

Abstract
Introduction
Multiple organ dysfunction is the main cause of death in severe acute pancreatitis. Primary mitochondrial dysfunction plays a central role in the development and progression of organ failure in critical illness. The present study investigated mitochondrial function in seven tissues during early experimental acute pancreatitis.
Methods
Twenty-eight male Wistar rats (463 ± 2 g; mean ± SEM) were studied. Group 1 (n = 8), saline control; Group 2 (n = 6), caerulein-induced mild acute pancreatitis; Group 3 (n = 7) sham surgical controls; and Group 4 (n = 7), taurocholate-induced severe acute pancreatitis. Animals were euthanased at 6 h from the induction of acute pancreatitis and mitochondrial function was assessed in the heart, lung, liver, kidney, pancreas, duodenum and jejunum by mitochondrial respirometry.
Results
Significant early mitochondrial dysfunction was present in the pancreas, lung and jejunum in both models of acute pancreatitis, however, the Heart, liver, kidney and duodenal mitochondria were unaffected.
Conclusions
The present study provides the first description of early organ-selective mitochondrial dysfunction in the lung and jejunum during acute pancreatitis. Research is now needed to identify the underlying pathophysiology behind the organ selective mitochondrial dysfunction, and the potential benefits of early mitochondrial-specific therapies in acute pancreatitis.
Keywords: rat, acute pancreatitis, mitochondrial dysfunction, multiple organ dysfunction syndrome
Introduction
Multiple organ dysfunction syndrome (MODS) is the main cause of death in both the early and late phases of severe acute pancreatitis.1,2 In the initial 2 weeks of severe acute pancreatitis, MODS is caused by exaggerated cytokine-mediated systemic inflammatory response syndrome (SIRS) which is thought to primarily affect the lungs,3–5 whereas in the latter phase MODS is secondary to sepsis thought to result from intestinal barrier failure.1,6 Mortality from severe acute pancreatitis correlates with the number of organs that fail,7 and the presence of MODS has also been shown to identify patients most at risk of death.8 The response of early organ failure to initial resuscitation and intensive care support is also predictive of outcome.1,3 For example, resolution of organ failure with resuscitation within 48 h confers good prognosis, whereas persistent organ failure does not.1,2 Development of novel therapies for acute pancreatitis associated MODS has been frustrated by a lack of understanding of the underlying pathophysiological processes during early severe acute pancreatitis.9
Recent research has linked mitochondrial dysfunction (MD) with MODS.10–13 Primary MD during SIRS, recently termed ‘cytopathic hypoxia’, is now thought to play a central role in the development and progression of MODS.14–16 This primary mitochondrial failure is postulated to lead to metabolic failure and eventual organ dysfunction.13,16 In addition to cytopathic hypoxia, disruption of the mitochondrial electron transport system (ETS, complexes I–IV, which are responsible for oxidative phosphorylation) in a disease state can result in the excess release of reactive oxygen species (ROS), usually in the form of superoxide (O2·-) from complexes I and III.17 Excess ETS-derived ROS cause further oxidative damage to the mitochondria, which can not only impair respirational flux but also lead to increased mitochondrial permeability with the concomitant release of cytochrome c, a well-defined apoptotic mediator.17 Thus, MD may kill cells by excessive ROS production, energy starvation and/or apoptosis.
Here, we hypothesise that MD occurs in distant organs early during the development of acute pancreatitis and before the development of MODS. Confirmation of MD in distant organ systems during the early phase of acute pancreatitis would support the proposal that mitochondrial-specific therapies may help prevent the subsequent development of MODS in severe acute pancreatitis as the disease progresses.
The aim of the present study was to characterise mitochondrial function in seven tissue homogenates using high-resolution respirometry during early acute pancreatitis in two different rodent models.
Methods
Ethical approval and study design
The present study was approved by the University of Auckland Animal Ethics Committee. Twenty-eight male Wistar rats (6–8 months old, 463 ± 2 g; mean ± SEM), fed a standard Harlan Teklad 2018 rodent diet (Madison, WI, USA) were randomised to four groups: Group 1 (n = 8), saline control; Group 2 (n = 6), caerulein-induced acute pancreatitis (CIP); Group 3 (n = 7) sham surgical controls (laparotomy only without induction of pancreatitis); and Group 4 (n = 7), taurocholate-induced acute pancreatitis (TIP, a laparotomy followed by intraductal infusion of taurocholate into the pancreas).18,19 All chemicals used in these experiments were supplied by Sigma Aldrich Pty Ltd (New South Wales, Australia), unless otherwise stated.
Caerulein-induced pancreatitis model
Mild CIP was induced by four subcutaneous (s.c.) injections of 20 µg/kg caerulein, a cholecystokinin analogue, in a total of 0.8 ml of 0.9% NaCl at hourly intervals.20,21 Analgesia was by buprenorphine (0.05 µg/kg, s.c.; Temgesic ®, Reckitt and Coleman, Hull, England). Saline controls were treated with 0.9% NaCl (normal saline) s.c. only.
Taurocholate-induced pancreatitis model
Balanced general anaesthesia was induced and maintained by isoflurane (2–5%; 2 l/min O2 via nasal cone) and buprenorphine (0.05 µg/kg, s.c.). Body temperature was maintained between 36–38°C by use of a warming plate. All animals received 5 ml of SC normal saline administered at the start of the experimental protocols, after which the common pancreatic duct was cannulated with a 24-G angiocath passed transduodenally through the Ampulla of Vater through a 1.5-cm abdominal midline incision. The rostral part of the animal was raised 60° to the horizontal for 5 min to allow the bile duct system to drain (∼0.1 ml). During the last 2 min of this procedure, the common hepatic duct was occluded at the hilum (Biemer atraumatic vascular clip; Aesculap, Center Valley, PA, USA).
Sodium taurocholate (4% w/v in 0.9% NaCl; 0.1 ml/100 g BW) was infused at 0.1 ml/min by a controlled infusion pump (Genie Precision Pump; Kent Scientific, Torrington, CT, USA). The Biemer clip and angiocath were removed upon completion of the infusion, and the common pancreatic duct was ligated to prevent reflux of taurocholate into the duodenum. The abdomen was then closed and the rat recovered on a warming plate.
In the post-operative phase, all animals were individually housed, had access to food and tap water ad libitum for 6 h and were monitored as per a standard post-operative recovery protocol; thereafter, they were rapidly anaesthetised under isoflurane. Blood was collected for biochemical assays by a euthanising cardiac puncture and stored at −80°C until analysis, whereas the various tissues analysed were then rapidly excised, chilled in ice-cold phosphate-buffered solution (PBS) buffer, and used fresh for the respective mitochondrial studies.
Mitochondrial respiration assay
We employed high-resolution respirometry with a previously published substrate-inhibitor protocol that permitted rapid analysis of ETS function and integrity in tissue homogenates as well as permeabilised cardiac fibres.22,23 We elected to use tissue homogenates because these provided a means for rapid simultaneous processing of the several tissues for measurement of tissue-specific mitochondrial respirational flux rate. Homogenates were also considered superior to mitochondrial isolation in this experimental setting as they sum the entire mitochondrial population present in tissue samples. This approach avoids the processing delays of enriched organelle preparations as well as the risk of any potential confounding bias that can result if swollen, more fragile and/or damaged mitochondrial subpopulations are lost through the processing steps required for pure mitochondrial isolation.23–25
The tissues studied were: the pancreas (tail and head separately), duodenum, mid-jejunum, lung (left lower lobe), heart (left ventricular endomyocardium), left kidney and liver (left lobe). All tissues with the exception of the left ventricular endomyocardium, were cut into small pieces (∼2 mm2), quickly blotted dry, weighed and placed into ice-cold respiration assay media [0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 110 mM sucrose and 1 mg/ml bovine serum albumin (BSA) in 20 mM HEPES, pH 7.1 at 30°C] containing one Complete™ protease inhibitor tablet (Roche, Basel, Switzerland) per 30 ml and 20 mg/ml fatty acid free BSA. These tissues were homogenised immediately before assay. The permeabilised left ventricular endomyocardial fibers were prepared according to a published methodology.22 Approximately 25 mg was placed into a droplet of ice-cold high-energy relaxing solution [10 mM EGTA-Ca2EGTA buffer (free Ca2+ concentration 0.1 µM), 9.5 mM MgCl2, 3 mM KH2PO4, 20 mM taurine, 5 mM ATP, 15 mM creatine phosphate, 49 mM K+ MES, 29 mM imidazole-HCl, pH 7.1] and dissected into fibre bundles of ∼0.5 × 1 mm. The dissected fibre bundles were transferred into 1 ml of fresh high-energy relaxing solution plus 50 µg saponin and gently stirred for 30 min at 4°C for permeabilisation. They were then washed three times in ice-cold respiration medium to remove the saponin and adenine nucleotides. Fibre bundles were then weighed after removing adherent liquid by blotting on lint-free lens tissue.
Mitochondrial respiration was measured in parallel 2-ml chambers using an OROBOROS® Oxygraph 2 K (Anton Paar, Graz, Austria). The respiratory measurements were performed at 30°C and the oxygen concentration at air saturation of the medium was 215 nmol O2 per ml at 95 kPa barometric pressure.26 For pancreatic and intestinal tissues, an additional 10 mg/ml of fatty acid-free BSA was added to the respiration medium.
A specially designed substrate/inhibitor titration approach allowed the step-by-step analysis of several mitochondrial complexes within the one experiment.27 This substrate-inhibitor titration protocol has previously been used in our laboratory28,29 and by others.30,31 This permitted tests of the: ‘state 2’ or leak state respiration (LEAK); oxidative phosphorylation flux with Complex I and Complex I and II substrates (OXP-I, and OXP-I,II); uncoupled electron transport chain flux (ETS); ETS flux on Complex II substrates alone (ETS-II); and cytochrome c oxidase flux (CCO). Therefore, this protocol provided rapid insight into the phosphorylation capacity of mitochondria and components of the ETS, as well as testing inner and outer membrane integrities, all in a single rapid experiment.
The protocol started with the addition of: (in mM unless stated) 10 glutamate and 5 malate (LEAK-I); followed by 1.25 ADP (OXP-1); 5 succinate (OXP-1,2), two to three additions of 0.5 µM carbonyl cyanide p-(trifluoro-methoxy) phenyl-hydrazone (FCCP, ETS-I,II), 1 µM; 1 µM rotenone (ETS-II); antimycin A, 5 µM N'-tetramethyl-p-phenylendiamine (TMPD) with 0.5 ascorbate (CCO), and finally 5 µM cytochrome c (CCOc). The addition of the metabolic inhibitors rotenone and antimycin provided an indication of non-specific oxidation. Figure 1 illustrates a typical tracing of mitochondrial respiratory flux obtained during a single representative experiment.
Figure 1.
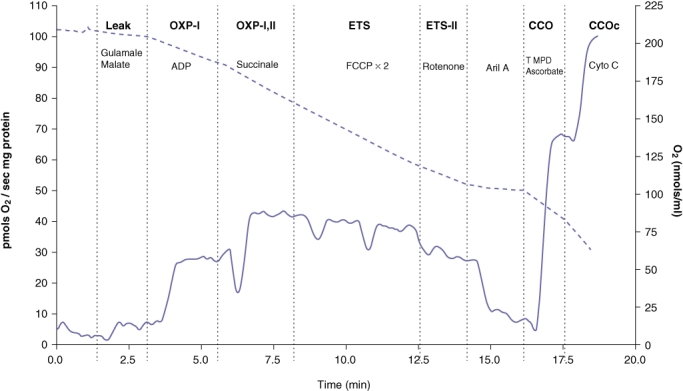
Representative trace generated by the OROBOROS® Oxygraph 2 K. Solid line represents mitochondrial respiration (left y-axis) and the dotted line represents the oxygen concentration (right y-axis). LEAK: state 2 respiration with glutamate and malate. OXP-I: oxidative phosphorylation with Complex I substrates (state 3 respiration) with the addition of adenosine diphosphate (ADP). OXP-I,II: state 3 respiration with succinate a Complex II substrate. ETS-I,II: uncoupled respiration with addition of carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP). ETS-I,II: addition of rotenone. CCO: addition of tetramethyl-p-phenylenediamine (TMPD)/ascorbate. CCOc addition of cytochrome C. Cyto C: cytochrome C. Ant A: antimycin A. Concomitant oxygen concentration (nmol/ml) is shown on the right y-axis
Total protein concentrations were determined for pancreas, intestine, liver, kidney and lung samples using Biuret reagent with BSA as standard and the respiration medium as blank. Flux rates were determined using DatLab 4 Analysis Software (OROBOROS®, Austria) and reported as weight-specific oxygen flux (pmol O2/mg protein/second) for all tissues except the heart, which was reported as pmol O2/mg wet weight/second.
Incubation of lung tissue with caerulein
In a separate experiment, we determined whether there might be any direct toxic effect of caerulein on rodent lung mitochondria. Lung tissue (n = 4) was incubated with 200 µg/kg caerulein at 37°C for 30 min in oxygenated assay medium. Control lung tissue (n = 4) was incubated similarly in the oxygenated assay medium. Mitochondrial respiration was measured as described above.
Histology and assays
Pancreatic tissue was fixed (10% neutral buffered formalin), and blinded histological severity scoring was performed by a consultant histopathologist on 5-µm thick longitudinal paraffin sections using haematoxylin and eosin stain. The score assessed leukocyte infiltration, pancreatic oedema, haemorrhage, fat necrosis and acinar necrosis using a published scoring system.32
All biochemical assays with the exception of amylase were performed using a Roche/Hitachi MODULAR® analytical system (Roche Diagnostics GmbH, Mannheim, Germany) in accordance with the manufacturer's instructions. Amylase was measured on a COBAS MIRA analyser (Roche) using a commercial reagent set (Pointe Scientific, Michigan, USA) according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism ™ version 4.00 for Macintosh (GraphPad Software, San Diego, California, USA). The Mann–Whitney U-test with a Bonferroni correction was used for the three relevant planned comparisons. A P-value < 0.05 was considered significant.
Results
All animals survived the study protocols. Both CIP and TIP models produced acute pancreatitis with the expected elevation in serum amylase (Table 1). There was no clinical or biochemical evidence of established MODS being present at this early time point in either model. The TIP model produced a more severe acute pancreatitis than the CIP model, as shown by the higher histology scores in the TIP group (Table 1). The serum creatinine and electrolyte measurements were unchanged in the CIP group compared with matched controls (saline-control group). The mild nature of the CIP model was also confirmed by the lack of any other biochemical derangement (Table 1). By contrast, the TIP model had a modest systemic derangement in various biochemical parameters (Table 1).
Table 1.
Diagnostic, histological, and physiological markers of severity in the different experimental groups
| Test | Saline control | Caerulein-induced mild pancreatitis | Sham surgery | Taurocholate-induced severe pancreatitis |
|---|---|---|---|---|
| Histological score1 | 1.0 ± 0.2a | 4.8 ± 0.5a,c | 0.7 ± 0.2b | 8.9 ± 0.3b,c |
| Amylase(U/l) | 1489 ± 152a | 5990 ± 458a,c | 1409 ± 150b | 12 848 ± 1 909b,c |
| Sodium(mmol/l) | 132.8 ± 1.4 | 137.0 ± 2.1 | 136.9 ± 1.3 | 130.0 ± 1.7 |
| Potassium(mmol/l) | 4.7 ± 0.2 | 4.6 ± 0.2 | 4.7 ± 0.1 | 5.2 ± 0.3 |
| Chloride(mmol/l) | 95.9 ± 1.2 | 99.0 ± 1.3 | 97.4 ± 1.9 | 91.0 ± 2.3 |
| Glucose(mmol/l) | 11.0 ± 0.9 | 11.2 ± 1.3 | 10.2 ± 1.2 | 14.3 ± 1.8 |
| Urea | 5.8 ± 0.2 | 6.3 ± 0.1 | 7.4 ± 1.2b | 13.3 ± 1.2b |
| Creatinine(µmol/l) | 31.8 ± 1.4 | 35.5 ± 1.3 | 36.3 ± 2.5b | 81.0 ± 14.4b |
| Calcium(mmol/l) | 2.52 ± 0.04 | 2.53 ± 0.03 | 2.56 ± 0.05 | 2.61 ± 0.05 |
| Albumin(g/l) | 34.6 ± 0.9 | 35.2 ± 0.8 | 35.4 ± 0.5 | 33.6 ± 0.8 |
| Bilirubin(µmol/l | <2 | 2 | <2 | 18.9 ± 9.4 |
| GGT(U/l) | <2 | <2 | <2 | 16 ± 2 |
| ALP(U/l) | 84 ± 7 | 72 ± 14 | 63 ± 3b | 105 ± 11b |
| AST(U/l) | 83 ± 12 | 72 ± 21 | 108 ± 4 | 5437 ± 2303 |
| ALT(U/l) | 50 ± 4 | 44 ± 15 | 56 ± 9b | 739 ± 328b |
Values are mean ± SEM;
Histology score is for pancreatic tissue. Values for bilirubin <2 and GGT < 2 were below the limit of detection and not quantified further by the Roche/Hitachi MODULAR® analytical system. Like-letters indicate P < 0.05 for the Mann–Whitney U-tests with Bonferroni's correction.
Effect of acute pancreatitis on the pancreatic mitochondria
Pancreatic MD was apparent in both acute pancreatitis models (Table 2), but its pattern differed between the two. Whereas CIP lead to a significant depression of glutamate oxidation (complex I dysfunction with decreased GM2 and GM3 along with increased (OXP-I/LEAK-I) in the pancreatic tail, it caused only an increase in the OXP-I,II/OXP-I ratio (indicating Complex I dysfunction relative to complex II) in the pancreatic head (Table 2). TIP significantly depressed flux through Complexes I and II in the pancreatic head (Table 2, Fig. 2). TIP induced no significant changes in mitochondrial function in the pancreatic tail in spite of the tail being oedematous.
Table 2.
Effect of surgery, caerulein-induced mild pancreatitis and taurocholate-induced severe pancreatitis on mitochondrial function (lung, jejunum and pancreas)
| Tissue | Saline control | Caerulein-induced mild pancreatitis | Sham surgery | Taurocholate-induced severe pancreatitis |
|---|---|---|---|---|
| Lung | ||||
| LEAK | 10.8 ± 0.9b | 8.1 ± 1.2 | 4.2 ± 0.3b | 4.8 ± 0.4 |
| OXP-I | 71.5 ± 7.8a,b | 42.8 ± 5.1a | 27.3 ± 1.7b | 26.8 ± 2.5 |
| OXP-I,II | 98.7 ± 11.2a,b | 61.6 ± 7.4a | 38.7 ± 3.0b | 38.0 ± 3.7 |
| ETS | 107.4 ± 14.9b | 67.7 ± 8.3 | 41.7 ± 4.2b | 42.1 ± 3.4 |
| ETS-II | 55.8 ± 9.5 | 41.9 ± 5.1 | 26.0 ± 2.2 | 28.8 ± 3.8 |
| CCO | 144.4 ± 22.9a,b | 73.6 ± 10.5a | 56.6 ± 5.0b | 59.3 ± 5.6 |
| CCOc | 199.7 ± 27.4b | 160.2 ± 43.7 | 86.8 ± 5.4b | 86.6 ± 12.1 |
| OXP-I,II/ OXP-I | 1.4 ± 0.0 | 1.4 ± 0.0 | 1.4 ± 0.0 | 1.4 ± 0.0 |
| ACR | 6.6 ± 0.4 | 5.6 ± 0.5 | 6.9 ± 0.8 | 5.6 ± 0.1 |
| Jejunum | ||||
| LEAK | 22.2 ± 3.7 | 18.8 ± 2.2 | 24.5 ± 1.8 | 13.7 ± 3.7 |
| OXP-I | 123.1 ± 21.3 | 98.2 ± 12.7 | 117.1 ± 8.8c | 44.4 ± 10.1c |
| OXP-I,II | 212.1 ± 37.2 | 202.8 ± 26.8 | 200.6 ± 52.9c | 73.0 ± 17.4c |
| ETS | 171.4 ± 31.2 | 183.0 ± 31.6 | 158.2 ± 20.6c | 53.7 ± 12.7c |
| ETS-II | 115.0 ± 21.5 | 110.5 ± 20.3 | 95.1 ± 14.4c | 37.9 ± 8.7c |
| CCO | 174.2 ± 21.1 | 194.1 ± 20.0 | 182.2 ± 16.3 | 132.4 ± 32.7 |
| CCOc | 337.7 ± 41.7 | 297.5 ± 30.8 | 353.2 ± 22.8 | 269.0 ± 58.8 |
| OXP-I,II/ OXP-I | 1.7 ± 0.1a | 2.1 ± 0.0a | 1.7 ± 0.1 | 1.7 ± 0.1 |
| ACR | 5.4 ± 0.48 | 5.2 ± 0.3 | 5.0 ± 0.5 | 4.4 ± 0.6 |
| Pancreas head | ||||
| LEAK | 31.9 ± 6.2b | 21.0 ± 2.9 | 11.4 ± 2.3b,c | 3.5 ± 0.5c |
| OXP-I | 137.0 ± 19.7 | 73.3 ± 8.4 | 57.6 ± 13.4c | 9.3 ± 5.2c |
| OXP-I,II | 240.3 ± 33.0 | 192.7 ± 24.1 | 134.0 ± 24.1c | 46.1 ± 11.3c |
| ETS | 232.5 ± 32.8 | 198.5 ± 25.0 | 139.0 ± 24.0c | 47.6 ± 12.7c |
| ETS-II | 177.9 ± 24.7 | 163.6 ± 20.8 | 111.4 ± 18.8 | 45.8 ± 11.8 |
| CCO | 411.1 ± 75.9 | 406.8 ± 86.2 | 249.5 ± 30.1 | 230.7 ± 64.7 |
| CCOc | 597.8 ± 108.7 | 541.8 ± 108.3 | 362.5 ± 61.8 | 324.9 ± 77.0 |
| OXP-I,II/ OXP-I | 1.8 ± 0.1a,b | 2.6 ± 0.1a | 2.6 ± 0.2b | 6.8 ± 1.7 |
| ACR | 4.6 ± 0.3 | 4.1 ± 0.9 | 5.2 ± 0.8 | 3.6 ± 2.1 |
| Pancreas tail | ||||
| LEAK | 40.0 ± 2.3a,b | 19.0 ± 2.6a | 18.2 ± 4.4b | 7.5 ± 1.4 |
| OXP-I | 146.7 ± 12.3a,b | 75.7 ± 12.6a | 70.9 ± 19.0b | 33.5 ± 6.8 |
| OXP-I,II | 247.1 ± 16.5b | 180.4 ± 27.6 | 140.4 ± 30.7b | 90.9 ± 12.1 |
| ETS | 240.3 ± 15.3a | 183.0 ± 27.5a | 139.4 ± 30.7 | 94.5 ± 13.4 |
| ETS-II | 177.3 ± 9.5 | 146.2 ± 20.9 | 108.3 ± 23.6 | 82.5 ± 12.3 |
| CCO | 313.5 ± 21.0 | 265.9 ± 37.9 | 219.1 ± 42.4 | 221.2 ± 35.6 |
| CCOc | 465.8 ± 28.6 | 441.4 ± 93.2 | 332.4 ± 87.4 | 307.7 ± 41.7 |
| OXP-I,II/ OXP-I | 1.7 ± 0.1a,b | 2.6 ± 0.4a | 2.3 ± 0.3b | 3.0 ± 0.4 |
| ACR | 3.7 ± 0.3 | 3.9 ± 0.5 | 3.9 ± 0.5 | 4.8 ± 0.8 |
Units are pmols O2/mg protein/second. LEAK: State 2 respiration with glutamate and malate. GM3: State 3 respiration with addition of adenosine diphosphate (ADP). Units are pmols O2/mg wet weight/second. OXP-I: State 3 respiration with addition of succinate. ETS: uncoupled respiration with addition of carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP). ETS-II: addition of rotenone. CCO: addition of tetramethyl-p-phenylenediamine (TMPD)/ascorbate. CCOc addition of cytochrome C. OXP-I,II/OXP-II: provides a measure of complex I contribution to respiration. ACR: acceptor control ratio. Like letters represent significant differences between those groups (P < 0.05, Bonferroni corrected) whereby
Caerulein pancreatitis vs. saline control
Saline control vs. sham surgery
Sham surgery vs. taurocholate pancreatitis.
Figure 2.
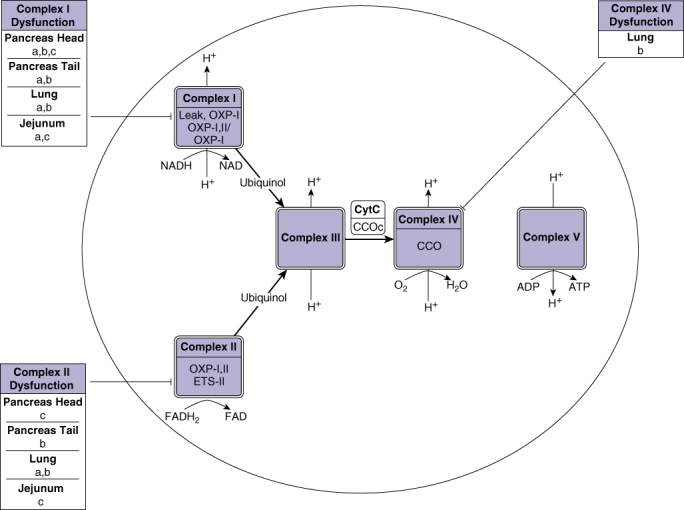
A summary of the effects of caerulein pancreatitis, sham surgery and taurocholate pancreatitis on the various respiratory chain complexes. For definitions of LEAK-I through to CCOc see legend for Figure 1. ‘-’ represents inhibition of the complex. Letters ‘a’, ‘b’ and ‘c’ represent significant inhibition of the complex whereby a: caerulein pancreatitis vs. saline control; b: sham surgery vs. saline control; c: taurocholate pancreatitis vs. sham surgery.
Effect of anaesthesia and surgery on mitochondrial function
The combination of a volatile anaesthetic and surgery depressed respiratory flux through Complex I, Complex II and isolated Complex IV (CCO) in the lung when compared with the non-operated saline control group (Table 2, Fig. 2) thus preventing the investigation of lung MD in the TIP model.
Sham surgery also impacted pancreatic mitochondrial function in spite of there being no significant difference between the pancreas histology scores from the saline control and sham surgical groups (Table 1). Depressed Complex I flux was seen in both the pancreatic head and tail while depression of Complex II was limited to the pancreatic tail. The heart, liver, kidney, duodenal and jejunal mitochondrial function were not altered in the sham surgical group (Table S1).
Effect of acute pancreatitis on distant organ mitochondrial function
MD secondary to pancreatitis in distant organs was restricted to the lung and jejunum (Table 2, Fig. 2). CIP depressed flux through Complexes I, II and IV in lung tissues. Respirational flux through Complex I was also depressed in jejunal homogenates when compared with the saline control group. TIP caused jejunal Complex I and II dysfunction when compared with sham surgery. However, there was no difference in the lung mitochondrial function between the TIP group vs. sham surgery, where the combination of the anaesthetic and surgery appeared to depress overall lung mitochondrial function in the sham group, potentially masking any effect of TIP. Neither model of acute pancreatitis at 6 h had any significant impact on mitochondrial respiration in the heart, liver, kidney or duodenum (Table S1).
No direct effect of caerulein on lung mitochondrial function
The lung appeared to be the most susceptible to MD induced by pancreatitis or surgery (Table 2). Therefore, in order to exclude a direct toxic effect of the caerulein on the lung, an additional experiment was performed in which lung tissue was incubated directly with 10 times the concentration of caerulein used in the in vivo study. The results show no significant alteration in lung mitochondrial function as a result of the direct effects of caerulein (Table 3).
Table 3.
Pulmonary mitochondrial respiration with and without incubation with caerulein
| Tissue | Control | Incubation with caerulein |
|---|---|---|
| Lung | ||
| LEAK | 2.6 ± 0.6 | 2.6 ± 0.7 |
| OXP-I | 13.0 ± 2.9 | 13.0 ± 2.9 |
| OXP-I,II | 21.7 ± 4.9 | 21.4 ± 4.8 |
| ETS | 25.6 ± 5.8 | 25.0 ± 5.7 |
| ETS-II | 14.8 ± 3.3 | 14.8 ± 3.3 |
| CCO | 53.2 ± 12.3 | 42.8 ± 9.3 |
| CCOc | 58.4 ± 15.6 | 58.9 ± 13.0 |
| OXP-I,II/ OXP-I | 1.7 ± 0.0 | 1.6 ± 0.0 |
| ACR | 5.1 ± 0.4 | 5.9 ± 1.1 |
Units are pmol O2/mg protein/second. For abbreviation descriptions see footnote Table 2
Discussion
The present study has characterised mitochondrial function in seven tissues 6 h after the induction of acute pancreatitis in two accepted rodent models of acute pancreatitis. The key findings were that the inflamed pancreas itself showed significant MD in both models and that in other organs distant from the pancreas the MD was highly selective in its distribution. This is the first study to document early and selective MD in the lungs and jejunum during early acute pancreatitis and before the development of MODS.
The first organ system to demonstrate dysfunction and to fail in clinical severe acute pancreatitis is the lung.33 In our own experience, 88% of patients with severe acute pancreatitis had established respiratory failure within the first week of hospitalisation.4 It has also been shown that about half of the deaths within the first week of severe acute pancreatitis can been attributed to respiratory failure.34–36 Experimental models of acute pancreatitis have also demonstrated early lung injury. It has been reported that within 3 h of caerulein administration in rats it was possible to demonstrate decreased surfactant synthesis, alveolar capillary endothelial injury and increased neutrophil infiltration, and that these effects were not as a result of a direct effect of caerulein on the lung.36,37
Here, we report for the first time that there is a significant depression of lung mitochondrial function, with reductions in Complex I and II respiratory flux occurring within 6 h of the induction of mild acute pancreatitis by caerulein. Healthy lung function and viability are known to be directly related to normal mitochondrial function,38,39 and failure of mitochondrial respiration and oxidative phosphorylation has been considered an important part of the complex pathophysiology of adult respiratory dysfunction syndrome.40 An early impairment of lung mitochondrial function during the development of acute pancreatitis would not only cause early deterioration in lung function through decreased ATP production10 but would also predispose the lung to further damage evoked by excessive ROS production and apoptosis,41 contributing to the later development of adult respiratory dysfunction syndrome.
Volatile anaesthetic and/or sham surgery confounded the investigation of lung MD in the TIP model. The sham-operated group showed substantially depressed mitochondrial respiration in the lung when compared with the saline control (non-operated) group. Attenuation of the lung mitochondria Complexes I, II and IV persisted 6 h after recovery from the brief anaesthetic and sham surgery. This depression of mitochondrial respiration in the lung may be as a result of a combined effect of the volatile anaesthetic and the surgical stress associated with bowel handling. The effect of the anaesthetic may be explained by the observation that lung epithelial cells are among the first to be exposed to highly concentrated volatile anaesthetics and may suffer from cytotoxic and genotoxic effects as a consequence.42 Volatile anaesthetics are reported to inhibit Complex I,43 open mitochondrial ATP-inhibited K+ channels which leads to mitochondrial depolarisation44 and cause depression of the mitochondrial respiratory chain sufficient to cause reversal of Complex V (F1/F0 ATPase),44 which involves ATP hydrolysis (as opposed to synthesis) in order to maintain membrane potentials. In addition to the effects of the anaesthetic, it has previously been reported that a mini-laparotomy and simple handling of the small bowel can cause significant lung injury with evidence of increased lung permeability and oxidative stress.45,46 Our finding of lung MD in the sham-operated group is consistent with these studies. Therefore, the CIP model was better suited to assessing lung MD in early acute pancreatitis.
Mitochondrial dysfunction in the jejunum within 6 h of the induction of mild-to-moderate acute pancreatitis in both models was an important and unexpected finding of the present study. Previous reports have suggested that the failure of intestinal function, and more specifically increased permeability and bacterial translocation, occurs over the course of the first week after the onset of severe acute pancreatitis.6,9 By contrast, the present study provides evidence for the first time that intestinal dysfunction, and more specifically jejunal MD, occurs within hours of the onset of mild to moderate acute pancreatitis. Our findings challenge conventional thinking by demonstrating that this early MD in the jejunum also occurs in less severe acute pancreatitis and occurs before the development of MODS or hypovolaemic shock. Consistent with our findings is previous work suggesting that intestinal mucosal injury after the induction of endotoxic shock was independent of hypoperfusion, and might be as a result of impaired mitochondrial respiration or cytopathic hypoxia.15 Interestingly, no evidence for mitochondrial dysfunction was found in the duodenum during the present study. It has previously been reported that the jejunum has areas more susceptible to ischaemia–reperfusion mucosal injury than the duodenum or ileum because of differences in vascular anatomy.47 This might be part of the explanation for our findings of increased MD in the jejunum but not the duodenum. Other factors, such as differences in microbes and intra-luminal content48 between the duodenum and jejunum, might also contribute to differences in mitochondrial function and this requires further investigation.
Impairment of intestinal barrier function has been shown to correlate strongly with subsequent MODS and septic complications as a result of translocation of bacteria and the priming of neutrophils.6,9 Intestinal permeability to bacteria was reported to correlate inversely with enterocyte ATP levels, thereby supporting the idea that the intestinal barrier function is ATP dependent.49 Early intestinal MD, with its concomitant decrease in ATP production and increase in ROS,10,41 may therefore underpin intestinal barrier failure, which then leads to not only the worsening of MODS by exacerbation of SIRS but also to the delayed septic complications. Thus our data suggests that jejunal mitochondria might represent a previously unrecognised and very early event in acute pancreatitis, long before increased intestinal permeability and bacterial translocation are thought to occur. As such, jejunal MD might therefore be understood as a primary step in the subsequent failure of the intestinal barrier function in MODS and thus offer a newly identified target for early intervention therapy.
There are very few reports of mitochondrial function in tissues outside of the pancreas in acute pancreatitis. These have been restricted to the investigation of isolated liver mitochondrial function in acute pancreatitis where hepatic MD has been reported in rodents with TIP.50,51 Interestingly, we did not find any change in the in situ mitochondrial function in liver homogenates during early pancreatitis in either model at 6 h, and this may reflect differences in experimental protocols. For example, we gave 5 ml of SC normal saline for resuscitation at the start of the protocol in the TIP model, and used tissue homogenates rather than isolated mitochondria to avoid any potential risk of mitochondrial extraction stress or artefact.
The development of novel therapies to prevent the progression of severe acute pancreatitis-associated MODS continues to be frustrated by a clear understanding of key pathophysiology during the early stages of severe acute pancreatitis.9 In the present study we have shown that the early inhibition of mitochondrial respiratory chain complexes is not global but occurs selectively in lung and jejunum during early acute pancreatitis. Failure of these two specific organ systems contributes significantly to the morbidity and mortality associated with severe acute pancreatitis.4,6,9,34 This early and selective MD seen in the lung and jejunum during the development of acute pancreatitis offers new insights and avenues to pursue in the underlying pathophysiological events during the early phase of acute pancreatitis. Further research is now needed to document the evolution of MD at multiple time points with disease progression, and to investigate if mitochondrial-specific therapies can prevent or aid recovery of lung and jejunal MD.11,13,16 Such an approach has not been considered for the treatment of acute pancreatitis.
Conclusions
The present study provides the first comprehensive description of mitochondrial function in multiple organs during early acute pancreatitis. We have identified a previously unrecognised and early organ-selective inhibition of mitochondrial function distant from the primary site of pancreatic inflammatory damage. These data highlight the need for further research to identify the underlying pathophysiology behind the selective MD in these organs, and the potential benefits of early mitochondrial-specific therapies in acute pancreatitis.
Funding
The present study was supported with funding for salary, consumables and equipment by the Royal Australasian College of Surgeons, the University of Auckland Research Committee, the Maurice & Phyllis Paykel Trust, Auckland Medical Research Council and Lottery Health New Zealand.
Conflicts of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Effect of surgery, caerulein-induced mild pancreatitis and taurocholate-induced severe pancreatitis on mitochondrial function (heart, liver, kidney and duodenum)
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Johnson C, Abu-Hilal MD. Organ failure as an indicator of severity of acute pancreatitis: time to revisit the Atlanta classification. Gastroenterology. 2005;128:1133–1135. doi: 10.1053/j.gastro.2005.02.059. [DOI] [PubMed] [Google Scholar]
- 2.Rau BM, Bothe A, Kron M, Beger HG. Role of early multisystem organ failure as major risk factor for pancreatic infections and death in severe acute pancreatitis. Clin Gastroenterol Hepatol. 2006;4:1053–1061. doi: 10.1016/j.cgh.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 3.Flint R, Windsor JA. Early physiological response to intensive care as a clinically relevant approach to predicting the outcome in severe acute pancreatitis. Arch Surg. 2004;139:438–443. doi: 10.1001/archsurg.139.4.438. [DOI] [PubMed] [Google Scholar]
- 4.Flint R, Windsor J, Bonham M. Trends in the management of severe acute pancreatitis: interventions and outcome. ANZ J Surg. 2004;74:335–342. doi: 10.1111/j.1445-1433.2004.02940.x. [DOI] [PubMed] [Google Scholar]
- 5.Davies MG, Hagen PO. Systemic inflammatory response syndrome. Br J Surg. 1997;84:920–935. doi: 10.1002/bjs.1800840707. [DOI] [PubMed] [Google Scholar]
- 6.Flint R, Windsor J. Role of intestine in the pathogenesis of acute pancreatitis. HPB. 2003;5:69–85. doi: 10.1080/13651820310001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neoptolemos JP, Raraty M, Finch M, Sutton R. Acute pancreatitis: the substantial human and financial costs. Gut. 1998;42:886–891. doi: 10.1136/gut.42.6.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKay CJ, Imrie CW. The continuing challenge of early mortality in acute pancreatitis. Br J Surg. 2004;91:1243–1244. doi: 10.1002/bjs.4750. [DOI] [PubMed] [Google Scholar]
- 9.Beger HG, Rau BM. Severe acute pancreatitis: clinical course and management. World J Gastroenterol. 2007;13:5043–5051. doi: 10.3748/wjg.v13.i38.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brealey D, Brand MD, Hargreaves I, Heales S, Land J, Smolenski R, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 11.Singer M, Brealey D. Mitochondrial dysfunction in sepsis. Biochem Soc Symp. 1999;66:149–166. doi: 10.1042/bss0660149. [DOI] [PubMed] [Google Scholar]
- 12.Piantadosi CA, Carraway MS, Haden DW, Suliman HB. Protecting the permeability pore and mitochondrial biogenesis. In: Derek J, Chadwick JG, editors. Sepsis: New Insights, New Therapies. Durham, NC: Novartis Foundation; 2007. pp. 266–276. [DOI] [PubMed] [Google Scholar]
- 13.Singer M. Metabolic failure. Critical Care Medicine. 2005;33(Suppl):S539–S542. doi: 10.1097/01.ccm.0000186080.13402.96. [DOI] [PubMed] [Google Scholar]
- 14.Dare A, Phillips A, Hickey AJ, Mittal A, Loveday B, Thompson N, et al. A systemic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic Biol Med. 2009;47:1517–1525. doi: 10.1016/j.freeradbiomed.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Lobo SM, De Backer D, Sun Q, Tu Z, Dimopoulos G, Preiser JC, et al. Gut mucosal damage during endotoxic shock is due to mechanisms other than gut ischemia. J Appl Physiol. 2003;95:2047–2054. doi: 10.1152/japplphysiol.00925.2002. [DOI] [PubMed] [Google Scholar]
- 16.Fink MP. Bench-to-bedside review: cytopathic hypoxia. Crit Care. 2002;6:491–499. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 18.Zhang XP, Ye Q, Jiang XG, Ma ML, Zhu FB, Zhang RP, et al. Preparation method of an ideal model of multiple organ injury of rat with severe acute pancreatitis. World J Gastroenterol. 2007;13:4566–4573. doi: 10.3748/wjg.v13.i34.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mittal A, Flint RJ, Fanous M, Delahunt B, Kilmartin PA, Cooper GJ, et al. Redox status of acute pancreatitis as measured by cyclic voltammetry: initial rodent studies to assess disease severity. Crit Care Med. 2008;36:866–872. doi: 10.1097/CCM.0B013E318165FA7F. [DOI] [PubMed] [Google Scholar]
- 20.Reinheckel T, Prause J, Nedelev B, Augustin W, Schulz HU, Lippert H, et al. Oxidative stress affects pancreatic proteins during the early pathogenesis of rat caerulein pancreatitis. Digestion. 1999;60:56–62. doi: 10.1159/000007589. [DOI] [PubMed] [Google Scholar]
- 21.Schild L, Matthias R, Stanarius A, Wolf G, Augustin W, Halangk W. Induction of permeability transition in pancreatic mitochondria by cerulein in rats. Mol Cell Biochem. 1999;195:191–197. doi: 10.1023/a:1006988625831. [DOI] [PubMed] [Google Scholar]
- 22.Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, et al. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol Cell Biochem. 1998;184:81–100. [PubMed] [Google Scholar]
- 23.Jullig M, Hickey AJ, Chai CC, Skea GL, Middleditch M, Costa S, et al. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics. 2008;8:2556–2572. doi: 10.1002/pmic.200700977. [DOI] [PubMed] [Google Scholar]
- 24.Carre JE, Singer M. Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim Biophys Acta. 2008;1777:763–771. doi: 10.1016/j.bbabio.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 25.Kudin AP, Malinska D, Kunz WS. Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim Biophys Acta. 2008;1777:689–695. doi: 10.1016/j.bbabio.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Gnaiger E. Mitochondrial Pathways and Respiratory Control. Innsbruck: OROBOROS MiPNet Publications; 2007. [Google Scholar]
- 27.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 28.Jullig M, Hickey A, Middleditch M, Crossman D, Lee S, Cooper G. Characterization of proteomic changes in cardiac mitochondria in streptozotocin-diabetic rats using iTRAQ isobaric tags. Proteomics Clin Appl. 2007;1:565–576. doi: 10.1002/prca.200600831. [DOI] [PubMed] [Google Scholar]
- 29.Jullig M, Hickey A, Chai C, Skea G, Middleditch M, Costa S, et al. Is the failing heart out of fuel or a worn engine running rich? A study of mitochondria in old spontaneously hypertensive rats. Proteomics. 2008;8:2556–2772. doi: 10.1002/pmic.200700977. [DOI] [PubMed] [Google Scholar]
- 30.Kuznetsov AV, Strobl D, Ruttman E, Konigsrainer A, Margreiter R, Gnaiger E. Evaluation of mitochondrial respiratory function in small biopsies of liver. Anal Biochem. 2002;305:186–194. doi: 10.1006/abio.2002.5658. [DOI] [PubMed] [Google Scholar]
- 31.Gnaiger E. Polarographic oxygen sensors, the oxygraph, and high-resolution respirometry to assess mitochondrial function. In: Dykens J, Will W, editors. Drug-Induced Mitochondrial Dysfunction. Hoboken, NJ: John Wiley & Sons, Inc; 2008. pp. 327–352. [Google Scholar]
- 32.Schmidt J, Lewandrowski K, Fernandez-del Castillo C, Mandavilli U, Compton CC, Warshaw AL, et al. Histopathologic correlates of serum amylase activity in acute experimental pancreatitis. Dig Dis Sci. 1992;37:1426–1433. doi: 10.1007/BF01296014. [DOI] [PubMed] [Google Scholar]
- 33.Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;190:117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 34.Steer ML. Relationship between pancreatitis and lung diseases. Respir Physiol. 2001;128:13–16. doi: 10.1016/s0034-5687(01)00259-6. [DOI] [PubMed] [Google Scholar]
- 35.Pastor CM, Matthay MA, Frossard JL. Pancreatitis-Associated Acute Lung Injury* New Insights. Chest. 2003;124:2341–2351. doi: 10.1378/chest.124.6.2341. [DOI] [PubMed] [Google Scholar]
- 36.Guice KS, Oldham KT, Johnson KJ, Kunkel RG, Morganroth ML, Ward PA. Pancreatitis-induced acute lung injury. An ARDS model. Ann Surg. 1988;208:71–77. doi: 10.1097/00000658-198807000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guice KS, Oldham KT, Wolfe RR, Simon RH. Lung injury in acute pancreatitis: primary inhibition of pulmonary phospholipid synthesis. Am J Surg. 1987;153:54–61. doi: 10.1016/0002-9610(87)90201-7. [DOI] [PubMed] [Google Scholar]
- 38.Fukuse T, Hirata T, Ohmasa M, Wada H. Mitochondrial respiratory function and reperfusion injury during pulmonary preservation. Transplant Proc. 2003;35:461–462. doi: 10.1016/s0041-1345(02)03978-7. [DOI] [PubMed] [Google Scholar]
- 39.Hirata T, Fukuse T, Hanaoka S, Matsumoto S, Chen Q, Wada H. Mitochondrial respiration as an early marker of viability in cardiac-arrested rat lungs. J Surg Res. 2001;96:268–276. doi: 10.1006/jsre.2000.6079. [DOI] [PubMed] [Google Scholar]
- 40.Sanders A, Baylin G. A common denominator in the etiology of adult respiratory distress syndrome. Med Hypotheses. 1980;6:951–965. doi: 10.1016/0306-9877(80)90047-x. [DOI] [PubMed] [Google Scholar]
- 41.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 42.Stephanova E, Valtecheva-Sarker R, Topouzova-Hristova T, Lalchev Z. Influence of volatile anaesthetics on lung cells and lung surfactant. Biotechnol Biotechnol Equip. 2007;21:393–398. [Google Scholar]
- 43.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH:ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–693. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bains R, Moe MC, Larsen GA, Berg-Johnsen J, Vinje ML. Volatile anaesthetics depolarize neural mitochondria by inhibiton of the electron transport chain. Acta Anaesthesiol Scand. 2006;50:572–579. doi: 10.1111/j.1399-6576.2006.00988.x. [DOI] [PubMed] [Google Scholar]
- 45.Thomas S, Ramamoorthy P, Balasubamanian KA. Surgical manipulation of the intestine and distant organ damage – protection by oral glutamine supplementation. Surgery. 2005;137:48–55. doi: 10.1016/j.surg.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura M, Motoyama S, Saito S, Minamiya Y, Saito R, Ogawa J. Hydrogen peroxide derived from the intestine through mesenteric lymph induces lung edema after surgical stress. Shock. 2004;21:160–164. doi: 10.1097/01.shk.0000105500.75189.cc. [DOI] [PubMed] [Google Scholar]
- 47.Anthony A, Pounder RE, Dhillon AP, Wakefield AJ. Vascular anatomy defines sites of indomethacin induced jejunal ulceration along the mesenteric margin. Gut. 1997;41:763–770. doi: 10.1136/gut.41.6.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez de la Cruz Moreno M, Oth M, Deferme S, Lammert F, Tack J, Dressman J, et al. Characterization of fasted-state human intestinal fluids collected from duodenum and jejunum. J Pharm Pharmacol. 2006;58:1079–1089. doi: 10.1211/jpp.58.8.0009. [DOI] [PubMed] [Google Scholar]
- 49.Wattanasirichaigoon S, Menconi M, Delude R, Fink M. Effect of mesenteric ischemia and reperfusion or hemorrhagic shock on intestinal mucosal permeability and ATP concent in rats. Shock. 1999;12:127–133. doi: 10.1097/00024382-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Coelho A, Machado M, Sampietre S, Leite K, Oliveira V, Pinotti H. Hepatic damage during acute pancreatitis in the rat. Braz J Med Biol Res. 1997;30:947–953. doi: 10.1590/s0100-879x1997000800006. [DOI] [PubMed] [Google Scholar]
- 51.Poplawski C, Dlugosz JW, Gabryelewicz A, Pawlicka E, Wroblewski E, Adrzejewska A. Hepatic mitochondrial and lysosomal alterations in acute experimental pancreatitis with ethanolic coetiology in rats. Dig Dis Sci. 1996;41:139. doi: 10.1007/BF02208596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.