz cuvette used in these experiments (18,19). Therefore, all affinities are reported as the apparent Kd, and the actual affinities may be higher.
We next tested the Cd2+ binding affinity of several helical peptides containing a cysteine residue at the same location, but with histidine residues inserted right next to the cysteine (Cys/His i+1), one helical turn away (Cys/His i+3 and Cys/His i+4), or with two histidines one turn away from the cysteine (Cys/His2 i+3,4; Fig. 2 A). As predicted, the histidines were able to coordinate metal with the cysteine and slow the rate of reaction with bimane at significantly lower Cd2+ concentrations compared with cysteine alone (Fig. 2, B–D, and Table 1). Insertion of a histidine right next to the cysteine increased the binding affinity to 27.8 μM (stabilization of binding by 1.03 kcal/mol). Placing the histidine residue one helical turn away at i+3 (13.5 μM, stabilized by 1.44 kcal/mol) or i+4 (16.2 μM, stabilized by 1.34 kcal/mol) caused an even greater stabilization of binding. Inserting two histidines at i+3 and i+4 provided the greatest increase in affinity (4.69 μM, stabilized by 2.07 kcal/mol). Zn2+ also binds cysteine, although (being a harder metal) not as well as Cd2+ (Fig. S1 and Table 1). The Cys Only peptide bound Zn2+ with 1.09 mM affinity. Inserting a histidine at position i+4 relative to the cysteine increased the affinity to 28.1 μM (stabilization of 2.05 kcal/mol).
Figure 2.
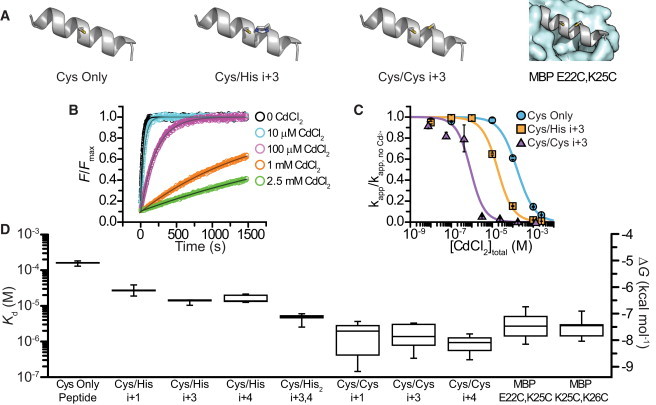
Cd2+ affinity of helical peptides with introduced minimal metal-binding sites. (A) Cartoons depicting peptides with a single cysteine (Cys-only), a cys-his pair (Cys/His i+3), and a cys-cys pair (Cys/Cys i+3) spaced at residues i and i+3 as well as a helix from MBP E22C,K25C. (B) Time course of fluorescence increase upon reaction of Cys/His i+3 peptide with bimane C3-maleimide at different [CdCl2]. (C) Concentration-response curve showing the rate (normalized to 0 Cd2+) of the reaction of Cys Only peptide, Cys/His i+3, and Cys/Cys i+3 with bimane C3-maleimide as a function of [CdCl2]total. Data were fit as indicated in Materials and Methods. (D) Box plot showing the apparent Kd and ΔG for Cd2+ binding to peptides or proteins containing a single cysteine, cys-his binding sites, and cys-cys binding sites.
We also examined the Cd2+ affinity for peptides with pairs of cysteines (Fig. 2, C and D). Because each cys-cys peptide had two reactive cysteines and two potential metal affinities (i.e., the affinity of two cysteines for metal and the affinity after one cysteine reacted), the actual reaction scheme is more complicated than the one shown in Fig. 1 B. To simplify our analysis, we examined only the initial portion of the reaction where the fluorescence increase was linear with respect to time. We assumed that no appreciable amount of reacted peptide had accumulated during this time, and therefore the measured rates and inferred Cd2+ affinities were solely the result of the reaction and binding of the unreacted cys-cys peptide. The addition of a second cysteine at position i+3 allowed Cd2+ to reduce the rate of reaction with bimane at much lower concentrations than for cysteine alone or any of the cys-his pairs tested (Fig. 2 C). The increased affinity compared with cys-his peptides may reflect the compatible binding properties of cysteine with the soft metal Cd2+. Cd2+ affinities of the cys-cys peptides (i+1, i+3, i+4) were increased by a factor of 88–168 (2.8–3.1 kcal/mol) over the Cys Only peptide (Fig. 2 D). Similar results were obtained in two mutants of the natively cysteine-free Escherichia coli MBP with cysteines inserted right next to each other (MBP K25C,K26C) or one turn away (MBP E22C,K25C; Fig. 2 A) in a known helical region (Fig. 2 D) (20).
Circular dichroism measurements for the above peptides showed characteristic minima at 208 nm and 220 nm, indicative of an α-helical structure (Fig. S2). However, the calculated percent helicity for a related peptide was 21% in aqueous buffers at 25°C. In the presence of 50% 2,2,2-trifluoroethanol (TFE), a solvent that promotes helix formation, the calculated helicity was 70% (15). To determine whether the introduced metal binding sites were helical, we used reagents to drive the folding equilibrium for our peptides toward α-helix (50% TFE) or random coil (4 M GuHCl). The increased Cd2+ binding stability of our peptides in aqueous buffer compared with cysteine alone more closely resembled the stabilization in TFE than stabilization in the presence of GuHCl (Fig. S2 B). We conclude that the metal-binding sites were substantially helical. In addition to these experiments, the observation that the cys-cys MBP mutants, which are known to be in a helical portion of the protein, bind Cd2+ with similar affinity to the cys-cys peptides provides further evidence that the metal-binding sites in the model peptides adopt a helical structure (Fig. 2 D).
We next investigated whether a minimal metal-binding site could be incorporated into a β-strand. As a model system, we used the β roll of the cysteine-free C-terminal domain of the mouse HCN2 ion channel (5,13). First, we introduced a cysteine at position 563 in the β roll (Fig. 3 A). The affinity was measured and was nearly identical to that measured for the Cys Only peptide (Fig. 3 B, Table 1). Introduction of a histidine residue at position 565 (residue i+2; Fig. 3 A) provided a 26-fold increase in Cd2+ binding (2.1 kcal/mol stabilization; Fig. 3 B and Table 1). We conclude that it is possible to incorporate small metal-binding sites into any regular secondary structure, α-helix, or β-strand.
Figure 3.
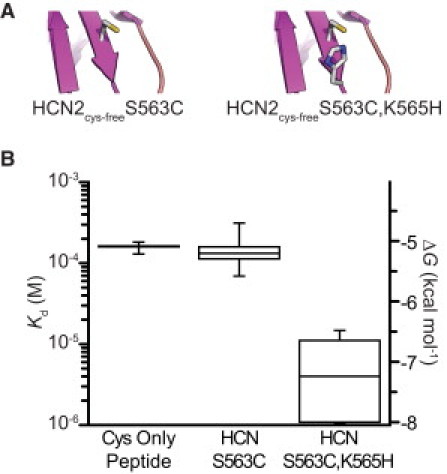
Cd2+ binding to a β-strand metal-binding site. (A) Cartoons depicting β-strand Cd2+-binding sites containing a single cysteine (HCN2cys-free S563C) and a cys-his pair (HCN2cys-free S563C,K565H). (B) Box plot showing the apparent Kd and ΔG for Cd2+ binding to Cys-only peptide, and the constructs depicted in A.
Labeling specific components in a protein mixture
A potential application of this technique would be to selectively label one protein in a mixture of other soluble proteins. To test this, we prepared an equimolar mixture of two proteins, a fusion of MBP and HCN2 with a single cysteine in the β roll (MBP-HCN2cys-free K570C, referred to as MBP-HCN-cys) and the HCN2 C-terminal region with a cys-his binding site in the β roll (HCN2cys-free S563C,K565H, referred to as HCN-cys,his; Fig. 4 A). The Cd2+ affinities of the individual proteins were 158 μM for HCN2cys-free K570C (Fig. 1 E) and 6 μM for HCN2cys-free S563C,K565H (Fig. 3 B).
Figure 4.
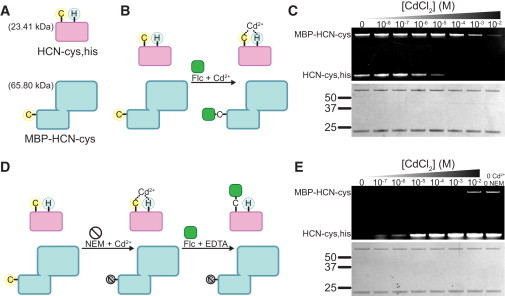
Application of protection of a cys-his motif to label individual components in a mixture of soluble proteins. (A) Cartoon showing HCN2 with a cys-his pair in the β roll (HCN2cys-free S563C,K565H; HCN-cys,his) and a fusion of MBP and HCN2 with a single cysteine (MBP-HCN2cys-free K570C; MBP-HCN-cys). (B) Schematic diagram representing an experiment in a mixture of proteins in solution in which MBP-HCN-cys is labeled with Flc while HCN-cys,his is protected from reaction with Flc by binding Cd2+. (C) SDS-PAGE showing Flc labeling of MBP-HCN-cys and HCN-cys,his at increasing [CdCl2]. A Coomassie stain of the same gel is shown below. (D) Schematic diagram representing an experiment in which HCN-cys,his is selectively labeled with Flc by first protecting it with Cd2+ while MBP-HCN-cys is blocked with NEM, and then removing Cd2+ and labeling the deprotected HCN-cys,his with Flc. (E) SDS-PAGE showing Flc labeling of MBP-HCN-cys and HCN-cys,his after reaction of the proteins with NEM in increasing [CdCl2]. The final lane shows labeling of both proteins in the absence of Cd2+ or NEM. A Coomassie stain of the same gel is shown below.
The first test was to label the MBP-HCN fusion and protect HCN-cys,his from labeling (Fig. 4 B). Because the Cd2+ affinity was ∼26-fold higher for HCN-cys,his than for MBP-HCN-cys, we expected to achieve specific labeling of MBP-HCN-cys by reacting the mixture with a fluorophore in the presence of low concentrations of Cd2+ that would protect HCN-cys,his but not MBP-HCN-cys. Flc was applied to the mixture in the presence of increasing concentrations of Cd2+. The reaction products were separated on an SDS-PAGE gel and the fluorescent bands were imaged to assess labeling intensity. As the [Cd2+] was increased above 1 μM, HCN-cys,his was protected from reaction with Flc as predicted, consistent with the measured affinity (Fig. 4 C). In contrast to this, MBP-HCN-cys was maximally labeled at concentrations below 1 mM. At 100 μM Cd2+, MBP-HCN-cys labeled 18 times more brightly than HCN-cys,his, even though the two proteins labeled with similar intensities in the absence of Cd2+.
We also performed a converse experiment (Fig. 4 D) in which we reacted the same mixture of proteins with NEM in increasing amounts of Cd2+. At low [Cd2+], we expected HCN-cys,his to be protected from NEM block and the cysteine in MBP-HCN-cys to be modified. After metal chelation with EDTA, the remaining unreacted cysteines were labeled with Flc. As predicted, HCN-cys,his was protected from NEM modification at lower [Cd2+], and therefore was available to react with Flc after Cd2+ was removed (Fig. 4 E). HCN-cys,his labeled at Cd2+ concentrations above 1 μM, whereas MBP-HCN-cys was blocked below 1 mM Cd2+. At 100 μM Cd2+, HCN-cys,his labeled 40 times more brightly than MBP-HCN-cys. HCN-cys,his labeling at 100 μM Cd2+ was 70% as bright as in the absence of Cd2+ or NEM, indicating that although undesired background reaction with fluorophore was virtually eliminated, the overall labeling intensity was not appreciably compromised (Fig. 4 E, far-right lane).
We repeated the above experiments using two different proteins: HCN2cys-free K570C, with a single cysteine in the β roll, and MBP K25C,K26C, with two adjacent cysteines in a surface-accessible helix (Fig. S3 A). The Cd2+ affinities of the individual proteins were 158 μM for HCN2 K570C (Fig. 1 E) and 3 μM for MBP K25C,K26C (Fig. 2 D). The proteins were copurified as a fusion on an amylose column and cleaved with thrombin to yield a 1:1 mixture. The results obtained were very similar to the mixture of proteins shown in Fig. 4 (Fig. S3).
Reducing background labeling in a cellular environment
To demonstrate the utility of our method for increasing the signal/background ratio in a cellular environment, we created a fusion protein with MBP K25C,K26C attached to the C-terminus of the single-pass membrane protein Syntaxin1A so that MBP would be located on the extracellular side of the membrane. The N-terminus of Syntaxin1A was fused to GFP so that relative levels of protein expression could be monitored (GFP-Syx-MBP; Fig. 5 A). Intact Xenopus laevis oocytes injected with RNA encoding GFP-Syx-MBP and water-injected oocytes were labeled extracellularly with 10 μM AlexaFluor 546 C5 maleimide (Alexa). Before the Alexa labeling was performed, some oocytes were blocked with NEM in the presence or absence of Cd2+. The results were imaged by confocal microscopy. This experiment is directly analogous to the scheme presented in Fig. 1 A.
Figure 5.
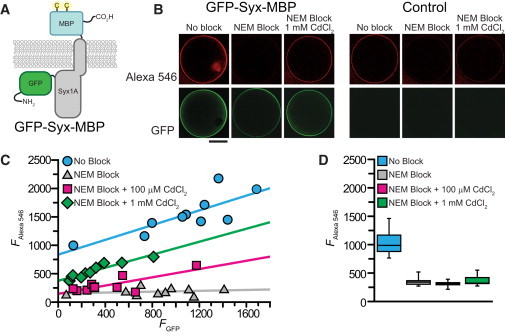
Application of the cysteine protection method to reduce the fluorescence background when labeling a protein in a cellular environment. (A) Cartoon depicting the fusion protein expressed in Xenopus laevis oocytes for extracellular labeling experiments. The protein is based on the single-pass membrane protein Syntaxin1A with GFP fused to the N-terminus (intracellular) and MBP K25C,K26C fused to the C-terminus (extracellular). (B) Confocal images of Xenopus laevis oocytes injected with mRNA encoding GFP-Syx-MBP or water (control). Oocytes were labeled with Alexa Fluor 546 C5 maleimide after incubation in buffer, buffer containing 100 μM NEM, or buffer containing 100 μM NEM + 1 mM CdCl2. Scale bar = 500 μm. (C) Plot showing the intensity of AlexaFluor 546 C5 maleimide labeling versus GFP fluorescence for oocytes injected with RNA encoding GFP-Syx-MBP with no block (cyan circles), after 100 μM NEM block (gray triangles), and after 100 μM NEM block in the presence of 100 μM (magenta squares) or 1 mM (green diamonds) CdCl2. (D) Box plot showing (from left to right) AlexaFluor 546 labeling intensity of water-injected oocytes with no block (cyan), after 100 μM NEM block (gray), and after 100 μM NEM block in the presence of 100 μM (magenta) or 1 mM (green) CdCl2.
Oocytes expressing GFP-Syntaxin-MBP labeled brightly with Alexa and also demonstrated bright GFP fluorescence (Fig. 5 B). Background Alexa labeling is illustrated by the degree of labeling in control (water-injected) oocytes (Fig. 5 B). It is evident from a comparison of these two conditions that a significant portion of the fluorescence intensity in the GFP-Syx-MPB-expressing oocyte arose from nonspecific background labeling. When GFP-Syntaxin-MBP oocytes were preblocked with NEM in the absence of Cd2+, there was very little labeling from subsequent exposure to Alexa (Fig. 5 B). At low [Cd2+], the cysteines on GFP-Syx-MBP were expected to bind Cd2+ and be protected from NEM block, and therefore free to react with Alexa upon Cd2+ removal. The background cysteines in the oocyte membrane were not expected to bind Cd2+ and should therefore have been blocked by NEM. Consistent with this prediction, GFP-Syx-MBP expressing oocytes that were blocked with NEM in the presence of 1 mM Cd2+ still labeled with Alexa (Fig. 5 B). In similarity to the GFP-Syx-MBP-expressing oocytes, labeling was blocked by NEM pretreatment in control oocytes (Fig. 5 B). The presence of 1 mM Cd2+ protected very little of this background labeling from block by NEM (Fig. 5 B).
In Fig. 5 C, the amount of oocyte membrane labeling by Alexa is plotted as a function of GFP fluorescence (i.e., GFP-Syx-MBP). With no NEM block, there is a clear linear relationship (R = 0.8 from linear regression to the data) between the intensity of Alexa labeling and GFP fluorescence (Fig. 5 C). The Y-intercept at zero GFP fluorescence indicates the background Alexa labeling when there was no GFP-Syx-MBP expression. This level is comparable to the amount of labeling by 10 μM Alexa in water-injected oocytes (Fig. 5 D). When the oocytes were pretreated with NEM (100 μM) before Alexa labeling, most of the labeling was blocked and Alexa fluorescence no longer correlated (R = 0.2) with GFP fluorescence (Fig. 5 C). The small amount of labeling that remained is likely due to Alexa accumulating in the membranes surrounding the oocyte, and is comparable to the amount of labeling in control oocytes that were preblocked with NEM (Fig. 5 D).
When 100 μM and 1 mM Cd2+ were present during NEM block, the increase in Alexa labeling once again correlated with the amount of GFP fluorescence (R = 0.8 and R = 0.9, respectively; Fig. 5 C). At 1 mM Cd2+, the slope was indistinguishable from that of the unblocked oocytes, but the intercept was reduced to 45% of its original value, indicating a reduction in background. When 100 μM Cd2+ was present during NEM application, the background (Y-intercept) was completely blocked to the same level observed with no Cd2+ present. The slope was marginally reduced, by a factor of 1.78, indicating that there was also a small reduction in the labeling of GFP-Syx-MBP. Blocking background in the presence of 100 μM Cd2+ increased the signal/background ratio more than threefold over oocytes with no NEM block. In contrast to this, control oocytes demonstrated very little additional fluorescence when Cd2+ was present during NEM block (Fig. 5 D).
Discussion
We have presented a new (to our knowledge) method that increases the specificity of site-directed fluorescent labeling of a desired protein. This method, which we call CyMPL, involves the introduction of cysteine-containing minimal metal-binding sites into regions of known secondary structure. These sites coordinate group 12 metal ions such as Cd2+ and Zn2+ with high affinity to protect a cysteine from reaction with thiol-modifying compounds. The fluorescence background can then be reduced through the covalent blockade of undesired cysteines. We demonstrated the ability of this method to label a single component in a mixture of proteins or to increase the signal/background ratio for the labeling of a membrane protein in Xenopus oocytes, where large background labeling results from the expression of unknown proteins. The specific applications presented here show the great promise of this method for specifically labeling individual cysteines or cysteine pairs. In every application of our method, the results were consistent with the binding affinities measured for the individual protein components. Therefore, these constants, which were remarkably similar for cysteines in a variety of protein backgrounds, can be used as guidelines to design a specific labeling protocol for virtually any protein of known secondary structure.
This technique has advantages over previously established methods for site-directed fluorescence labeling. Although many of these methods provide specificity, they involve the insertion of entire structured protein motifs ranging from six (e.g., polyhistidine and tetracysteine motifs used for FlAsH) or 18 amino acids (zinc finger domains) to 238 amino acids (GFP) or more. Our method requires as few as two point mutations (fewer if one uses native amino acids) inserted into existing secondary structural elements (i.e., α-helix or β-strand). When coupled with small, cysteine-reactive fluorophores with short linkers, this method provides specific labeling of a protein with minimal structural perturbation.
The efficacy of our method relies on the selective increase in metal-binding affinity of a desired cysteine. This is accomplished by placing histidine or cysteine residues on the same side of an α-helix or β-strand close to the cysteine intended for labeling. A large increase in Cd2+ affinity was demonstrated with the use of cys-his pairs in α-helical structures; however, an even greater increase in affinity resulted from the use of cys-cys pairs (although cys-his pairs with Zn2+ displayed great promise). When cys-cys pairs are used, the protein of interest will have two reactive cysteines after metal chelation. These cysteines can be reacted either with two molecules of fluorophore or with a bifunctional fluorophore such as dibromobimane (21). Indeed, dibromobimane reacts readily with cys-cys pairs in all of our dicysteine peptides to produce a single product labeled with only one bimane molecule (Fig. S4). Although our chosen examples all used maleimide-linked fluorophores and nonfluorescent blockers, the method should readily extend to the use of methanethiosulfonate reagents, iodoacetamides, and other linkages. The use of methanethiosulfonate reagents as nonfluorescent blockers in a labeling experiment would allow for the removal of the blocker with a reducing agent after labeling (if a nonreducible fluorophore is used to label). This would reduce any effect the blocking agent might have on the protein of interest or other cellular processes.
Acknowledgments
We thank Greg Martin, Shellee Cunnington, Kevin Black, and Gay Sheridan for technical assistance; Dottie Hanck for comments on the manuscript; and Justin Taraska for providing the EGFP-Syntaxin clone and helpful input. We also thank the members of David Baker's laboratory for the use of their spectropolarimeter, and Mary Ann Gawinowicz of Columbia University for performing the mass spectrometry.
This work was supported by the Howard Hughes Medical Institute and the National Institutes of Health (grants EY10329 to W.N.Z. and F32EY018981 to M.C.P.).
Supporting Material
References
- 1.Lakowicz J.R. Springer; New York: 2006. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 2.Taraska J.W., Zagotta W.N. Fluorescence applications in molecular neurobiology. Neuron. 2010;66:170–189. doi: 10.1016/j.neuron.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taraska J.W., Puljung M.C., Zagotta W.N. Short-distance probes for protein backbone structure based on energy transfer between bimane and transition metal ions. Proc. Natl. Acad. Sci. USA. 2009;106:16227–16232. doi: 10.1073/pnas.0905207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsien R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 5.Taraska J.W., Puljung M.C., Zagotta W.N. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat. Methods. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandtner W., Bezanilla F., Correa A.M. In vivo measurement of intramolecular distances using genetically encoded reporters. Biophys. J. 2007;93:L45–L47. doi: 10.1529/biophysj.107.119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaietta G., Deerinck T.J., Ellisman M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 8.Ratner V., Kahana E., Haas E. A general strategy for site-specific double labeling of globular proteins for kinetic FRET studies. Bioconjug. Chem. 2002;13:1163–1170. doi: 10.1021/bc025537b. [DOI] [PubMed] [Google Scholar]
- 9.Islas L.D., Zagotta W.N. Short-range molecular rearrangements in ion channels detected by tryptophan quenching of bimane fluorescence. J. Gen. Physiol. 2006;128:337–346. doi: 10.1085/jgp.200609556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng J., Zagotta W.N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 2000;28:369–374. doi: 10.1016/s0896-6273(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 11.Smith J.J., Conrad D.W., Hellinga H.W. Orthogonal site-specific protein modification by engineering reversible thiol protection mechanisms. Protein Sci. 2005;14:64–73. doi: 10.1110/ps.04965405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuiper J.M., Pluta R., Poolman B. A method for site-specific labeling of multiple protein thiols. Protein Sci. 2009;18:1033–1041. doi: 10.1002/pro.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zagotta W.N., Olivier N.B., Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 14.Marqusee S., Robbins V.H., Baldwin R.L. Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. USA. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenfield N., Fasman G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 1969;8:4108–4116. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- 16.Gordon S.E., Zagotta W.N. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- 17.Rulísek L., Vondrásek J. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 1998;71:115–127. doi: 10.1016/s0162-0134(98)10042-9. [DOI] [PubMed] [Google Scholar]
- 18.Leverrier P., Montigny C., Champeil P. Metal binding to ligands: cadmium complexes with glutathione revisited. Anal. Biochem. 2007;371:215–228. doi: 10.1016/j.ab.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Ma Z., Wong K.Y., Horrigan F.T. An extracellular Cu2+ binding site in the voltage sensor of BK and Shaker potassium channels. J. Gen. Physiol. 2008;131:483–502. doi: 10.1085/jgp.200809980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spurlino J.C., Lu G.Y., Quiocho F.A. The 2.3-A resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 21.Kosower N.S., Kosower E.M., Ranney H.M. Bimane fluorescent labels: labeling of normal human red cells under physiological conditions. Proc. Natl. Acad. Sci. USA. 1979;76:3382–3386. doi: 10.1073/pnas.76.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.