

Abstract
A series of organochalcogenanes was synthesized and evaluated as protein tyrosine phosphatases (PTPs) inhibitors. The results indicate that organochalcogenanes inactivate the PTPs in a time- and concentration-dependent fashion, most likely through covalent modification of the active site sulfur-moiety by the chalcogen atom. Consequently, organochalcogenanes represent a new class of mechanism-based probes to modulate the PTP-mediated cellular processes.
Introduction
The prospection of tellurium and selenium compounds exhibiting biological activity has been increased in the last decades, especially after a series of studies that have demonstrated the biological potential of these exotic compounds.1 Antioxidant activity,2 anti-inflammatory properties,3,4 neuroprotective and convulsant effects,5 cancer prevention,6 apoptotic events,7 and immunomodulator activities8 are some of the biological properties that have been documented for selenium and tellurium-containing compounds. The development of small selenium- and tellurium-containing molecules as enzymatic inhibitors is based on the reactivity and high affinity of selenium and tellurium atoms towards thiol-dependent enzymes such as caspases,9 tyrosine kinase10 and cysteine (papain, cathepsins) proteases.11 A particular class of selenium and tellurium compounds that has been less explored in enzymatic inhibition is the hypervalent organochalcogenanes. Recent investigations have shown that organoselenanes and organotelluranes are very potent inhibitors of cysteine cathepsins, a thiol-dependent enzyme.12 The affinity between the sulfur-moiety from the catalytic site of these enzymes and chalcogen atom (especially tellurium) makes favorable the formation of a σY-S-Enz (Y = Se and Te, S-Enz = thiol-dependent enzyme) bound in the inhibitory process. Due to their distinct molecular arrangement and charge distribution, the chalcogen, present in these hypervalent compounds, accommodates a positive charge and consequently, become more electrophilic than their chalcogenides congeners. In this way, based on the reactivity of selenium- and tellurium-containing compounds and their molecular interaction with different enzymes, the investigation of hypervalent chalcogenanes as inhibitors of other thiol-dependent enzymes is warranted. Protein tyrosine phosphatases (PTPs) constitute a large family of cysteine-dependent enzymes that catalyze the hydrolysis of phosphotyrosine residues in proteins.13 PTPs, together with protein tyrosine kinases, play a central role in cell signaling by regulating the phosphorylation status and, in turn, the functional properties, of target proteins in various signal transduction pathways.14 Dysfunction in PTP activity has been linked to the etiology of several human diseases, including cancer, diabetes and obesity, and autoimmune disorders.15 Consequently, there is intense interest in developing small molecule PTP inhibitors that not only serve as powerful tools to delineate the physiological roles of these enzymes in vivo, but also as excellent leads for the development of new therapeutic agents.
Herein, we described a study of the potential use of hypervalent selenium(IV) and tellurium(IV) (selenanes and telluranes, respectively) as inhibitors of the PTPs (Figure 1). Besides the presence of chalcogen atoms (Se or Te), these compounds were designed to contain halogen (Cl or Br) atoms and a chiral center. The presence of a methyl group at the benzylic carbon generates an asymmetric center, which offers the possibility of differential recognition of the enantiomers by the enzyme (PTP) and also a good comparison with the achiral congener. The halogens bounded to chalcogen were chosen to evaluate the possible influence of the leaving group stability to the activity of these compounds. In this way, with simple structural modifications a SAR could be established.
Figure 1.

Selenanes telluranes evaluated as inhibitors of Protein Tyrosin Phosphatase.
Results and discussion
Synthesis of organoselenanes and organotelluranes (1–12)
The synthesis of organoselenanes involved a short and versatile synthetic route, which employed classic reactions of selenium chemistry (Scheme 1).11
Scheme 1.

Synthesis of organoselenanes 1–2, 5–6 and 9–10.11a
Similar synthetic approaches were used to prepare the organotelluranes congeners. The achiral organotelluranes 3 and 4 were synthesized from bromo-benzylic ether 1a. A bromo-lithium exchange reaction of 1a followed by capture with an electrophilic tellurium specie (BuTeTeBu) led to telluride 1c in 79% yield. The desired achiral organotelluranes 3 and 4 were obtained (Scheme 2-A) by a tellurium oxidation reaction with SO2Cl2 or Br2. In order to acquire the chiral organotelluranes (7–8, 11–12) a chemo-enzymatic methodology was developed. In this way, (RS)-1-phenylethanol 1g was submitted to enzymatic kinetic resolution (EKR) through enantioselective transesterification reaction mediated by Candida antartica lipase-B (CAL-B). This reaction led to alcohol (S)-1g and the ester (R)-1h in high enantiomeric excess (> 99%) and yield (45%), for each product. After hydrolysis of (R)-1h, the BuTe- group was introduced to each enantiomer of 1g. An ortho-lithiation reaction of (S)- or (R)-1g followed by its capture with BuTeTeBu led to the telluro-alcohols (S)- or (R)-1i in good yields (46%). O-Methylation of the (S)- and (R)-1i and, finally, reaction of tellurides (S)-and (R)-1j with SO2Cl2 or Br2 led to organotelluranes 7–8, 11–12 (Scheme 2-B).
Scheme 2.

Reagents and conditions: (i) a: t-BuLi, THF (−78-0 °C, 30 min); b: BuTeTeBu (r.t., 3 h); (ii) SO2Cl2 or Br2, THF (0 °C, 15 min); (iii) NaBH4, MeOH (r.t., 1h); (iv) vinyl acetate, CAL-B, hexane (32 °C, 7 h); (v) K2CO3, MeOH, H2O (r.t. overnight); (vi) a: n-BuLi. TMEDA, pentane (reflux, 24 h), b: BuTeTeBu (0 °C - r.t., 2 h); (vii) a: NaH, THF (0 °C, 30 min), b: MeI (r.t., 2 h).
Assessment of organoselenanes and organotelluranes as PTP inhibitors
The PTPs share a conserved active site and a common catalytic mechanism that features a highly nucleophilic Cys residue.13 This active site Cys displays an unusually low pKa of ~5,16 and is situated at the bottom of the phosphotyrosine-binding pocket (i.e. the active site) such that its Sγ atom is poised 3 A from the phosphorus atom of phosphotyrosine.17 In the catalytic mechanism, the active site Cys initiates a nucleophilic attack on the phosphorus atom, leading to the formation of a thiophosphoryl enzyme intermediate. Hydrolysis of this covalent enzyme intermediate then completes the catalytic cycle.
Given the reactivity of selenium and tellurium atoms towards thiol-dependent enzymes, we reasoned that the organochalcogenanes may also display inhibitory activity against the PTPs. To determine whether organoselenanes and organotelluranes (1–12) can function as mechanism-based PTP inhibitors, we first examined for their effect on PTP activity using para-nitrophenyl phosphate (pNPP) as a substrate (Supporting Information). All 12 compounds inactivated PTP1B and the PTP from Yersinia YopH in a time-dependent first order process (Table 1).
Table 1.
Rate constants for onset inactivation of the PTPs by organochalcogenanes 1–12.
| Structure | Inactivator Code | PTP1Ba (kobs, min−1) | YopHb (kobs, min−1) |
|---|---|---|---|
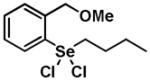
|
1 | 0.46 ± 0.15 | 0.25 ± 0.17 |
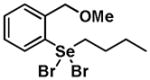
|
2 | 0.48 ± 0.12 | 0.39 ±0.22 |
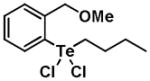
|
3 | 0.53 ± 0.25 | 0.89 ± 0.21 |
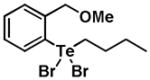
|
4 | 0.30 ± 0.19 | 0.92 ± 0.19 |
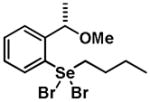
|
5 | 0.22 ± 0.25 | 0.18 ± 0.12 |
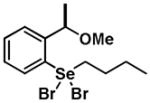
|
6 | 0.21 ± 0.18 | 0.09 ± 0.14 |
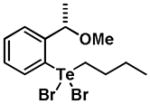
|
7 | 0.43 ± 0.25 | 0.74 ± 0.22 |
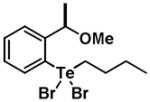
|
8 | 0.31 ± 0.15 | 0.59 ± 0.10 |
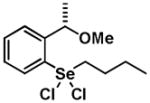
|
9 | 0.20 ± 0.16 | 0.39 ± 0.20 |
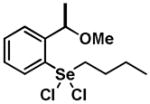
|
10 | 0.20 ± 0.23 | 0.30 ± 0.11 |
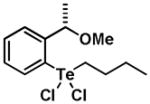
|
11 | 0.46 ± 0.19 | 1.07 ± 0.46 |
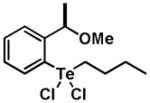
|
12 | 0.60 ± 0.39 | 0.65 ± 0.46 |
[inactivator] = 0.05mM;
[inactivator] = 0.1mM
These assays were very important to identify the relevance of the chalcogen atom for the profile of the organochalcogenanes as inhibitor of PTPs. As we can see in Table 1, the values of kobs showed that organotelluranes are more potent than organoselenanes for inhibition of PTP1B and the YopH. However, the contributions of the halogens and a possible stereochemistry discrimination of these compounds were not clear from the observed SAR towards the PTPs.
Inactivation of the PTPs by organoselenanes and organotelluranes appeared irreversible as extensive dialysis and/or buffer exchange of the reaction mixture failed to recover enzyme activity.
Since organotelluranes displayed higher inhibitory profile than organoselenanes, 3 was chosen as a model inhibitor, to perform a more detailed kinetic analysis in the PTP1B inactivation. Analysis of the pseudo-first-order rate constant as a function of inhibitor concentration showed that compound 3-mediated PTP1B inactivation displayed saturation kinetics (Figure 2), yielding values for the equilibrium binding constant KI and the inactivation rate constant ki of 1.9 ± 0.17 mM and 17.2 ± 0.9 min−1, respectively. These results suggest that 3 is an active site-directed affinity agent whose mode of action likely involves at least two steps: binding to the PTP active site followed by covalent modification of the active site Cys residue. It is worthwhile to point out that the kinetic parameters KI and ki for compound 3 compare very favorably to those determined for previously described activity-based probes for the PTPs, including α-bromobenzyl phosphonate18 and aryl vinyl sulfonates.19
Figure 2.

Kinetic analysis of PTP1B inactivation by 3 at 25 °C and pH 7. Panel on the left: time and concentration dependence of inhibitor 3-mediated PTP1B inactivation. Compound 3 concentrations were as follows: ⬢ 6 μM, □ 10 μM, ▴ 18 μM, ▽ 33 μM, ◆ 59 μM, ○ 106 μM, ⬢ 190 μM, ▵ 343 μM, ▼ 617 μM, ⋄ 1111 μM, and ■ 2000 μM. B. Concentration dependence of the pseudo-first-order rate constants kobs for 3-mediated PTP1B inactivation.
In summary, the results highlight the potential for developing hypervalent chalcogenated based small molecule probes to modulate PTP activity in signaling and in diseases. Among the organochalcogenanes used as inhibitor of PTPs, organotelluranes showed to be more potent than organoselenanes for inhibition of PTP1B and the YopH. The general reactivity of the organotelluranes toward the PTPs should facilitate the design of novel activity-based PTP probes. Additionally, PTP isozyme-specific organotelluranes based inhibitors could be developed by introducing specificity determinants into the aryl group to increase potency and selectivity.
Supplementary Material
Acknowledgments
L.W. and Z.-Y.Z. were supported in part by the National Institutes of Health Grants CA126937 and CA152194. L.P. and L.H.A. thank CNPq, CAPES and FAPESP for financial support.
Footnotes
Supporting Information Available: Experimental details and characterization data (1H, 13C and 125Te NMR, IR and mass spectrometry) for all compounds is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ba LA, Doring M, Jamier V, Jacob C. Org Biomol Chem. 2010;8:4203–4216. doi: 10.1039/c0Ob00086h. [DOI] [PubMed] [Google Scholar]
- 2.a) McNaughton M, Engman L, Birminngham A, Powis G, Cotgreave IA. J Med Chem. 2004;47:233–239. doi: 10.1021/jm030916r. [DOI] [PubMed] [Google Scholar]; b) Kanda T, Engman L, Cotgreave IA, Powis GJ. J Org Chem. 1999;64:8161–8169. doi: 10.1021/jo990842k. [DOI] [PubMed] [Google Scholar]; c) Wieslander E, Engman L, Svensjo E, Erlansson M, Johansson U, Linden M, Andersson CM, Brattsand R. Biochem Pharmacol. 1998;55:573–584. doi: 10.1016/s0006-2952(97)00517-0. [DOI] [PubMed] [Google Scholar]
- 3.a) Duntas LH. Horm Met Res. 2009;41:443–447. doi: 10.1055/s-0029-1220724. [DOI] [PubMed] [Google Scholar]; b) Bhabak KP, Mugesh G. Chem Asian J. 2009;4:974–983. doi: 10.1002/asia.200800483. [DOI] [PubMed] [Google Scholar]
- 4.Brodsky M, Halpert G, Albeck M, Sredni B. J Inflamm. 2010;7:3. doi: 10.1186/1476-9255-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Pinton S, da Rocha JL, Zeni G, Nogueira CW. Neuroscience Letters. 2010;472:56–60. doi: 10.1016/j.neulet.2010.01.057. [DOI] [PubMed] [Google Scholar]; b) Prigol M, Pinton S, Schumacher RF, Nogueira CW, Zeni G. Arch Toxicol. 2010;84:373–378. doi: 10.1007/s00204-009-0507-y. [DOI] [PubMed] [Google Scholar]; c) Prigol M, Schumacher RF, Nogueira CW, Zeni G. Toxicology Letters. 2009;189:35–39. doi: 10.1016/j.toxlet.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 6.a) Schrauzer GN. Crit Rev Biotechnol. 2009;29:10–17. doi: 10.1080/07388550802658048. [DOI] [PubMed] [Google Scholar]; b) Gundimeda U, Schiffman JE, Gottlieb SN, Roth BI, Gopalakrishna R. Carcinogenesis. 2009;30:1553–1561. doi: 10.1093/carcin/bgp164. [DOI] [PubMed] [Google Scholar]; c) Fleming J, Ghose A, Harrison PR. Nutrition and Cancer. 2001;40:42–49. doi: 10.1207/S15327914NC401_9. [DOI] [PubMed] [Google Scholar]; d) Ganther HE. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]; e) Combs GF, Jr, Gray WP. Pharmacol Ther. 1998;79:179–192. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 7.a) Sanmartin C, Plano D, Palop JA. Mini-Reviews in Medicinal Chemistry. 2008;8:1020–1031. doi: 10.2174/138955708785740625. [DOI] [PubMed] [Google Scholar]; b) Sinha R, El-Bayoumy K. Curr Cancer Drug Target. 2004;4:13–28. doi: 10.2174/1568009043481614. [DOI] [PubMed] [Google Scholar]; c) Zhou N, Xiao H, Li T-K, Nur-E-Kamal A, Liu LF. J Biol Chem. 2003;278:29532–29537. doi: 10.1074/jbc.M301877200. [DOI] [PubMed] [Google Scholar]
- 8.Carmely A, Meirow D, Peretz A, Albeck M, Bartoov B, Sredni B. Hum Reprod. 2009;24:1322–1329. doi: 10.1093/humrep/den481. [DOI] [PubMed] [Google Scholar]
- 9.Okun E, Arumugam TV, Tang SC, Gleichmann M, Albeck M, Sredni B, Mattson MP. J Neurochem. 2007;102:1232–1241. doi: 10.1111/j.1471-4159.2007.04615.x. [DOI] [PubMed] [Google Scholar]
- 10.Hayun R, Shpungin S, Malovani H, Albeck M, Okun E, Nir U, Sredni B. Ann N Y Acad Sci. 2007;1095:240–250. doi: 10.1196/annals.1397.028. [DOI] [PubMed] [Google Scholar]
- 11.Albeck A, Weitman H, Sredni B, Albeck M. Inorg Chem. 1998;37:1701–1712. [Google Scholar]
- 12.a) Piovan L, Alves MFM, Juliano L, Bromme D, Cunha RLOR, Andrade LH. J Braz Chem Soc. 2010;21:2108–2118. [Google Scholar]; b) Cunha RLOR, Gouvea IE, Feitosa GPV, Alves MFM, Bromme D, Comasseto JV, Tersariol ILS, Juliano L. Biol Chem. 2009;390:1205–1212. doi: 10.1515/BC.2009.125. [DOI] [PubMed] [Google Scholar]; c) Cunha RLOR, Urano ME, Chagas JR, Almeida PC, Bincoleto C, Tersariol ILS, Comasseto JV. Bioorg Med Chem Lett. 2005;15:755–760. doi: 10.1016/j.bmcl.2004.11.012. [DOI] [PubMed] [Google Scholar]; d) Persike DS, Cunha RLOR, Juliano L, Silva IR, Rosim FE, Vignoli T, Dona F, Cavalheiro EA, Fernandes MJS. Neurobiol Dis. 2008;31:120–126. doi: 10.1016/j.nbd.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z-Y. Prog Nucleic Acid Res & Mol Biol. 2003;73:171–220. doi: 10.1016/s0079-6603(03)01006-7. [DOI] [PubMed] [Google Scholar]
- 14.Tonks NK. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ZY. Curr Opin Chem Biol. 2001;5:416–423. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhang ZY, Dixon JE. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 17.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Zhou B, Liang F, Wang WQ, Huang Z, Zhang ZY. Proc Natl Acad Sci USA. 2004;101:7943–7948. doi: 10.1073/pnas.0402323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Zhou B, Yang H, He Y, Jiang ZX, Kumar S, Wu L, Zhang ZY. J Am Chem Soc. 2008;130:8251–8260. doi: 10.1021/ja711125p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.