

Abstract
Treacher Collins syndrome (TCS) is a congenital disorder of craniofacial development arising from mutations in TCOF1, which encodes the nucleolar phosphoprotein Treacle. Haploinsufficiency of Tcof1 perturbs mature ribosome biogenesis, resulting in stabilization of p53 and the cyclin G1–mediated cell-cycle arrest that underpins the specificity of neuroepithelial apoptosis and neural crest cell hypoplasia characteristic of TCS. Here we show that inhibition of p53 prevents cyclin G1–driven apoptotic elimination of neural crest cells while rescuing the craniofacial abnormalities associated with mutations in Tcof1 and extending life span. These improvements, however, occur independently of the effects on ribosome biogenesis; thus suggesting that it is p53-dependent neuroepithelial apoptosis that is the primary mechanism underlying the pathogenesis of TCS. Our work further implies that neuroepithelial and neural crest cells are particularly sensitive to cellular stress during embryogenesis and that suppression of p53 function provides an attractive avenue for possible clinical prevention of TCS craniofacial birth defects and possibly those of other neurocristopathies.
Treacher Collins syndrome (Online Mendelian Inheritance in Man (OMIM) number 154500) is an autosomal dominant disorder of facial morphogenesis. The clinical features of TCS include abnormalities of the external ears and middle ear ossicles, which often result in conductive hearing loss1, lateral downward slanting of the palpebral fissures with colobomas of the lower eyelids, hypoplasia of the facial bones and cleft palate2,3. TCS results from mutations in TCOF1 that lead to nonsense-mediated mRNA decay or truncations of the encoded protein Treacle4–7. This nucleolar phosphoprotein is structurally similar to Nopp140, which mediates pre-ribosomal ribonucleoprotein export from the nucleus and ribosomal protein import from the cytoplasm8. Treacle colocalizes with upstream binding factor (UBF) and RNA polymerase I in nucleolar organizing regions, where it functions in ribosomal DNA gene transcription9. Treacle also interacts directly with human Nop56p, a component of the ribonucleoprotein complex that 2′-O-methylates pre-ribosomal RNA (pre-rRNA) during the early stages of pre-rRNA processing10. Although the full function of Treacle remains to be elucidated, these studies imply that it has a key role in ribosome biogenesis, which is essential for proper cell growth and proliferation.
Indeed, we previously demonstrated in vivo, using a mouse model of TCS, that haploinsufficiency of Tcof1 compromizes the production of mature ribosomes in neuroepithelial and neural crest cells11. Insufficient ribosome biogenesis is associated with diminished cell proliferation, increased neuroepithelial apoptosis and deficient formation of migrating cranial neural crest cells. These observations directly underscore the characteristic hypoplasia of cranioskeletal elements in TCS, because most of the bone, cartilage and connective tissue in the head and face are derived from the neural crest. Thus, the facial dysmorphology associated with TCS arises very early during embryogenesis as a consequence of defects in the formation and proliferation of neural crest cells.
Individuals diagnosed with severe TCS typically undergo, over several years, multiple major reconstructive surgeries that are rarely fully corrective. Although stem cell therapy holds promise for treating degenerative diseases, it is unlikely to benefit the reconstructive repair of severe craniofacial malformations. Consequently, prevention provides the best hope for improving the prognosis of at-risk or affected individuals. Identifying potential avenues for rescue and/or repair depends, however, on appreciation of the developmental origins and etiology of congenital craniofacial birth defects.
RESULTS
TCS correlates with activation of p53-dependent transcription
To understand the molecular mechanism underlying TCS, we performed genome-wide transcriptional analyses of embryonic day 8.5 (E8.5) wild-type and Tcof1+/− embryos using microarrays. This embryonic stage coincides temporally with the increase in neuroepithelial apoptosis that underpins the TCS phenotype11. Because our mouse model of TCS is heterozygous for Tcof1, using this embryonic stage provided an internal control for establishing appropriate thresholds for changes in gene expression. A nine-way independent pair-wise comparison between each wild-type and mutant embryo showed that loss of one copy of Tcof1 decreased Tcof1 expression in mutant embryos by ~40%. We therefore used log2 mean values equivalent to a gross fold change of ±1.5 as a threshold to identify genes that were upregulated or downregulated in Tcof1+/− embryos.
The complete results from the microarray analysis will be published elsewhere; from the genes upregulated by a factor of ≥1.5, however, Ccng1 (5.79), Trp53inp1 (4.44), Pmaip1 (2.79), Perp (2.46) and Wig1 (2.14) were notable (Supplementary Table 1 online). Ccng1 (encoding cyclin G1) is a p53-responsive gene that inhibits growth, arresting cells at the G1 phase of the cell cycle12. Trp53inp1 is a p53-induced proapoptotic gene that also mediates G1 cell-cycle arrest13. Pmaip1 (Noxa) constitutes a p53-inducible member of the Bcl2 family that is important in mitochondrial fragmentation and caspase activation14. Perp encodes a tetraspan membrane protein and is a proapoptotic target of p53 (ref. 15). Wig1 encodes a zinc-finger protein that binds double-stranded RNA as part of a p53-dependent stress response16. Thus, these five genes are all recognized targets and mediators of p53-dependent transcription that have been linked to diverse cellular processes such as cell-cycle regulation, apoptosis, senescence and DNA repair (reviewed in ref. 17). These results suggest that there is a strong correlation between p53-dependent cell-cycle arrest and apoptosis and the development of TCS.
To validate both the microarray results and this association, we performed real-time quantitative RT-PCR analysis on the five genes. In agreement with the microarray results, Ccng1 (9.40), Trp53inp1 (8.38), Pmaip1 (4.39), Perp (2.14) and Wig1 (3.17) showed a ≥1.5-fold upregulation of expression in Tcof1+/− embryos relative to their wild-type littermates (Supplementary Table 1), confirming that the neuroepithelial apoptosis and deficiency in migrating neural crest cells characteristic of TCS are associated with p53-mediated cell-cycle arrest.
p53 is essential for normal cell growth, and, consequently, its activity is tightly regulated. Our microarray analyses indicated, however, that the Trp53 gene itself was not upregulated. We therefore focused on p53 protein, which, owing to continuous degradation, has a short half-life of 20–30 min and is thus normally maintained in small, often undetectable amounts18. Immunoblot analysis showed that p53 concentrations were 2.28-fold higher in E10.5 Tcof1+/− embryos than in wild-type littermates (Fig. 1). The increase in stability of p53 was indicative of altered post-transcriptional control of p53 and correlated with development of the TCS phenotype; however, definitive proof of an association required demonstration of a tissue-specific link between p53 stabilization and the neuroepithelial apoptosis characteristic of Tcof1+/− embryos.
Figure 1.

Immunoblot analysis of p53. (a) Analysis of the 53-kDa protein in E10.5 Tcof1+/+ embryos (lanes 1 and 2) as compared to E10.5 Tcof1+/− littermates (lanes 3 and 4). α-tubulin was used as a protein loading control. (b) Bar graph denistometry quantification of p53 protein levels in Tcof1+/+ and mutant Tcof1+/− littermates. Statistical significance was determined with the one-tailed Student's t-test with P = 0.006.
We therefore performed immunostaining of E8.5–E9.5 embryos with an antibody to p53 (Fig. 2). p53 expression was barely detectable in wild-type embryos (Fig. 2a) but was considerably higher in Tcof1+/− embryos (Fig. 2d), where the increased staining was confined to the neuroepithelium. Quantification of the fluorescence intensity as a measure of protein in individual neuroepithelial cells indicated that p53 concentrations were, on average, 1.8-fold higher in Tcof1+/− embryos than in control wild-type littermates. This value correlated well with the enhancement observed in immunoblot analyses. Counterstaining with DAPI showed that the enhanced p53 signal was restricted to the nuclei of neuroepithelial cells (Fig. 2b,c,e,f). This observation provides a direct tissue-specific correlation between nuclear stabilization of p53 protein, activation of p53-dependent transcription, and the induction of neuroepithelial apoptosis in Tcof1+/− embryos. The connection between p53 activity and neuroepithelial apoptosis also explains the deficiency in migrating neural crest cells observed in TCS.
Figure 2.

Immunostaining of p53. Immunostaining of E8.5 embryos for p53 protein (green) in Tcof1+/+ embryos (a–c) and inTcof1+/− embryos (d–f). Counterstaining with DAPI (blue; b,e) shows that p53 is specifically stabilized in the nucleus of neuroepithelial cells in Tcof1+/− embryos (f). Scale bar, 40 mm. (g) Bar graph of mean fluorescence denistometry quantification of p53 protein levels in individual neuroepithelial cells of Tcof1+/+ and Tcof1+/− littermates. Statistical significance was determined with the one-tailed Student's t-test with P < 0.001.
Tcof1 is required for cell-cycle progression
Tcof1 is highly expressed in the lateral edges of the neuroepithelium of E8.5 embryos, possibly corresponding to cells in the G1 and S phases of the cell cycle11. Successful progression from G1 to S is crucial for generating and delaminating neural crest cells in the neural tube19 and Snail1, a mediator of the G1–S cell-cycle transition, is downregulated in Tcof1+/− embryos11. We therefore postulated that Tcof1 plays an essential part in neural crest cell formation and proliferation by acting as a key cell-cycle regulator. The above results suggested that haploinsufficiency of Tcof1 results in p53-mediated activation of Ccng1 and, consequently, G1 cell-cycle arrest, neuroepithelial apoptosis and a deficiency in neural crest cell formation.
To validate this mechanism as the developmental basis of TCS, we performed in situ hybridization for Ccng1 on E8.5 wild-type and Tcof1+/− embryos (Fig. 3). Ccng1 was either absent or expressed in small amounts in wild-type embryos (Fig. 3a,c), but it was markedly upregulated along the whole anterior-posterior axis of the neural tube in Tcof1+/− embryos (Fig. 3b). Sections of stained embryos showed that the induced Ccng1 expression was restricted to the neuroepithelial cells (Fig. 3d). Ccng1 overexpression has been shown to inhibit growth, arresting cells at the G1 phase of the cell cycle12. The upregulation of Ccng1 in Tcof1+/− embryos implies that neuroepithelial cells arrest in the G1 phase of the cell cycle. The restriction in cell-cycle progression from G1 to S phase would explain the observation of decreased proliferation in the neural tube of Tcof1+/− embryos, as measured by 5-bromodeoxyuridine incorporation11. G1 phase arrest would also compromise the generation of neural crest cells and account for the high level of neuroepithelial cell–specific apoptosis.
Figure 3.

Expression of Ccng1 and inhibition of apoptosis. (a–d) In situ hybridization for Ccng1 in whole (a,b) and sectioned (c,d) E8.5 embryos. (e,f) TUNEL staining (green) for apoptosis in E8.75 Tcof1+/+ and Tcof1+/− littermates. (g,h) Intraperitoneal injection of pregnant dams with pifithrin-α in wild-type and Tcof1+/− embryos. Scale bars, 150 μm (a,b,e–g); 40 μm (c,d).
p53 inactivation reduces neuroepithelial apoptosis in TCS
The relationship between p53 activation and TCS raised the possibility of preventing the onset of TCS by blocking p53 function. We therefore explored the efficacy of pifithrin-α, a chemical inhibitor of p53-dependent transcription and apoptosis20, in suppressing neuroepithelial cell death in vivo. Pregnant C57BL/6 wild-type females that had been mated with DBA Tcof1+/− males were given pifithrin-α once daily from E6.5 to E8.5. This regime was temporally designed to coincide with the onset of neuroepithelial apoptosis in our mouse model of TCS. Control matings were also established in which pregnant females were either untreated or given the PBS carrier instead of pifithrin-α. Control and treated embryos were recovered at E8.5, which corresponds with the onset of neural crest cell formation and migration, and assayed for apoptosis by a whole-mount TUNEL assay. Untreated and PBS-treated wild-type embryos typically showed little cell death (Fig. 3e and data not shown), whereas untreated and PBS-treated Tcof1+/− embryos showed a massive increase in neuroepithelial apoptosis (Fig. 3f and data not shown). When treated with pifithrin-α, neuroepithelial apoptosis was substantially reduced in 67% (n = 12/18) of Tcof1+/− embryos (Fig. 3h) as compared with untreated Tcof1+/− embryos (Fig. 3g) and, consequently, more closely resembled the levels of cell death observed in wild-type embryos (Fig. 3e). Although not abrogated completely, the apoptosis levels achieved with pifithrin-α suggest that inhibition of p53 may provide a therapeutic avenue for preventing onset of the TCS phenotype.
Next, we initiated the genetic rescue of TCS by using p53-mutant mice. Progeny from intercrosses of Tcof1+/− and Trp53+/− mice were assayed for apoptosis at E8.5 with an antibody to caspase-3. In wild-type (Tcof+/+Trp53+/+) embryos, apoptosis was negligible (Fig. 4a); by contrast, activated capsase3 was detected throughout the neural tube of Tcof1+/−Trp53+/+ embryos (Fig. 4b). Loss of one copy of p53 considerably reduced the amount of neuroepithelial apoptosis in Tcof1+/− embryos, as judged by decreased caspase3 immunostaining in the neural tube of the compound heterozygote (Tcof1+/−Trp53+/−) embryos (Fig. 4c and Supplementary Fig. 1a online). Loss of two copies of p53 further abrogated caspase-3 activity as compared with Tcof1+/−Trp53+/− embryos (Supplementary Fig. 1a,b) such that apoptosis was commensurate with that of the wild type (compare Fig. 4a and Supplementary Fig. 1b). Quantification of the number of caspase-3–positive cells highlighted the marked suppression of apoptosis caused by dose-dependent inhibition of p53 (Supplementary Fig. 1c). These results correlate with the reduced neuroepithelial cell death observed in Tcof1+/− embryos treated with pifithrin-α (Fig. 3h). Thus, although haploinsufficiency of Tcof1 compromises neural crest cell formation through the apoptotic depletion of the neuroepithelial cells, the inhibition of p53 activity by both pharmocological and genetic approaches is sufficient to suppress neuroepithelial cell death in Tcof1+/− embryos in a dose-dependent manner.
Figure 4.
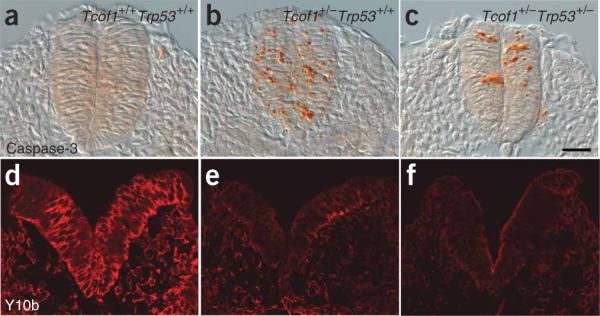
Analyses of apoptosis and ribosome biogenesis. (a–c) Immunostaining of sections of E9.0–E9.5 embryos for caspase3 activity (orange) in wild-type Tcof1+/+ embryos (a), Tcof1+/− mutants (b) and in Tcof1+/−Trp53+/− littermates (c). (d–f) Immunostaining of sections of E8.5 embryos with the mature 28S rRNA-specific antibody Y10b (red) in Tcof1+/+ embryos (d), Tcof1+/− mutants (e) and in Tcof1+/− Trp53+/− embryo littermates (f). Scale bar, 50 μm.
Haploinsufficiency of Tcof1 is associated with tissue-specific depletion of mature ribosome biogenesis and increased cell death in neuroepithelial cells11. Because p53 is a negative regulator of ribosome biogenesis21, we determined whether the reduction in apoptosis observed in rescued Tcof1+/− embryos through loss of p53 activity resulted in enhanced ribosome biogenesis. Immunostaining with Y10b antibody, which labels 28S rRNA, revealed a similar and marked depletion in mature ribosome biogenesis in E8.5 Tcof1+/−Trp53+/+ and Tcof1+/−Trp53+/− embryos, as compared with wild-type littermates (Tcof1+/+Trp53+/+; Fig. 4d–f). Thus, the production of mature ribosomes was neither altered nor restored to wild-type levels through suppression of p53 function. This finding distinguishes p53-dependent neuroepithelial apoptosis from deficient mature ribosome biogenesis as the primary cause of TCS craniofacial anomalies; it also heightens the potential for p53 inhibition as a therapy for preventing TCS.
p53 inactivation rescues TCS patterning defects
To evaluate the efficacy of p53 inhibition in preventing the pathogenesis of TCS, we examined whether blocking p53 would rescue the neural crest cell deficiencies observed in Tcof1+/− embryos. In situ hybridization with a Sox10 probe labeled the discrete segregated streams of migrating cranial neural crest cells in E9.0 wild-type (Tcof1+/+Trp53+/+) embryos (Supplementary Fig. 2a online). In Tcof1+/− embryos, hypoplasia of cranial neural crest cells was evident from diminished Sox10 staining combined with smaller narrower streams of labeled cells migrating into the frontonasal process and branchial arches (Supplementary Fig. 2b). Loss of a single Trp53 allele was sufficient to restore the migrating cranial neural crest cell phenotype in Tcof1+/−Trp53+/− embryos to a wild-type pattern (Supplementary Fig. 2c). Thus, partial inactivation of p53 is sufficient to reduce neuroepithelial apoptosis and, as a consequence, to minimize neural crest cell hypoplasia in Tcof1 haploin-sufficient embryos.
To substantiate p53 inhibition as a method for rescuing TCS, we examined whether loss of p53 could improve the development and patterning of the neural crest-derived sensory ganglia and craniofacial skeleton in Tcof1+/− embryos. We used anti-neurofilament immunostaining to label the cranial sensory ganglia in E10.5 embryos (Supplementary Fig. 2). In contrast to wild-type embryos, the cranial ganglia were hypoplastic in Tcof1+/−Trp53+/+ littermates (Supplementary Fig. 2d,e). In particular, the ophthalmic and maxillary nerve branches projecting from the trigeminal ganglion were absent in Tcof1+/−Trp53+/+ embryos (Supplementary Fig. 2e). Loss of one allele of Trp53 was sufficient to rescue the cranial ganglia abnormalities in Tcof1+/−Trp53+/− embryos (Supplementary Fig. 2f). These observations show that, although haploinsufficiency of Tcof1 causes severe craniofacial malformations, inhibition of p53 activity is sufficient to rescue the TCS-associated hypoplasia and disorganization of the sensory ganglia as early as E10.5.
p53 inactivation suppresses craniofacial abnormalities
To establish p53 inhibition as a viable therapeutic approach for preventing TCS, we characterized the head and facial phenotype of Tcof1+/− embryos in which p53 function was pharmocologically or genetically blocked (Fig. 5). Because administration of pifithrin-α to pregnant C57BL/6 females for 3 consecutive days from E6.5 to E8.5 reduced neuroepithelial cell–specific apoptosis in the Tcof1+/− progeny (Fig. 3h), we postulated that reducing the apoptotic elimination of neural crest precursor cells in E8.5 Tcof1+/− embryos might translate into normal development of the neural crest–derived structures that are hypoplastic in late-gestation Tcof1+/− embryos (compare Fig. 5a and 5b). Pregnant C57BL/6 females mated with DBA Tcof1 heterozygous males were given pifithrin-α from E6.5 to E8.5, and the progeny were analyzed at E17.5. Comparison of bone and cartilage staining of untreated E17.5 Tcof1+/− embryos (Fig. 5b) and pifithrin-α–treated E17.5 Tcof1+/− embryos (n = 10; Fig. 5d) showed only minor rescue. The degree of hypoplasia in the frontal, nasal, premaxilla and maxilla bones and the development of microphthalmia were only slightly lessened in pifithrin-α–treated E17.5 Tcof1+/− embryos. The cranial vault also remained shortened and domed (Fig. 5d), indicating that short-term pifithrin-α treatment does not sufficiently rescue the long-term, TCS-associated defects.
Figure 5.

Embryonic skeletal analyses and prevention of TCS. Craniofacial skeletons of E17.5 embryos were stained for bone (alizarin red) and cartilage (alcian blue). (a) Wild-type Tcof1+/+ embryo, showing a normal craniofacial skeleton. (b) Mutant Tcof1+/− embryo, showing severe frontonasal hypoplasia including the nasal, frontal, premaxilla, maxilla and mandibular bones, and doming of the skull and severe microphthalmia. (c) Tcof1+/+ embryo treated with pifithrin-α for 3 d in utero, showing a normal craniofacial skeleton. (d) Tcof1+/− embryo treated with pifithrin-α for 3 d in utero, showing slightly reduced frontonasal hypoplasia and less severe microphthalmia. (e) Tcof1+/+ embryo treated with pifithrin-α for 12 d in utero, showing a normal craniofacial skeleton. (f) Tcof1+/− embryo treated with pifithrin-α for 12 d in utero, showing substantially reduced frontonasal hypoplasia with considerable restoration of normal patterning of the frontal, nasal, premaxilla, maxilla and mandibular bones, and little evidence of any persistent calvarial doming or microphthalmia. (g) Wild-type Tcof1+/+Trp53+/+ embryo, showing a normal craniofacial skeleton. (h) Tcof1+/−Trp53+/− littermate embryo, showing a completely normal craniofacial skeleton that is indistinguishable from wild-type. Scale bar, 2 mm.
We therefore increased the duration of daily pifithrin-α treatment from E6.5 to E17.5 and observed a partial but noticeable rescue of TCS-associated cranioskeletal defects in E17.5 Tcof1+/− embryos (n = 2). The longer duration of pifithrin-α treatment markedly reduced the extent of cranial doming, and the cranial vault was considerably more elongated (compare Fig. 5d and 5f). Patterning of the frontal, nasal, premaxilla and maxilla bones closely resembled that of wild-type E17.5 embryos (Fig. 5e). The severity of microphthalmia was also markedly improved. Thus, sustained inhibition of p53 function through long-term treatment with pifithrin-α is necessary to rescue TCS-associated cranioskeletal abnormalities. We note, however, that it was difficult to obtain sufficient numbers of long-term pifithrin-α–treated E17.5 Tcof1+/− embryos for analysis because the stress accompanying repetitive intraperitoneal injections in pregnant females often resulted in spontaneous abortion of the pregnancy. The fact that embryos from long-term pifithrin-α–treated mice can survive indicates that pifithrin-α is not generally toxic, but rather the occasional lethality is due to repetitive stress associated with repeated daily handling, restraint and multiple injections.
We used a genetic approach to investigate further whether inhibition of p53 function could rescue TCS cranioskeletal abnormalities at E17.5. This approach was based on our observation that the inhibition of Trp53 reduced caspase3-mediated neuroepithelial apoptosis and consequently restored the deficit in the number of migrating cranial neural crest cells. Progeny from Tcof1+/− × Trp53+/− matings were analyzed at E17.5. Seventy-five per cent of E17.5 Tcof1+/−Trp53+/− embryos (n = 4) showed rescued development of the cranioskeleton, including the premaxilla, maxilla, frontal, nasal and temporal bones (Fig. 5h), such that the phenotype of the compound heterozygote was indistinguishable from that of the wild-type littermate (Fig. 5g). Thus, genetic loss of Trp53 can rescue the TCS-associated cranioskeletal abnormalities characteristic of late-gestation Tcof1+/− embryos.
Genetic inactivation of Trp53 produces viable Tcof1+/− pups
All newborn Tcof1+/− embryos on a mixed DBA × C57BL/6 background die within 24 h of birth from an inability to feed or breathe, a direct consequence of cleft palate and the severe frontonasal hypoplasia in which the nasal cavities fail to form11. Although genetic loss of Trp53 rescued cranioskeletal abnormalities in Tcof1+/− at E17.5, support for using p53 inhibition as a therapy for preventing the development of TCS required the generation of viable postnatal Tcof1+/− embryos. We therefore allowed the progeny of Tcof1+/− × Trp53+/− matings to proceed to birth. No craniofacial anomalies were observed in Tcof1+/+ × Trp53+/+ mice on a pure DBA, pure C57BL/6, or mixed DBA × C57BL/6 background (data not shown). Similarly, no craniofacial anomalies were associated with loss of one (Tcof1+/+ × Trp53+/−; Fig. 6a,b) or two (Tcof1+/+ × Trp53−/−; Fig. 6c,d) copies of Trp53 on a mixed DBA × C57BL/6 background. Genetic inhibition of Trp53 conferred postnatal viability in all Tcof1+/−Trp53+/− newborns (n = 10). Half of these mice were viable beyond 6 months and showed no evidence of the characteristic cranioskeletal dysmorphology of TCS (Fig. 6e,f). Thus, the craniofacial phenotype of 50% of Tcof1+/−Trp53+/− mice was indistinguishable from that of their wild-type littermates. However, the remaining 50% of Tcof1+/−Trp53+/− newborns showed some doming of the head and limited frontonasal hypoplasia (data not shown). Nonetheless, these individuals were still viable postnatally. Although these mice survived beyond weaning, three individuals were killed at 3 months owing to fluid accumulation and the remaining two died of unknown causes. Collectively, these results demonstrate the applicability of p53 inhibition in the therapeutic prevention of TCS; however, they also highlight the limitation of partial loss of p53 activity.
Figure 6.
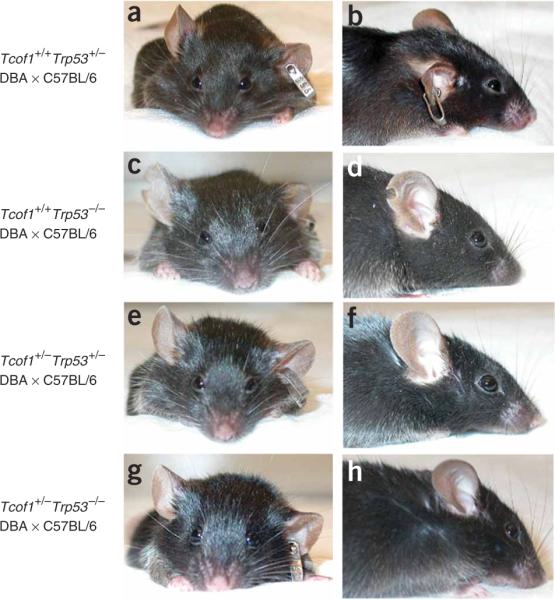
Genetic prevention of postnatal TCS craniofacial anomalies. Shown are frontal and lateral views highlighting restoration of normal craniofacial morphology through loss of p53 function. (a,b) Tcof1+/+Trp53+/− mouse. (c,d) Tcof1+/+Trp53−/− mouse. (e,f) Tcof1+/−Trp53+/− mouse. (g,h) Tcof1+/−Trp53−/− mouse. All mice were on a mixed DBA × C57BL/6 background.
We therefore evaluated the effect of complete loss of p53 on the development of TCS by mating viable Tcof1+/−Trp53+/− and Trp53+/− mice to generate Tcof1+/−Trp53−/− newborns (n = 11). All Tcof1+/−Trp53−/− pups survived beyond weaning with a craniofacial morphology that was indistinguishable from that of wild-type and littermate controls (Fig. 6g,h). Although these mice were viable, two were killed at 6 months owing to the presence of tumors and two died of unknown causes. These characteristics are common in Trp53−/− mice22 and are not TCS phenotypes associated with the rescue experiments. Thus, complete loss of p53 function can prevent the development of TCS in 100% of Tcof1+/− mice.
To understand how p53 mediates the neuroepithelial apoptosis underlying the origins of TCS and how its inhibition prevents the pathogenesis of craniofacial anomalies, we investigated the effect of dose-dependent blocking of p53 on its transcriptional targets Wig1, Ccng1, Trp53inp1, Pmaip1 and Perp, which are all markedly upregulated in Tcof1+/− mutant embryos. As determined by quantitative RT-PCR, the expression of each of these genes was substantially reduced in E8.5–E9.0 Tcof1+/−Trp53−/− embryos (n = 3) as compared with Tcof1+/− mutants, and was almost identical to Tcof1+/+ wild-type embryos (Supplementary Fig. 3). This finding is commensurate with the inhibition of apoptosis and rescue of the TCS facial phenotype, and highlights the dependency of activation of these genes on p53. We also noted that, in Tcof1+/−Trp53+/− embryos (n = 3), only the expression of Ccng1 (P = 0.035) and Perp (P = 0.013) was significantly reduced as compared with Tcof1+/− embryos (Supplementary Fig. 3). Because Tcof1+/−Trp53+/− newborns are viable postnatally, this finding implies that Ccng1 and Perp might be essential downstream targets mediating the p53-dependent induction of neuroepithelial apoptosis in Tcof1+/− mutant embryos. Perp-null mutant mice die postnatally and show extensive epithelial blistering, a phenotype not observed in Tcof1+/−Trp53+/− or Tcof1+/−Trp53−/− newborn pups23. By contrast, Ccng1-null mutant mice are developmentally normal and postnatally viable24. Ccng1 also has a role in stabilizing p53 (ref. 25). These observations suggest that Ccng1 is the candidate most likely to mediate the p53-dependent effects underlying the pathogenesis of TCS.
We therefore examined the effect of p53 inhibition on the expression of Ccng1 in our TCS embryo rescue experiments. In E8.5–E9.5 wild-type embryos, Ccng1 expression was not detectable (Supplementary Figs. 4a and 5a online). In Trp53+/− and Trp53−/− embryos (Supplementary Figs. 4b,c and 5b,c), similarly low Ccng1 expression was observed. In contrast, Tcof1+/− embryos showed increased Ccng1 expression along the whole the neural tube (Supplementary Figs. 4d and 5d). Loss of either one or two copies of Trp53 in Tcof1+/− embryos (Supplementary Figs. 4e,f and 5e,f) revealed dose-dependent repression of Ccng1 expression and restoration of normal neural tube morphology (compare Supplementary Fig. 4a and 4f). Thus, inhibition of p53 suppresses the ectopic expression of Ccng1 and prevents the neuroepithelial apoptosis underlying the etiology and pathogenesis of TCS.
To confirm that Ccng1 is a crucial mediator of p53-dependent apoptosis in the manifestation of TCS craniofacial anomalies, we tested whether Ccng1 overexpression alone would be sufficient to induce neuroepithelial apoptosis. Using electroporation, we transfected the neuroepithelium of HH stage-9 chick embryos with constructs expressing control enhanced GFP (EGFP) or Ccng1 (Supplementary Fig. 6 online). In control experiments (n = 6), EGFP was overexpressed on one side of the neural tube only and, 24 h after electroporation, showed strong fluorescence; counterstaining with DAPI and TUNEL to label nuclei and dying cells, respectively, revealed low background apoptosis (Supplementary Fig. 6a,c,e,g,i). By contrast, Ccng1 over-expression (n = 6) in the neural tube led to a marked and significant (P < 0.001) increase in the number of apoptotic neuroepithelial cells (Supplementary Fig. 6b,d,f,h,i). Thus, ectopic expression of Ccng1 alone in the neural tube is sufficient to induce neuroepithelial apoptosis. Ccng1, a transcriptional target of p53, thus has a primary role in mediating the cell-cycle arrest and apoptotic effects of p53-dependent activation in the etiology and pathogenesis of TCS.
DISCUSSION
Our analysis of the pathogenesis of TCS has shown that haploinsufficiency of Tcof1 (Treacle) leads to nuclear stabilization of p53 protein and activation of p53-dependent transcriptional targets such as Ccng1 in the neuroepithelium. This activation directly correlates with the cell-cycle arrest and caspase3-mediated apoptosis of neural crest cell progenitors in the neuroepithelium of Tcof1+/− embryos, events that account for the neural crest cell deficiencies underlying TCS (Supplementary Fig. 7 online).
The association between p53 and TCS enhances our understanding of the pathogenesis of this neurocristopathy. Trp53 mediates various stress-induced responses and regulates important functions in diverse cellular processes17. The involvement of p53 in ribosome biogenesis is, however, particularly intriguing in the etiology of TCS. We propose that the p53-induced Ccng1-mediated cell-cycle arrest observed in the pathogenesis of TCS reflects a nucleolar stress response caused by deficient mature ribosome biogenesis (Supplementary Fig. 7). p53 represses RNA polymerase I activity by preventing the interaction between UBF and promoter selectivity factor (SL1), and thereby directly interferes with transcriptional initiation at the promoter of rRNA21. Thus, p53 can inhibit cell proliferation through its role as a negative regulator of ribosome biogenesis. By contrast, Treacle is a positive regulator of ribosome biogenesis and thus cell proliferation11 through its ability to bind UBF and to promote RNA polymerase I transcriptional activity9. Collectively, these findings indicate that the pathogenesis of TCS arises initially through compromised mature ribosome biogenesis as a direct result of Tcof1 haploinsufficiency. This deficiency may be further compounded by the nucleolar stress response activation of a p53-dependent checkpoint, which leads to stabilization of p53 and cyclin G1–mediated cell-cycle arrest and apoptosis.
The opposing effects of Treacle and p53 on ribosome biogenesis and thus cell proliferation provided an opportunity to explore the therapeutic prevention of TCS. Pharmacological and genetic inhibition of p53 function repressed activation of Ccng1 and diminished apoptosis in our mouse model of TCS, as assessed by TUNEL and activated caspase-3 staining (Supplementary Fig. 7). The reduced apoptosis rescued the deficiency of Sox10-labeled migrating neural crest cells in Tcof1+/− embryos and correlated with restoration of the cranial sensory ganglia phenotype to a wild-type pattern. Rescue of the neural crest cell deficiency resulted in the prevention of cranioskeletal anomalies typical of TCS in about 50% of late-gestation Tcof1+/− embryos. Inhibition of p53 also prevented TCS and its associated postnatal complications in a dose-dependent manner: 50% of Tcof1+/−Trp53+/− mice and 100% of Tcof1+/−Trp53−/− mice showed a craniofacial phenotype that was grossly indistinguishable from wild-type, with no evidence of frontonasal hypoplasia, cleft palate, microphthalmia or doming of the skull. In addition, whereas all Tcof1+/− embryos die within 24 h of birth, all Tcof1+/−Trp53+/− and Tcof1+/−Trp53−/− mice survived beyond weaning. Thus, inhibition of p53 is sufficient to rescue the craniofacial defects characteristic of TCS and to prevent its associated neonatal lethality.
The pharmacological and genetic inhibition of Trp53, which was so successful in inhibiting neuroepithelial apoptosis, occurred without altering ribosome biogenesis. Mature ribosome production in Tcof1+/−Trp53+/− embryos remained equivalent to that in Tcof1+/−Trp53+/+ and was markedly lower than in wild-type Tcof1+/+Trp53+/+ littermates. Thus, it is not necessarily the deficiency in ribosome biogenesis that is responsible for the pathogenesis of TCS; rather, it is the nucleolar stress activation of a Trp53-dependent checkpoint and subsequent Ccng1-mediated cell-cycle arrest that cause the increase in neuroepithelial apoptosis responsible for the characteristic hypoplasia of the neural crest and subsequently the craniofacial skeleton. Thus, inhibition of Trp53 either genetically or pharmacologically provides an exciting therapeutic route for preventing TCS.
Tcof1 is widely expressed during embryogenesis11,26 and Tcof1−/− embryos die between implantation and gastrulation. Thus, Tcof1 is also important in cell types other than neuroepithelial and neural crest cells. Our analyses have demonstrated, however, that haploinsufficiency of Tcof1 primarily affects neuroepithelial and neural crest cell development. One logical explanation for this observation is that, during early embryogenesis, the neuroepithelium proliferates rapidly while at the same time generating a completely new cell population, the neural crest. These processes require substantial transcriptional and metabolic activity and correlate temporally with mitochondrial maturation and the commencement of oxidative phosphorylation during embryogenesis27. Our results show that neuroepithelial and neural crest cells are particularly sensitive to cellular stress that disrupts these processes. We consider that this lower threshold sensitivity in neuroepithelial cells helps to explain the high incidence of neural crest cell–related cranio-facial anomalies that constitute roughly a third of all congenital birth defects. The p53 checkpoint control mechanism may have evolved to monitor ribosome production in the nucleolus and to activate a cell-cycle inhibitory response when confronted by nucleolar stress or various defects in ribosome biogenesis. This response mechanism seems to function efficiently because the incidence of TCS is rare.
Treacher Collins syndrome is one of the most severe examples of a group of conditions known as `first arch syndrome', which collectively describes congenital anomalies broadly affecting the eyes, ears, mandible and palate. Our work has implications for other congenital craniofacial neurocristopathies that fall under the first arch syndrome definition in which a similar deficiency in the number of migrating neural crest cells causes cranioskeletal hypoplasia. Activation of a p53 checkpoint together with decreased proliferation and G1 cell-cycle arrest may represent a common mechanism that leads to neural crest cell deficiencies and could account for the high frequency (30%) of craniofacial anomalies in congenital birth defects. Indeed, Splotch (Pax3−/−) mutant mice, which are embryonic lethal and show spina bifida, have been shown to be rescued during early embryogenesis by p53 inhibition28. Although that analysis examined only organogenesis-stage embryos and not postnatal mice, it lends credence to the idea that suppression of p53-mediated cell-cycle arrest and neuroepithelial apoptosis may be broadly successful for rescuing or preventing neurocristopathies and congenital craniofacial birth defects.
A caveat to this idea is the association between loss of p53 function and the development of spontaneous tumors: 75% of Trp53−/− mice develop neoplasms within 6 months, and tumors appear as early as 10 weeks22. Therefore, the connection between Tcof1 and Trp53 in ribosome biogenesis has important implications in our understanding of cancer and tumorigenesis. The extent to which ribosome function is directly modulated by tumor suppressors and oncogenes, and whether this modulation represents a cause or consequence of cancer progression, is under increasing scrutiny29. If Trp53 inhibition can rescue haploinsufficiency of Tcof1, is the reverse also true? Despite extensive searching, we have not identified individuals with TCS that also develop cancer or related tumors (although such individuals would be extremely rare because the incidence of TCS alone is 1 in 50,000). Where tumors are associated with enhanced ribosome biogenesis, however, Tcof1 might be a potential modifier of tumor progression. It will be interesting to examine the effects of Tcof1 overexpression on ribosome biogenesis and to determine whether there is any prevention or delay in the onset of tumorigenesis in mouse cancer models that are also deficient for Tcof1.
Collectively, our data show that appropriate titration of p53 function is key to preventing the pathogenesis of TCS without risking the onset of tumorigenesis. In support of this idea, Trp53+/− mice rarely show tumor formation. In addition, none of the viable postnatal Tcof1+/−Trp53+/− rescued mice showing normal craniofacial morphology has so far developed tumors. Similarly, `Super Trp53' transgenic mice are normal and show only an enhanced response to DNA damage30. The absence of abnormalities in Super Trp53 transgenic mice correlates with their generation by the introduction of large genomic DNA segments containing the whole Trp53 gene, which increases the gene dosage of p53 only minimally. The nature of the BAC transgene accurately reproduces the behavior of the endogenous gene. In Tcof1+/− embryos, by contrast, there is abnormal activation of p53 protein (more than twofold higher than wild type) specifically in the neuroepithelium during early embryogenesis. Thus, whereas low expression of p53 may enhance the repair response, high expression of p53 pushes cells down the apoptotic pathway, which accounts for the phenotypic differences between Super Trp53 and Tcof1+/− mice. Therefore, reducing but not completely eliminating p53 function is required to prevent TCS without inducing spontaneous tumors.
The causative mutations in many individuals with TCS remain unidentified, and mutations in components of the p53 pathway may underlie the phenotype in those individuals. TCS is not thought to be genetically heterogeneous, however, because all of the multigenerational TCS families analyzed so far show linkage to the chromosome 5q32 locus. Nonetheless, to further our understanding of the etiology and pathogenesis of TCS, it will be crucial to identify direct binding targets of Tcof1 by proteomic approaches. It will also be essential to pursue downstream transcriptional targets of p53, such as Ccng1, that do not show a tumorigenesis risk as potential avenues for preventing neurocristopathies such as TCS and other congenital craniofacial birth defects.
METHODS
Mice
All experiments involving higher vertebrates were approved by the Institutional Animal Care and Use Committee of the Stowers Institute for Medical Research. We bred congenic DBA Tcof1+/− mice and C57BL/6 wild-type mice to generate mixed DBA × C57BL/6 F1 progeny as described11. We obtained Trp53+/− mice on a C57BL/6 background22 from the Mouse Models of Human Cancers Consortium of the National Cancer Institute. We bred congenic DBA Tcof1+/− mice with wild-type C57BL/6 mice to obtain embryos as described11. We also bred congenic DBA Tcof1+/− mice with Trp53+/− mice on a C57BL/6 background to generate compound Tcof1+/−Trp53+/− mice, which we subsequently backcrossed to Trp53+/− to generate Tcof1+/−Trp53−/− mice. We obtained mutant embryos by timed mating; the morning of the vaginal plug was considered to be E0.5.
Genotyping
We extracted DNA from individual embryos or pups from yolk sacs or tails and PCR genotyped the Tcof1 progeny as described26. We performed genotyping of the Trp53 progeny by PCR analysis of genomic DNA using the following primers in a duplex reaction: wild-type forward primer Exon 4F, 5′-AAAGGTCCCAGTCCTCTCTTTGCT-3′ targeted forward primer Neo 3F, 5′-ATAGCGTTGGCTACCCGTGATATTG-3′ and common reverse primer Exon 6R, 5′-CTGTCTTCCAGATACTCGGGATAC-3′. The expected amplicon sizes were 479 bp (wild-type allele) and 392 bp (mutant allele). DNA samples were denatured at 95 °C for 5 min and then processed through 30 amplification cycles (95 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s). We lengthened the final extension step to 5 min.
Pharmacological inhibition of p53
We gave C57BL/6 wild-type female mice bred to DBA Tcof1 heterozygote male mice pifithrin-α (Alexis Biochemicals) at 2.2 mg per kg (body weight) in PBS by intraperitoneal injection. Female mice were treated at E6.5 for either 3 or 12 d, and embryos were subsequently analyzed at E8.5 and E17.5.
Cell death analysis
Embryos were fixed at E8.5 in 4% paraformaldehyde before whole-mount TUNEL staining with an FITC Cell Death Detection Kit (Roche) used in accordance with the manufacturer's instructions. We also assessed apoptosis by immunostaining with monoclonal antibody to cleaved caspase3 (Cell Signaling, dilution 1:200). In brief, embryos fixed in 4% paraformaldehyde were processed in paraffin and sectioned at 12 μm in transverse planes. Caspase3 incorporation was revealed by a standard antigen retrieval method with citrate buffer as described11. We analyzed cryosections of electroporated avian embryos by TUNEL using a rhodamine Cell Death Detection Kit (Roche) in accordance with the manufacturer's instructions.
Skeletal analysis and in situ hybridization
The craniofacial skeleton of E17.5 embryos and pups were fixed in 99% ethanol and stained for bone and cartilage as described31. E8.5–E10.5 embryos were fixed in 4% paraformaldehyde and processed in a graded series of methanol for whole-mount in situ hybridization as described32.
Immunohistochemistry
We fixed E8.5–E10.5 embryos in 4% paraformaldehyde and processed them in a graded series of ethanol for immunohistochemical analysis. We used whole-mount immunohistochemistry to label the cranial sensory ganglia and projections in E10.5 embryos using a 1:50 to 1:100 dilution of antibody to neurofilament (2H3; Developmental Studies Hybridoma Bank), together with a 1:200 dilution of a horseradish peroxidase (HRP)-coupled goat antibody to mouse IgG (Jackson Immunoresearch). We also performed immunohistochemistry on paraffin-processed transverse sections (10 μm) prepared from E8.5 embryos to examine rRNA integrity with Y10b monoclonal antibody as described11. We examined endogenous p53 protein with monoclonal antibody to p53 (Cell Signaling; dilution 1:200). p53 activity was revealed by using a standard antigen retrieval method with citrate buffer (pH 6.0, 95 °C for 10 min); the p53 signal was detected with biotin-conjugated antibody to mouse IgG (Vectastain; 1:200) and a streptavidin-AlexaFluor–conjugated secondary antibody (Molecular Probes; 1:200). We counterstained all sections with DAPI. We used Axiovision software on a Zeiss Axioplan instrument to photograph the immunostained sections and to quantify fluorescence intensity in individual cells as an approximate measure of relative protein.
Immunoblot analysis
p53 protein was analyzed in protein (29.7 μg) extracted from individual E10.5 embryos with monoclonal antibody to p53 (Cell Signaling; 1:1,000). We normalized the p53 signal to α-tubulin, which we detected with monoclonal antibody to α-tubulin mouse ascites fluid (Sigma; 1:200,000). The p53 and α-tubulin signals were detected by HRP-conjugated anti–mouse IgG (Amersham Biosciences; 1:5,000 and 1:10,000, respectively) and chemiluminescence (ECL and Pierce). We analyzed each band by densitometry with ImageJ software and measured against the α-tubulin value to quantify relative p53 protein in control and mutant samples.
RNA extraction, amplification and hybridization of microarray probes
We extracted total RNA from three E8.5 wild-type and three E8.5 Tcof1+/− littermate embryos by using the RNeasy Mini Protocol for Isolation of Total RNA from Animal Tissues (Qiagen) in accordance with the manufacturer's protocol. We determined RNA quality and quantity with a bioanalyzer. To generate targets for microarray analysis, we amplified total RNA (100 ng) from each wild-type and mutant embryo by using Two-Cycle Target Labeling (Affymetrix) in accordance with the manufacturer's instructions. We hybridized biotinylated target cRNAs (20 μg) from each wild-type and Tcof1+/− embryo to separate GeneChip Mouse Genome 430 2.0 arrays (Affymetrix) by standard procedures.
Data analysis
The GeneChip Mouse Genome 430 2.0 array provides 45,000 probe sets to analyze >39,000 transcripts and variants from >34,000 well-characterized mouse genes (http://www.affymetrix.com). Each transcript is represented by 11 pairs of oligonucleotide (25-nucleotide) probes, and each pair includes a perfect match and mismatch probe. We scanned the microarrays with an Affymetrix GeneChip Scanner 3000 and analyzed the raw data with GeneChip Operating Software Version 1.2.0.037. We used the `Simpleaffy' bioconductor package33 to assess the quality of the RNA samples. The scaling factors used to normalize the target values for the three wild-type and three Tcof1+/− mutant arrays were within threefold of one another (range 0.783–0.962), and all arrays showed control parameters for average background (<62) and percentage present calls (range 46.6–50.7%) that were within the recommended limits. The RNA quality was demonstrated to be acceptable from the 3′/5′ ratios of Gapdh and Actb (β-actin) genes.
An R script34 computed ratios and log2 ratios of expression for the nine possible pair-wise comparisons between the three wild-type and three Tcof1+/− mutant embryos. We further analyzed these data interactively by using a Microsoft Access database to sort and filter the Affymetrix probes. The Access `Query by Example' feature was used for interactive filtering. Affymetrix annotation information about the probes, including gene name and gene ontology assignments, was joined to the expression results to aid the analysis.
Quantitative real-time PCR analysis
From 1 μg of total RNA extracted from individual E8.5 wild-type, Tcof1+/−, Tcof1+/−Trp53+/− and Tcof1+/−Trp53−/− rescued embryos, we synthesized first-strand cDNAs by using a QuantiTect Reverse Transcription Kit (Qiagen) in accordance with the manufacturer's instructions. We performed quantitative RT-PCR in triplicate for each sample using an iCycler iQ Real Time PCR Detection System (Bio-Rad) and QuantiTect SYBR Green PCR Kit (Qiagen). The cycling parameters for all genes were 95 °C for 15 min, followed by 50 cycles of 94 °C for 15 s, 55 °C for 30 s and 72 °C for 30 s. We verified the specificities of the target and Gapdh assays by the absence of signal in reactions without reverse transcriptase and reactions without template. Efficiencies of the target and Gapdh assays were shown to be within 10% of each other by a relative standard curve method. Data were collected at the extension step, and a melting curve (55–95 °C) was generated at the end of each PCR cycle. Each 25-μl reaction mixture contained 12.5 μl of 2× QuantiTect SYBR Green PCR Master Mix, 2.5 μl of 10× Primer Assay and ~12.5 ng of cDNA template (1/80 of the RT reaction). Each Primer Assay contained specific primers derived from gene sequences contained in the NCBI Reference Sequence database. QuantiTect Primer Assays (Qiagen) were obtained for Gapdh (NM_001001303), Ccng1 (NM_009831), Pmaip1 (NM_021451), Trp53inp1 (NM_021897) and Wig1 (NM_009517). RT2-PCR Primer Assay (SuperArray) was obtained for Perp (NM_022032) owing to a lack of availability at Qiagen. Data were analyzed with a threshold set in the linear range of amplification, and the cycle threshold (CT) values were used to determine the fold change (FC), as described by the comparative CT (ΔΔCT) method35: ΔCT = CT (target gene) − CT (Gapdh) and FC = 2−(ΔCT mut sample − ΔCT wt sample).
Electroporation
We prepared chick eggs for electroporation as described36. Py-DF30Y (control GFP vector) and CyclinG1-pyDF30 (overexpression vector containing subcloned cyclin G1–EGFP12,37) were dissolved in water at 1–2 μg/μl, and 0.1% fast green solution was added to visualize injection into HH stage-9 chick neural tubes. We applied five 50-ms pulses of 20 V each by using a CUY-21 electroporator and gold electrodes. Eggs were resealed and incubated for 24 h at 37 °C under humidified conditions.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Trainor and Dixon groups for comments on the manuscript; R. McCay and M. Morgan for maintenance of mutant mouse lines and administration of pifithrin-α; T. Yao and J. Ren for technical assistance with protein analysis and p53 antibody activity; D. Stark for confocal imaging expertise; S. Das for the Ccng1 riboprobe, D. Quelle and M. Horne for Ccng1 expression vectors; and E. Rubel for the Y10b antibody. Research in the Trainor laboratory is supported by the Stowers Institute for Medical Research, March of Dimes (6FY05-82), National Institute of Dental and Craniofacial Research (RO1 DE 016082-01) and the Hudson Foundation. Research in the Dixon laboratory is supported by the National Institutes of Health (P50 DE 016215) and the Medical Research Council, UK (G81/535).
Footnotes
Accession codes Gene Expression omnibus accession code for microarray data, GSE10167.
Note: Supplementary information is available on the Nature Medicine website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Phelps PD, Poswillo D, Lloyd GA. The ear deformities in mandibulofacial dysostosis (Treacher Collins syndrome) Clin. Otolaryngol. 1981;6:15–28. doi: 10.1111/j.1365-2273.1981.tb01782.x. [DOI] [PubMed] [Google Scholar]
- 2.Rovin S, Dachi SF, Borenstein DB, Cotter WB. Mandibulofacial dysostosis, a familial study of five generations. J. Pediatr. 1964;65:215–221. doi: 10.1016/s0022-3476(64)80522-9. [DOI] [PubMed] [Google Scholar]
- 3.Fazen LE, Elmore J, Nadler HL. Mandibulo-facial dysostosis. (Treacher-Collins syndrome) Am. J. Dis. Child. 1967;113:405–410. doi: 10.1001/archpedi.1967.02090190051001. [DOI] [PubMed] [Google Scholar]
- 4.Edwards SJ, Gladwin AJ, Dixon MJ. The mutational spectrum in Treacher Collins syndrome reveals a predominance of mutations that create a premature-termination codon. Am. J. Hum. Genet. 1997;60:515–524. [PMC free article] [PubMed] [Google Scholar]
- 5.Gladwin AJ, et al. Treacher Collins syndrome may result from insertions, deletions or splicing mutations, which introduce a termination codon into the gene. Hum. Mol. Genet. 1996;5:1533–1538. doi: 10.1093/hmg/5.10.1533. [DOI] [PubMed] [Google Scholar]
- 6.The Treacher Collins Syndrome Collaborative Group Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 1996;12:130–136. doi: 10.1038/ng0296-130. [DOI] [PubMed] [Google Scholar]
- 7.Wise CA, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc. Natl. Acad. Sci. USA. 1997;94:3110–3115. doi: 10.1073/pnas.94.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier UT, Blobel G. Nopp140 shuttles on tracks between nucleolus and cytoplasm. Cell. 1992;70:127–138. doi: 10.1016/0092-8674(92)90539-o. [DOI] [PubMed] [Google Scholar]
- 9.Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. USA. 2004;101:10709–10714. doi: 10.1073/pnas.0402492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayano T, et al. Proteomic analysis of human Nop56p-associated pre-ribosomal ribonucleoprotein complexes. Possible link between Nop56p and the nucleolar protein treacle responsible for Treacher Collins syndrome. J. Biol. Chem. 2003;278:34309–34319. doi: 10.1074/jbc.M304304200. [DOI] [PubMed] [Google Scholar]
- 11.Dixon J, et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao L, et al. Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol. Cancer Res. 2003;1:195–206. [PubMed] [Google Scholar]
- 13.Tomasini R, et al. TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J. Biol. Chem. 2003;278:37722–37729. doi: 10.1074/jbc.M301979200. [DOI] [PubMed] [Google Scholar]
- 14.Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006;66:9235–9244. doi: 10.1158/0008-5472.CAN-06-0702. [DOI] [PubMed] [Google Scholar]
- 15.Reczek EE, Flores ER, Tsay AS, Attardi LD, Jacks T. Multiple response elements and differential p53 binding control Perp expression during apoptosis. Mol. Cancer Res. 2003;1:1048–1057. [PubMed] [Google Scholar]
- 16.Mendez-Vidal C, Wilhelm MT, Hellborg F, Qian W, Wiman KG. The p53-induced mouse zinc finger protein wig-1 binds double-stranded RNA with high affinity. Nucleic Acids Res. 2002;30:1991–1996. doi: 10.1093/nar/30.9.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 18.Zaika A, Marchenko N, Moll UM. Cytoplasmically `sequestered' wild type p53 protein is resistant to Mdm2-mediated degradation. J. Biol. Chem. 1999;274:27474–27480. doi: 10.1074/jbc.274.39.27474. [DOI] [PubMed] [Google Scholar]
- 19.Burstyn-Cohen T, Kalcheim C. Association between the cell cycle and neural crest delamination through specific regulation of G1/S transition. Dev. Cell. 2002;3:383–395. doi: 10.1016/s1534-5807(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 20.Komarov PG, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 21.Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000;20:5930–5938. doi: 10.1128/mcb.20.16.5930-5938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 23.Ihrie RA, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120:843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Jensen MR, et al. Reduced hepatic tumor incidence in cyclin G1-deficient mice. Hepatology. 2003;37:862–870. doi: 10.1053/jhep.2003.50137. [DOI] [PubMed] [Google Scholar]
- 25.Kimura SH, Nojima H. Cyclin G1 associates with MDM2 and regulates accumulation and degradation of p53 protein. Genes Cells. 2002;7:869–880. doi: 10.1046/j.1365-2443.2002.00564.x. [DOI] [PubMed] [Google Scholar]
- 26.Dixon J, Brakebusch C, Fassler R, Dixon MJ. Increased levels of apoptosis in the prefusion neural folds underlie the craniofacial disorder, Treacher Collins syndrome. Hum. Mol. Genet. 2000;9:1473–1480. doi: 10.1093/hmg/9.10.1473. [DOI] [PubMed] [Google Scholar]
- 27.Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat. Rev. Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Cao I, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman MH. The Atlas of Mouse Development. Vol. 507. Academic Press; London: 1992. [Google Scholar]
- 32.Nagy A, Gertsenstein M, Vintersten K, Behringer RR. Manipulating the Mouse Embryo. Cold Spring Harbor Laboratory; Cold Spring Harbor: 2003. Ch.16. [Google Scholar]
- 33.Wilson CL, Miller CJ. Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics. 2005;21:3683–3685. doi: 10.1093/bioinformatics/bti605. [DOI] [PubMed] [Google Scholar]
- 34.R Development Core Team A language and environment for statistical computing. The R Project for Statistical Computing. 2006 http://www.R-project.org.
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Krull CE. A primer on using in ovo electroporation to analyze gene function. Dev. Dyn. 2004;229:433–439. doi: 10.1002/dvdy.10473. [DOI] [PubMed] [Google Scholar]
- 37.Horne MC, et al. Cyclin G1 and cyclin G2 comprise a new family of cyclins with contrasting tissue-specific and cell cycle-regulated expression. J. Biol. Chem. 1996;271:6050–6061. doi: 10.1074/jbc.271.11.6050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.