

Abstract
The Cip/Kip protein family, which includes p27, p21, and p57, modulates the activity of cyclin-dependent kinases (Cdks). A domain within these proteins, termed the kinase inhibitory domain (KID), is necessary and sufficient for Cdk inhibition. The KID consists of a cyclin-binding subdomain (termed D1) and a Cdk-binding subdomain (termed D2) joined by a 22-residue linker subdomain (termed LH). Before binding the Cdks, D1 and D2 are largely unstructured and the LH subdomain exhibits nascent helical characteristics. Curiously, although the sequence of the linker subdomain is not highly conserved within the family, its nascent helical structure is conserved. In this study, we explored the role of this structural conservation in interactions with cyclin-dependent kinase 2 (Cdk2) and cyclin A. We constructed chimeric p27-KID molecules in which the p27 LH subdomain was replaced with the corresponding segments of either p21 or p57. The chimeric molecules bind and inhibit Cdk2 in a manner similar to wild-type p27-KID. However, the extent of enthalpy/entropy compensation associated with these interactions was dramatically different, indicating different extents of LH subdomain folding upon binding. Our results indicate that the different LH subdomains, despite their sequence and thermodynamic differences, play similar roles in binding and inhibiting Cdk2/cyclin A.
Introduction
The activity of cyclin-dependent kinases (Cdks), the master regulators of the cell division cycle (1), is regulated through post-translational modifications and protein-protein interactions. Phosphorylation within the activation loop activates Cdks when they are bound to regulatory subunits termed cyclins (2,3). Phosphorylation of Cdks within an N-terminal β-strand is inhibitory and these modifications are reversed by specific phosphatases at critical stages of cell division (4–6). Certain isolated Cdks and many Cdk/cyclin complexes associate with additional regulatory proteins, which are reported to either promote the assembly of Cdks with their cyclin partners (Cdk4 and Cdk6 paired with D-type cyclins) and thus promote catalytic activity (7,8), or to inhibit catalytic activity (Cdk2 paired with cyclin A or cyclin E) (8). There are two families of Cdk regulators (termed CKRs): the Ink4 family that includes p15Ink4b (9), p16Ink4a (10), p18Ink4c (11), and p19Ink4d (11); and the Cip/Kip family that includes p21Waf1/Cip1/Sdi1/Cap20 (p21) (12–14), p27Kip1 (p27) (15–17), and p57Kip2 (p57) (18,19). The Ink4 family inhibits Cdk activity by binding to Cdk4 and Cdk6 and inhibiting the formation of complexes with the D-type cyclins (20). The Cip/Kip proteins bind to and inhibit activated Cdk2/cyclin A and Cdk2/cyclin E complexes, causing cell cycle arrest at the G1 to S transition during cell division. Paradoxically, Sherr and co-workers (7) reported that p21 and p27 promoted the assembly of Cdk4/cyclin D and Cdk6/cyclin D complexes and that these CKR-containing complexes were catalytically active.
p21, p27, and p57 lack stable tertiary structure in isolation and are termed intrinsically disordered proteins (IDPs) (21–23). As has been observed for many IDPs (24,25), the Cip/Kip proteins perform their biological functions by folding upon binding to their Cdk/cyclin targets (24). Despite their classification as IDPs, the Cip/Kip proteins are not completely disordered. For example, some segments within p21 (27), p27 (25,28), and p57 (19) exhibit partially populated secondary structure (reviewed in (29)); these structural elements were termed intrinsically folded structural units (IFSUs) (28). IFSUs occur within the N-terminal, kinase inhibitory domain (KID) that is conserved between these three proteins (25,29). The KID consists of three subdomains: the cyclin-binding subdomain (termed D1) containing the conserved RxL motif; the Cdk-binding subdomain (termed D2), which also exhibits a pattern of conserved residues; and a linker subdomain that joins D1 and D2 (termed LH; Fig. 1 A). The KID of p27 (p27-KID) extensively folds upon binding to Cdk2/cyclin A (25). In the structure of p27-KID bound to Cdk2/cyclin A (30), subdomain D1 adopts an extended conformation and residues within the RxL motif specifically bind to a conserved groove on the surface of cyclin A; individual segments within subdomain D2 fold into a β-hairpin, a β-strand (which forms an intermolecular sheet with Cdk2), and a 310 helix while making extensive contacts with Cdk2; and subdomain LH forms an ∼40 Å-long α-helix that serves as a spacer, which positions subdomains D1 and D2 to independently bind cyclin A and Cdk2, respectively. The IFSUs observed for p27-KID correspond to partially folded forms of secondary structures that are fully folded in the Cdk2/cyclinA bound state. For example, a partially populated β-hairpin and a single turn of helix were observed within subdomain D2 for free p27-KID (28), and these structures are fully folded when p27-KID is bound to Cdk2 within the Cdk2/cyclin A complex. The most prominent IFSU in p27 before binding to Cdk2/cyclin A is a 22 residue long α-helix within subdomain LH that is folded to the extent of ∼30% in isolation (25). Of importance, this IFSU has also been observed in p21 (27) and p57 (19). Subdomain D1, which exhibits the most extensive sequence conservation within the Cip/Kip family and mediates specific recognition of cyclin A, is highly disordered before binding to the Cdk2/cyclinA complex. Interestingly, although the helical character and length of subdomain LH appear to be recapitulated in p21 and p57, the sequence of this subdomain is poorly conserved between p21, p27, and p57 (e.g., only three residues within subdomain LH are conserved as identities (Fig. 1 A)). This situation suggests that subdomain LH plays a structural role as a linker between subdomains D1 and D2 but that, otherwise, residues within this helical segment do not play specific roles in the function of p27.
Figure 1.
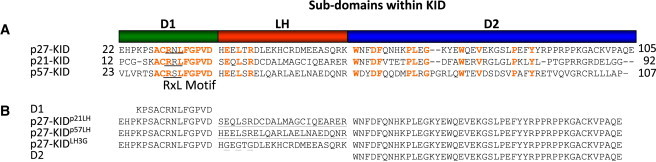
(A) Alignment of the sequences for the kinase inhibitory domains of human p27, p21, and p57. Identical residues are red. The RxL motif is underlined. (B) Sequences of the p27-KID mutants used in this study. The mutated residues or domains are underlined.
In this study, we prepared chimeric p27-KID variants in which subdomain LH was replaced with that from either p21 or p57 (termed p27-KIDp21LH and p27-KIDp57LH; Fig. 1 B) to probe relationships between the sequence and structure of subdomain LH and its role in Cdk inhibition. By replacing only the segment corresponding to subdomain LH, the functions associated with the conserved subdomains D1 and D2 were preserved, allowing the influence of the different LH subdomains on the overall function of p27-KID to be studied. In addition to these naturally derived chimeras, we also created a variant in which the three residues that are conserved as identities in subdomain LH (Glu-39, Leu-41, and Arg-43) were mutated to glycine (termed p27-KIDLH3G) to investigate the role of the conserved residues in function (Fig. 1 B). Furthermore, the importance of the linking function of subdomain LH was probed by studying the Cdk2 inhibitory function of the isolated D2 subdomain, which, in the context of p27-KID, interacts directly with Cdk2 (within the Cdk2/cyclin A complex) and inhibits ATP binding, and the separate D1 and D2 subdomains in combination (Fig. 1 B). In addition to using kinase activity assays to monitor inhibitory function, we used circular dichroism (CD) and thermal denaturation to study the structure of isolated p27-KID variants and the thermal stability of their complexes with Cdk2/cyclin A, respectively. Furthermore, isothermal titration calorimetry (ITC) was used to determine the values of thermodynamic parameters associated with interactions between the p27-KID variants and the Cdk2/cyclin A complex. We also used NMR spectroscopy to study the structural features of the ternary complexes of the p27-KID variants with Cdk2/cyclin A. The results of these experiments provide insights into the role of the partially structured LH subdomain in the biochemical function of a prototype intrinsically disordered protein, p27.
Materials and Methods
Preparation of proteins and peptides
p21, p27, and p57 subdomain LH peptides were expressed as glutathione-S-transferase—6× histidine fusion proteins in Escherichia coli and purified using the protocol given in the Supporting Material. The LH3G LH subdomain and the D1 peptides were prepared by solid-phase synthesis (Hartwell Center, St Jude Children's Research Hospital, Memphis, TN). The preparation of D2, KID variants, Cdk2/cyclin A, and the complexes used in this study are detailed in the Supporting Material. For the NMR experiments, the p27-KID variants were 2H, 15N, and ILV-1H/13C-methyl labeled as described in the Supporting Material.
ITC experiments
ITC studies were performed at 25°C using a VP-ITC (GE Healthcare, Piscataway, NJ) calorimeter. Either p27-KIDwt or its variants were titrated into Cdk2/cyclin A that was contained in the calorimeter cell (experimental details are described in the Supporting Material).
Thermal denaturation experiments
The thermal denaturation of ternary complexes consisting of p27-KIDwt or one of the LH subdomain variants bound to Cdk2/cyclin A was monitored from 15°C to 95°C using an AVIV model number 202-1 A circular dichroism spectropolarimeter equipped with a thermoelectric temperature controller (AVIV, Lakewood, NJ), as described in the Supporting Material.
NMR experiments
[1H,15N] TROSY (31) and [1H,13C] HMQC (32) spectra were recorded for unbound p27-KIDwt, p27-KIDwt/Cdk2, p27-KIDwt/cyclin A, D2/Cdk2/cyclin A, and p27-KID variant/Cdk2/cyclin A ternary complexes at 308 K using a Bruker Avance 800 MHz spectrometer (Bruker, Billerica, MA) equipped with a 1H and 13C detect, triple resonance cryogenic probe as described in the Supporting Material. The strategy for residue assignment is also reported in the Supporting Material.
Kinase activity/inhibition assays
The inhibition activity of the p27 variants was investigated by determining their ability to inhibit the phosphorylation of histone H1 by Cdk2/cyclin A. The experimental procedure is described in the Supporting Material.
Results
The LH subdomains exhibit nascent helical character
The secondary structure of the p21, p27, p57, and LH3G subdomain LH peptides (p21LH, p27LH, p57LH, and p27LH3G), and p27-KIDwt, p27-KIDp21LH, p27-KIDp57LH, and p27-KIDLH3G, were analyzed using CD (Fig. 2, A and B). The subdomain LH peptides were predominantly disordered based on the observation of minimum ellipticity values at ∼200 nm; however, two of the peptides (p27LH and p57LH) exhibited partial α-helical character based on the observation of reduced negative ellipticity near 200 nm and the appearance of a second albeit weak ellipticity minimum near 222 nm. In contrast, p21LH exhibited very weak α-helical character. These results are consistent with past observations of helicity within the LH subdomain of p27 (25,33) and the kinase inhibitory domain of p57 (19). Previous studies showed that the kinase inhibitory domain of p21 exhibited weak α-helical character (27) but the isolated p21LH peptide only partially recapitulated this feature (Fig. 2 A). The p21LH may exhibit reduced helicity due to sequence variations, including a glycine residue at position 40 (Fig. 1 A), as suggested by past secondary structure prediction (25). The p27LH3G peptide is significantly less helical relative to p27LH, probably because Gly residues generally exhibit reduced helical propensity. In summary, the three subdomain LH peptides exhibited α-helical features that are populated to different extents.
Figure 2.
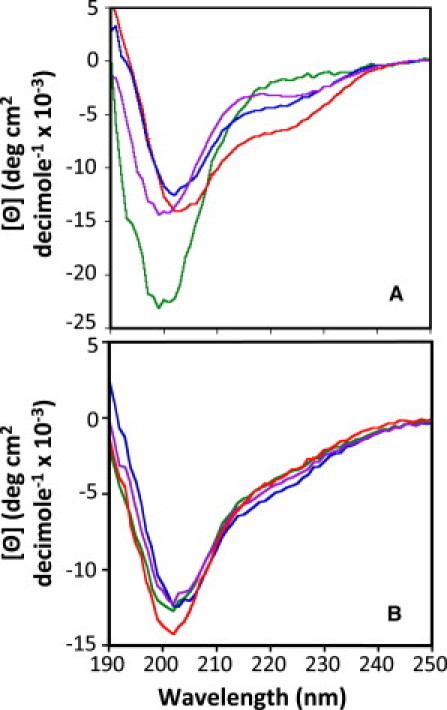
(A) CD spectra of the linker helix peptides of p27, p21, p57, and LH3G. Each sample contained 60 μM peptide in a buffer containing 20 mM sodium phosphate, pH 7.0 and 1 mM DTT. The spectra for p27, p21, p57, and LH3G linker helix domain peptides are red, green, blue, and purple, respectively. (B) CD spectra of the p27-KIDwt and the LH subdomain variants. Each sample contained 20 μM protein in the same buffer used for the linker helix peptides. Spectra were recorded at 25°C. The spectra for p27-KIDwt, p27-KIDp21LH, p27-KIDp57LH, and p27-KIDLH3G are red, green, blue, and purple, respectively.
The chimeric p27-KID subdomain LH variants exhibited nascent α-helical features very similar to those observed for p27-KIDwt. For example, the mean residue molar ellipticity values of the chimeric mutants at 222 nm ([Θ222]) (Fig. 2 B) were similar to that of p27-KIDwt, indicating that they have similar helical contents. These results suggest that, in the context of the chimeric variants, the LH subdomains of p21 and p57 exhibited structural features similar to those observed previously in their natural context within p21-KID and p57-KID, respectively (19,27). Therefore, these reagents are suitable to probe the role of the different LH subdomains in interactions with Cdk/cyclin complexes.
The p27-KIDLH3G variant exhibited structural features similar to those of the other constructs under study (Fig. 2 B). This contrasts with our observations with the isolated p27LH3G peptide (Fig. 2 A) and suggests that this variant subdomain becomes partially helical in the context of the p27-KIDLH3G construct. This may also reflect limitations in using CD to detect small differences in helicity within polypeptides that are predominantly disordered. The observation of these structural similarities establishes that any differences in the interactions between p27-KIDLH3G and the Cdk2/cyclin A complex, relative to those of p27-KIDwt, are not a consequence of gross structural differences between the wild-type and mutant LH subdomains.
Binding thermodynamics determined by ITC
We determined the values of the thermodynamic parameters for the binding of p27-KIDwt and the LH subdomain variants to Cdk2/cyclin A using ITC. Raw titration data (Fig. 3) shows that each of the binding reactions is exothermic and that the associated Kd values are approximately equal. ITC instruments can very accurately measure the enthalpy change (ΔH°) and stoichiometry (n) associated with protein-protein interactions. However, given the signal/noise limitations of current ITC instruments, it is difficult to accurately determine Kd (and thus ΔG°) values for very tight interactions (Kd < 5 nM), as was observed for p27-KID and the LH subdomain variants under study here binding to Cdk2/cyclin A. Therefore, the Kd and ΔG° values for the binding reactions reported in Table 1 are considered to be estimates of upper limits of the actual values. In contrast, the Kd and ΔG° values, in addition to values for ΔH° and n, for the D1 and D2 peptides binding to Cdk2/cyclin A (Table 1) are well determined by the raw data and therefore accurate.
Figure 3.
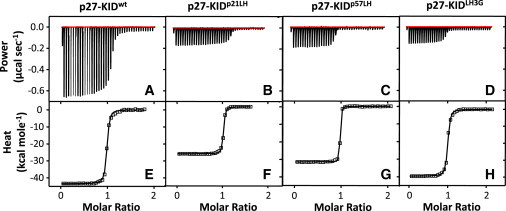
Titration calorimetry data and binding isotherms, respectively, for (A and E) p27-KIDwt, (B and F) p27-KIDp21LH, (C and G) p27-KIDp57LH, and (D and H) p27-KIDLH3G binding to Cdk2/cyclin A. The isotherms (E–H) were fit to a 1:1 binding model using procedures and software provided by the manufacturer.
Table 1.
Thermodynamic parameters for p27-KIDwt and LH subdomain variants binding to Cdk2/cyclin A determined by ITC at 25°C
| Interaction | Kd(nM) | ΔG∗(kcal mol−1) | ΔH(kcal mol−1) | −TΔS†(kcal mol−1) |
|---|---|---|---|---|
| p27-KIDwt + Cdk2/cyclin A | 3.3 ± 1.6 | −11.6 ± 0.3 | −43.8 ± 1.4 | + 32.2 ± 1.2 |
| p27-KIDp21LH + Cdk2/cyclin A | 2.8 ± 1.4 | −11.7 ± 0.3 | −27.9 ± 1.3 | + 16.2 ± 1.3 |
| p27-KIDp57LH + Cdk2/cyclin A | 0.5 ± 0.1 | −12.7 ± 0.2 | −31.1 ± 0.5 | + 18.4 ± 0.4 |
| p27-KIDLH3G + Cdk2/cyclin A | 2.0 ± 0.7 | −11.9 ± 0.2 | −39.7 ± 0.1 | + 27.8 ± 0.2 |
| D1 + Cdk2/cyclin A | 42.2 ± 5.2 | −10.0 ± 0.1 | −16.1 ± 0.2 | + 6.1 ± 0.3 |
| D2 + Cdk2/cyclin A | 83.0± 17.9 | −9.7 ± 0.1 | −31.6 ± 1.7 | + 21.9 ± 1.5 |
In all the experiments above, the stoichiometry of binding (n) was equal to 1.0 ± 0.1. The errors reported above are the standard deviations of the mean based on triplicate measurements.
Calculated using the equation: ΔG = −RT ln(1/Kd).
Calculated using the equation: −TΔS = ΔG − ΔH.
Among the proteins under study here, p27-KIDwt exhibited the largest binding enthalpy upon association with Cdk2/cyclin A (ΔH° = −43.8 ± 1.4 kcal mol−1; Table 1). The ΔH° value for p27-KIDLH3G was very similar to this value [ΔΔH° (ΔH°variant − ΔH°wt) = +4.1 kcal mol−1), indicating that the conserved residues of the LH subdomain contribute only about −4 kcal mol−1 (∼9.4%) toward the overall enthalpy of binding. In contrast, the ΔH° values for p27-KIDp21LH and p27-KIDp57LH were significantly less favorable (ΔΔH° = +15.9 kcal mol−1 and +12.7 kcal mol−1, respectively), suggesting that residues in the chimeric LH subdomains, and possibly others within the flanking D1 and D2 subdomains, interact differently (much less favorably) with Cdk2/cyclin A than do the wild-type subdomains. We showed previously and demonstrated again here that p27-KID extensively folds upon binding to Cdk2/cyclin A, which gives rise to a large and unfavorable entropy change upon binding (−TΔS° = +32.2 kcal mol−1; Table 1). The magnitude of the change in the values of −TΔS° (−TΔΔS°) for the p27 variants (relative to that for p27-KIDwt) were similar but opposite in sign with respect to the corresponding ΔΔH° values, indicating that the reduction in favorable interactions (between p27 variants and Cdk2/cyclin A) is offset by reduced ordering upon binding—an example of enthalpy/entropy compensation (34). These observations suggest that the LH subdomains of the p27 variants, within their complexes with Cdk2/cyclin A, are more dynamic and interact less extensively with Cdk2/cyclin A than does the wild-type LH subdomain.
We also used ITC to analyze the binding of subdomains D1 and D2 to Cdk2/cyclin A to understand the contributions of these interactions to the thermodynamics of p27-KIDwt and the LH subdomain variants binding to Cdk2/cyclin A. The crystal structure of p27-KID bound to Cdk2/cyclin A and our previous thermodynamic analyses of this system (25) showed that subdomains D1, LH, and D2 of p27 interact in a structurally independent manner with different surfaces of Cdk2/cyclin A (Fig. S1 A). Therefore, the ΔH° value for p27-KID binding to Cdk2/cyclin A approximately equals the sum of the ΔH° values for binding of the individual domains (e.g., ΔH°KID ≈ ΔH°D1 + ΔH°LH + ΔH°D2). Interactions between individual subdomains are possible but, based on a previous NMR and molecular dynamics study of isolated p27-KID (28), we believe that these interactions are very weak. Therefore, we have adopted this formalism to estimate and compare values of ΔH°LH for the different KID constructs. For p27-KID, this value is +3.9 kcal mol−1; this corresponds to an unfavorable contribution to the overall binding enthalpy of the KID. The corresponding values for the variant LH subdomains are ΔH°p21LH = +19.8 kcal mol−1, ΔH°p57LH = +16.6 kcal mol−1, and ΔH°LH3G = +8.0 kcal mol−1. These values correspond to significantly more unfavorable interactions with Cdk2/cyclin A than was observed for the wild-type LH subdomain. However, the ΔG° values for the binding of all of the KID constructs are similar, indicating that the lack of favorable contributions from the LH subdomains is accompanied by reduction of the unfavorable ΔS° values associated with binding.
The Kd values for D1 and D2 binding to Cdk2/cyclin A were also determined through analysis of ITC data; these values (42.2 nM and 83.0 nM, respectively; Table 1) indicated that these individual subdomains bind more weakly to their targets, cyclin A and Cdk2, respectively, than does the intact KID to Cdk2/cyclin A (Kd = 3.3 nM).
The structures of the ternary complexes of p27-KID variants with Cdk2/cyclin A are similar
Lacy et al. (25), determined that p27-KIDwt binds to the Cdk2/cyclin A complex in a sequential manner with subdomain D1 interacting with cyclin A first followed by folding of the LH, and finally the folding of D2 as it binds to Cdk2. p27-KIDwt adopts a molecular staple-like conformation in the ternary complex (Fig. S1 A). We were interested in determining whether the identical D1 and D2 subdomains within the p27-KID variants adopted similar conformations upon interacting with Cdk2/cyclin A. Two-dimensional (2-D) [1H,15N] TROSY and [1H,13C] HMQC spectra were recorded for isolated p27-KIDwt and several binary and ternary complexes, including p27-KIDwt/Cdk2, p27-KIDwt/cyclin A, p27-KIDwt/Cdk2/cyclin A, D2/Cdk2/cyclin A, p27-KIDp21LH/Cdk2/cyclin A, p27-KIDp57LH/Cdk2/cyclin A, and p27-KIDLH3G/Cdk2/cyclin A. These experiments were performed using 2H/15N-, ILV-1H/13C-methyl-labeled p27-KID species. A representative 2-D [1H,15N] TROSY spectrum of the p27-KIDwt ternary complex is shown in Fig. 4 A. Backbone resonance assignments for p27-KIDwt bound to Cdk2/cyclin A were previously published (35). The 2-D TROSY spectra for the various ternary complexes were very similar (Fig. S2), indicating that p27-KIDwt and the p27-KID variants interacted with Cdk2/cyclin A in a similar manner. For example, resonances for a subset of residues in subdomains D1 (C29, R30, N31, and L32) and D2 (G72, E75, Q77, and Y88), which served as reporters of the bound state conformation, exhibited very similar chemical shift values in the different ternary complexes (Fig. 4 A). The 2-D [1H, 13C] HMQC spectra for these ternary complexes were analyzed in a similar manner and, again, exhibited similar resonance patterns (Fig. 4 B, Fig. S3). A limited number of 1H/13C methyl resonances were assigned through comparison of 2-D [1H, 13C] HMQC spectra for p27-KIDwt individually bound to either Cdk2 or cyclin A (L32 and V36 in subdomain D1, and V79 and L70/L84 in subdomain D2; Fig. 4 B; see Supporting Material for the details of the assignment strategy). Together, this analysis of backbone amide and side-chain methyl resonances indicated the D1 and D2 subdomains of the p27-KID variants interacted with Cdk2/cyclin A in a manner similar to that of p27-KIDwt.
Figure 4.
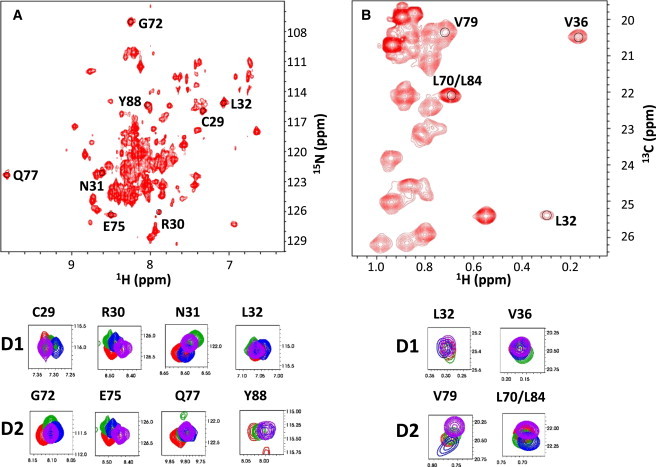
(A) [1H,15N] TROSY spectrum of U-2H/15N, ILV-1H/13C-methyl-labeled p27-KID/Cdk2/Cyclin A complex. The panels below the spectrum show overlaid, expanded views of resonances from [1H,15N] TROSY spectra for representative residues within subdomains D1 and D2 of p27-KIDwt (red), p27-KIDp21LH (green), p27-KIDp57LH (blue), or p27-KIDLH3G (purple) bound to Cdk2/cyclin A. (B) [1H,13C] HMQC spectrum of U-2H/15N, ILV-1H/13C-methyl-labeled p27-KID/Cdk2/Cyclin A complex. The panels below the spectrum show overlaid, expanded views of resonances from [1H,13C] HMQC spectra for representative residues within subdomains D1 and D2 of p27-KIDwt (red), p27-KIDp21LH (green), p27-KIDp57LH (blue), or p27-KIDLH3G (purple) bound to Cdk2/cyclin A.
Thermal stability of complexes containing the p27 LH subdomain variants and Cdk2/cyclin A
The ability of the subdomain LH variants to stabilize Cdk2/cyclin A against thermal denaturation was determined through the analysis of thermal denaturation curves measured using CD (36) (Fig. S4). Thermal denaturation of the various ternary complexes was irreversible due to precipitation of Cdk2 and cyclin A at temperatures above the unfolding transition and, therefore, apparent thermal denaturation temperatures (Tmapp) are reported (Table 2). Our results show that the ternary complex containing p27-KIDwt (Tmapp = 82.3 ± 0.5°C), which was characterized previously (37), was slightly more thermally stable than those that contained p27-KIDp21LH and p27-KIDp57LH (with Tmapp values of 80.7 ± 0.2°C and 80.1 ± 0.1°C, respectively). The ternary complex that contained p27-KIDLH3G was the least stable, with a Tmapp value of 74.1 ± 0.6°C. The relative instability of the latter ternary complex may in part be due to the loss of favorable contacts between the side chain of Leu-41 of p27 (mutated to Gly) and hydrophobic residues within a hydrophobic groove on the surface of cyclin A (30) (Fig. S1 B). In addition, mutation of Glu-39 and Arg-43 to Gly may eliminate favorable electrostatic interactions among a network of charged residues on the solvent-exposed face of the LH subdomain that spans from cyclin A to Cdk2 (Fig S1 B). These binding deficiencies may be compounded by the reduced helical content of this mutant LH subdomain, as shown by CD of the LH subdomain peptide (Fig. 2 A). In summary, with the exception of p27-KIDLH3G, the results of thermal denaturation studies suggest that the different p27-KID constructs interacted with and stabilized Cdk2/cyclin A similarly.
Table 2.
Melting temperatures of ternary complexes formed by either p27-KIDwt or the LH subdomain variants bound to the Cdk2/cyclin A complex
| Complex | Tmapp (°C) |
|---|---|
| p27-KIDwt/Cdk2/cyclin A∗ | 82.3 ± 0.5 |
| p27-KIDp21LH/Cdk2/cyclin A† | 80.7 ± 0.2 |
| p27-KIDp57LH/Cdk2/cyclin A∗ | 80.1 ± 0.1 |
| p27-KIDLH3G/Cdk2/cyclin A† | 74.1 ± 0.6 |
The error reported is the standard deviation from the mean of three independent experiments.
The error reported is the standard deviation from the mean of two independent experiments.
Activity of the p27 LH subdomain variants as inhibitors of Cdk2
The Cdk2 inhibitory activity of the p27 LH subdomain variants was determined by measuring the concentration dependence of inhibition of Histone H1 phosphorylation (Fig. 5, Fig. S5). The IC50 value for p27-KIDwt was 0.2 nM, whereas values for the LH subdomain variants ranged from 1.7 nM to 3.5 nM. The effective IC50 value for the subdomain D2 peptide was markedly higher (125 nM) and, importantly, was unable to completely inhibit Cdk2 even at concentrations above 10 μM. These results indicated that substitution of the wild-type LH subdomain in p27-KIDwt with those from p21 and p57, or mutation of three conserved residues within this subdomain to Gly, reduced inhibitory potency by approximately one order of magnitude (corresponding to increased IC50 values). In contrast, deletion of subdomains D1 and LH was associated with reduced inhibitory potency and an inability to completely inhibit Cdk2 within the subdomain D2/Cdk2/cyclin A complex.
Figure 5.

Cdk2/cyclin A inhibition curves for p27-KIDwt and the LH subdomain variants. The inhibition curves for p27-KIDwt, p27-KIDp21LH, p27-KIDp57LH, and p27-KIDLH3G are red, green, blue, and purple, respectively.
Discussion
Dating back to 1894 and the ideas of Emil Fischer (38), the classic theory of protein structure-function relationships postulated that a distinct three-dimensional structure was critical for protein function. Although this concept certainly applies to myriad folded proteins, especially enzymes, it is now well established that many functional proteins lack secondary and/or tertiary structure in isolation under physiological conditions (29). These so-called IDPs exist in all organisms, from prokaryotes to eukaryotes (39–41). Bioinformatics analyses of eukaryotic genomes predict that approximately one-third of all proteins are either completely disordered or contain lengthy disordered segments (40). Furthermore, IDPs are involved in a plethora of cellular processes including but not limited to transcription, translation, signal transduction, microtubule and actin dynamics, chaperone activity, and cell cycle regulation (42). The Cip/Kip cyclin-dependent kinase regulators, p21, p27, and p57, are among the best studied IDPs (29) and therefore serve as a model system for detailed analysis of relationships between polypeptide disorder and function.
The conserved kinase inhibitory domain (KID) within p21, p27, and p57 mediates interactions with Cdk/cyclin complexes. Residues within the D1 and D2 subdomains are highly conserved (Fig. 1 A), are disordered (19,25,27,33), and mediate specific interactions with conserved surfaces on the cyclin and Cdk subunits, respectively, of Cdk/cyclin complexes. Although the sequences are not highly conserved, the length and nascent helicity of the LH subdomain, which connects D1 and D2, are conserved within p21, p27, and p57 (25) (Fig. 1 A). However, how these features contribute to Cip/Kip protein function is poorly understood. To understand the role of the LH subdomain in p27 function, we created chimeric variants in which the D1 and D2 subdomains of p27 were connected by the LH subdomain of either p21, p27, or p57. Analysis using CD showed that the isolated LH subdomains exhibit different degrees of nascent helicity (Fig. 2 A). However, within the limits of CD analysis, these variations in secondary structure appeared to not influence the disordered features of the D1 and D2 subdomains within the chimeric KID constructs (Fig. 2 B). Finally, a fourth variant, p27-KIDLH3G, was prepared by connecting the p27 D1 and D2 subdomains with an LH subdomain in which three conserved residues were mutated to glycine (Fig. 1 A and B). The availability of these closely related chimeric KID variants allowed the role of the LH subdomain in p27 function to be studied using biochemical and biophysical methods.
The p27-KIDwt and the LH subdomain variants were shown to function similarly. For example, they exhibited similar Kd values for binding to Cdk2/cyclin A (Table 1) and generally exhibited similar IC50 values for inhibition of Cdk2 catalytic activity (Table 3). Furthermore, the D1 and D2 subdomains within all natural LH subdomain constructs bound similarly to Cdk2/cyclin A (Fig. 4) and they thermally stabilized Cdk2/cyclin A to similar extents (Table 2). Mutation of conserved residues within the LH subdomain did alter thermal stabilization (Table 2), probably due to disruption of favorable hydrophobic interactions between Leu-41 and the surface of cyclin A and favorable electrostatic interactions between Glu-39 and Arg-43 on the solvent exposed face of the LH subdomain helix (Fig. S1 B) and due to reduced helicity. Taken together, these results indicate that the sequence differences between the LH subdomains tested herein did not significantly affect the overall mode of interaction of the chimeric p27 KID variants with the Cdk2/cyclinA complex.
Table 3.
IC50 values for inhibition of Cdk2 by p27-KIDwt and the LH subdomain variants
| Variant | IC50 (nM) | 95% confidence intervals of IC50 (nM)∗ |
|---|---|---|
| p27-KIDwt | 0.2 | 0.2–0.3 |
| p27-KIDp21LH | 1.7 | 1.1–2.7 |
| p27-KIDp57LH | 3.5 | 2.3–5.1 |
| p27-KIDLH3G | 2.6 | 1.7–3.9 |
| D2 | 125.0†‖ | 45.9–340.8 |
Error is reported as the 95% confidence interval of the IC50 from a curve fit of a triplicate data set.
This variant only partially inhibits kinase activity. 25% residual kinase activity observed at inhibitor concentrations as high as 17.7 μM.
It is interesting, however, that although the ΔG° values for the p27-KID variants binding to Cdk2/cyclin A were very similar the extent of enthalpy/entropy compensation among them was highly variable. For example, with p27-KIDwt, ΔH° was very large and favorable (∼−44 kcal mol−1), which counterbalanced the large and unfavorable –TΔS° term (∼+32 kcal mol−1). In contrast, with p27-KIDp21LH, the favorable ΔH° value was reduced to ∼−28 kcal mol−1 and the unfavorable –TΔS° value was reduced to ∼+16 kcal mol−1. In both cases, the sum of these two values, giving ΔG°, was approximately the same (∼−12 kcal mol−1). The ΔH° values for the other two LH subdomain variants were between those for p27-KIDwt and p27-KIDp21LH and were counterbalanced by –TΔS° values that gave approximately equal ΔG° values.
We previously determined that extensive folding of p27-KIDwt upon binding to Cdk2/cyclin A is associated with a large and unfavorable value of –TΔS° (25). There are several contributions to the overall ΔS° associated with binding (43), including a favorable contribution to ΔS° associated with burial of hydrophobic residues (ΔS°HE), an unfavorable contribution associated with the change in configurational entropy due to a loss of rotational and translational degrees of freedom (ΔS°rt), and a highly unfavorable contribution associated with reduced conformational entropy due to folding coupled with binding (ΔS°other). We propose that the variations in the values of ΔH° and –TΔS° in the p27-KID constructs with different LH subdomains arise due to different extents of LH subdomain folding (manifested as reduced values of unfavorable ΔS°other) when the otherwise identical D1 and D2 subdomains bind to Cdk2/cyclin A. Less extensive folding of some LH subdomains is accompanied by smaller and less favorable ΔH° values probably due to the formation of fewer, enthalpically favorable helical H-bonds and side chain-side chain contacts. We suggest that the primary role of the Cip/Kip LH subdomain, with regard to interactions with Cdk2/cyclin A, is to covalently link subdomains D1 and D2 to achieve high inhibitory potency. The D2 subdomain alone binds weakly to Cdk2/cyclin A (Table 1) and is a poor Cdk2 inhibitor (Table 3) relative to p27-KIDwt. Similarly, the D1 subdomain binds relatively weakly to cyclin A (Table 1). However, covalent linkage of these two subdomains by the different LH subdomains examined herein similarly enhanced ΔG° values. This may explain why evolution has allowed divergence of LH subdomain sequence within the paralogs, p21, p27, and p57, whereas those of the D1 and D2 subdomains, which are required for specific recognition of Cdk/cyclin complexes, are conserved. Our analysis, however, did not address the possible influence of sequence variations within subdomain LH on the kinetics of Cdk2/cyclin A binding, and does not rule out the possibility that alteration of the p27 LH subdomain influences binding kinetics. We previously showed (25) that the isolated subdomain D1 binds to cyclin A significantly faster than isolated D2 subdomain binds to Cdk2. However, when the D2 subdomain was tethered to D1 via the LH subdomain, the entire KID bound as rapidly as did the isolated D1 subdomain. The alterations made within the LH subdomain could conceivably alter this kinetic behavior in such a way that affects both association and dissociation rates without affecting binding thermodynamics, as discussed previously by Lumb and co-workers (33).
A dual mode of recognition, with subdomain D1 binding to cyclins and subdomain D2 binding to Cdks within Cdk/cyclin complexes, is required for specific recognition of Cdk/cyclin complexes that directly regulate cell division (25). The results presented herein provide further insight into the requirement for the dual mode of recognition. The thermodynamics of subdomain D2/Cdk interactions may be incompatible with achieving a sufficiently low Kd value to complete Cdk/cyclin inhibition (Table 3), as is required for G1 to S phase arrest during cell cycle. The linkage of the D2 subdomain to the D1 subdomain, which promotes rapid association with cyclin A within the Cdk2/cyclin A complex (25), may be required to tether p27 to these complexes and, therefore, enhance the overall ΔG of binding. The thermodynamic gain associated with linkage via the LH subdomains examined herein is modest (the Kd value for subdomain D2 alone is ∼80 nM, whereas that for the KIDs studied herein ranged from 0.5 nM to 3.3 nM; Table 1). Nonetheless, this gain, which has been achieved through natural selection and evolution, may reflect the cellular conditions under which p27 must function, for example the nuclear concentrations of the Cdk/cyclin complexes which it targets.
Acknowledgments
We acknowledge Cheon-Gil Park (St. Jude) for assistance with isotope labeled protein expression and purification and Jack Sublett (St. Jude) for assistance with kinase assays.
This work was supported in part by National Institutes of Health (Cancer Center Support Core grant CA-21765 to St. Jude Children's Research Hospital and R01CA082491-08 to R.W.K.) and the American Lebanese Syrian Associated Charities.
Supporting Material
References
- 1.Morgan D.O. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 2.Solomon M.J. The function(s) of CAK, the p34cdc2-activating kinase. Trends Biochem. Sci. 1994;19:496–500. doi: 10.1016/0968-0004(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 3.Harper J.W., Elledge S.J. The role of Cdk7 in CAK function, a retro-retrospective. Genes Dev. 1998;12:285–289. doi: 10.1101/gad.12.3.285. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe N., Broome M., Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Booher R.N., Holman P.S., Fattaey A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 1997;272:22300–22306. doi: 10.1074/jbc.272.35.22300. [DOI] [PubMed] [Google Scholar]
- 6.Millar J., McGowan C., Russell P. cdc25 M-phase inducer. Cold Spring Harb. Symp. Quant. Biol. 1991;56:577–584. doi: 10.1101/sqb.1991.056.01.065. [DOI] [PubMed] [Google Scholar]
- 7.Cheng M., Olivier P., Sherr C.J. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherr C.J., Roberts J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 9.Hannon G.J., Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 10.Serrano M., Hannon G.J., Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 11.Hirai H., Roussel M.F., Sherr C.J. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell. Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper J.W., Adami G.R., Elledge S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 13.Harper J.W., Elledge S.J., Wei N. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.el-Deiry W.S., Harper J.W., Vogelstein B. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 15.Polyak K., Kato J.Y., Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 16.Polyak K., Lee M.H., Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 17.Toyoshima H., Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 18.Matsuoka S., Edwards M.C., Elledge S.J. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 19.Adkins J.N., Lumb K.J. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57Kip2. Proteins. 2002;46:1–7. doi: 10.1002/prot.10018. [DOI] [PubMed] [Google Scholar]
- 20.Russo A.A., Tong L., Pavletich N.P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature. 1998;395:237–243. doi: 10.1038/26155. [DOI] [PubMed] [Google Scholar]
- 21.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 22.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 23.Uversky V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 24.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lacy E.R., Filippov I., Kriwacki R.W. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 2004;11:358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- 26.Reference deleted in proof.
- 27.Kriwacki R.W., Hengst L., Wright P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sivakolundu S.G., Bashford D., Kriwacki R.W. Disordered p27(Kip1) exhibits intrinsic structure resembling the Cdk2/Cyclin A-bound conformation. J. Mol. Biol. 2005;353:1118–1128. doi: 10.1016/j.jmb.2005.08.074. [DOI] [PubMed] [Google Scholar]
- 29.Galea C.A., Wang Y., Kriwacki R.W. Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits. Biochemistry. 2008;47:7598–7609. doi: 10.1021/bi8006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russo A.A., Jeffrey P.D., Pavletich N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 31.Pervushin K., Riek R., Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller L. Sensitivity enhanced detection of weak nuclei using heteronuclear multiple quantum coherence. J. Am. Chem. Soc. 1979;101:4481–4484. [Google Scholar]
- 33.Bienkiewicz E.A., Adkins J.N., Lumb K.J. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41:752–759. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 34.McWherter C.A., Haas E., Scheraga H.A. Conformational unfolding in the N-terminal region of ribonuclease A detected by nonradiative energy transfer. Biochemistry. 1986;25:1951–1963. doi: 10.1021/bi00356a018. [DOI] [PubMed] [Google Scholar]
- 35.Grimmler M., Wang Y., Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 36.Pace C.N., Scholtz J.M. Measuring the conformational stability of a protein. In: Creighton T.E., editor. Protein Structure: A Practical Approach. IRL Press; New York: 1989. pp. 299–321. [Google Scholar]
- 37.Bowman P., Galea C.A., Kriwacki R.W. 2006. Thermodynamic characterization of interactions between p27(Kip1) and activated and non-activated Cdk2: intrinsically unstructured proteins as thermodynamic tethers. Biochim. Biophys. Acta. 2011;1764:182–189. doi: 10.1016/j.bbapap.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Fischer E. Influence of the configuration on the action of enzymes. [Einfluss der configuration auf die wirkung der enzyme] Ber. Dt. Chem. Ges. 1894;27:2985–2993. [Google Scholar]
- 39.Tompa P., Dosztanyi Z., Simon I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J. Proteome Res. 2006;5:1996–2000. doi: 10.1021/pr0600881. [DOI] [PubMed] [Google Scholar]
- 40.Dunker A.K., Obradovic Z., Brown C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 41.Ward J.J., Sodhi J.S., Jones D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 42.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 43.Spolar R.S., Record M.T., Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.