

Abstract
Background
Heavy alcohol consumption causes cerebellar degeneration, and the underlying mechanism is unclear. Chronic alcoholism is usually associated with thiamine deficiency (TD) which is known to induce selective neurodegeneration in the brain. However, the role of TD in alcohol-induced cerebellar degeneration remains to be elucidated. The double-stranded RNA-activated protein kinase (PKR) is a potent antiviral protein. Viral infection or binding to dsRNA causes PKR autophosphorylation and subsequent phosphorylation of the α-subunit of eukaryotic translation factor-2α, leading to inhibition of translation or apoptosis. PKR can also be activated by cellular stresses.
Methods
In this study, we used an in vitro model, cultured cerebellar granule neurons (CGNs), to investigate the interaction between TD and ethanol and evaluate the contribution of their interaction to neuronal loss. TD was induced by treatment with amprolium in association with ethanol. Cell viability was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide assay. PKR expression/phosphorylation and subcellular distribution was analyzed with immunoblotting and immunocytochemistry.
Results
Thiamine deficiency caused death of CGNs but ethanol did not. However, TD plus ethanol induced a much greater cell loss than TD alone. TD-induced PKR phosphorylation and ethanol exposure significantly promoted TD-induced PKR phosphorylation as well as its nuclear translocation. A selective PKR inhibitor not only protected CGNs against TD toxicity, but also abolished ethanol potentiation of TD-induced loss of CGNs.
Conclusions
Ethanol promoted TD-induced PKR activation and neuronal death. PKR may be a convergent protein that mediates the interaction between TD and ethanol.
Keywords: Alcohol, Cerebellum, Malnutrition, Neurodegeneration, Oxidative Metabolism, PKR
Heavy alcohol consumption causes damage to the central nervous system through numerous mechanisms. One of the proposed mechanisms is the reduced availability of an essential nutrient, thiamine, to the brain (Martin et al., 2003; Singleton and Martin, 2001). Thiamine, also known as vitamin B1, is an essential nutrient and plays an important role in metabolic and cellular function. In the human body, high concentrations of thiamine are found in skeletal muscles, heart, liver, kidney, and brain (Martin et al., 2003). Thiamine diphosphate is the active form of thiamine and serves as a cofactor for several enzymes involved in carbohydrate catabolism. In addition to their critical role in carbohydrate and energy metabolisms, thiamine and thiamine-utilizing enzymes regulate brain excitability and synthesis of neurotransmitters, nucleic acids, fatty acids, and steroids (Martin et al., 2003).
Several mechanisms for alcoholism-induced thiamine deficiency (TD) have been proposed, and these include: (i) inadequate nutritional intake in alcoholics; (ii) decreased absorption of thiamine from the gastrointestinal tract and reduced uptake into cells due to alcohol exposure; and (iii) impaired utilization of thiamine in the cells due to alcohol exposure (Gastaldi et al., 1989; Hoyumpa, 1980; Martin et al., 2003). Wernicke–Korsakoff syndrome (WKS) is a devastating neurological disorder characterized by oculomotor disturbances, ataxia, and mental confusion; it is believed that TD plays an important role in WKS pathogenesis (Harper, 2006). TD-induced WKS can be replicated in animal models using pyrithiamine alone or in combination with alcohol (He et al., 2007; Langlais et al., 1996; Zimitat et al., 1990). Chronic alcohol consumption is sometimes associated with WKS (Victor et al., 1989). The conventional idea is that WKS develops as a result of TD, and alcohol per se has no direct deleterious effects on the brain. The concept is challenged by subsequent studies in animals and humans showing that ethanol adversely affects brain structure and function probably due to direct neurotoxicity (Harper and Matsumoto, 2005; Ron, 1982). It is unclear whether the brain damage seen in chronic alcoholism is caused by one of these factors or by the association of both.
Alcohol causes neuronal loss in specific regions of the human brain, such as the superior frontal association cortex, hypothalamus, and cerebellum (Harper, 1998). The cerebellum is particularly susceptible to alcohol exposure, and chronic alcohol consumption is frequently accompanied by cerebellar degeneration (Baker et al., 1999; Martin et al., 2003). The shrinkage and degeneration of Purkinje cells in ethanol-dependent individuals is associated with ataxia of the lower limbs (Andersen, 2004; Sullivan et al., 2000). Interestingly, the degeneration of Purkinje cells is more marked in patients suffering from Wernicke’s encephalopathy (Baker et al., 1999). The etiology of alcoholic cerebellar degeneration is still a matter of debate. As the cerebellum is also sensitive to TD (Martin et al., 2003; Mulholland, 2006), some speculate that TD in alcoholics may be the cause of cerebellar damage (Charness, 1993; Diamond and Messing, 1994; Harper and Kril, 1990; Joyce, 1994; Maschke et al., 2005). However, animal experiments provide evidence of direct neurotoxicity of ethanol on cerebellar neurons (Fadda and Rossetti, 1998; Rajgopal et al., 2003; Tavares et al., 1987). Cerebellar atrophy may occur in alcoholics without malnourishment (Nicolás et al., 2000). The relationship between ethanol and TD and the contribution of their interaction to neurotoxicity remain to be elucidated.
The double-stranded RNA (dsRNA)-activated protein kinase (PKR) is a serine/threonine protein kinase ubiquitously expressed in mammalian cells (Williams, 1999, 2001). PKR is initially identified as an interferon-induced protein that is activated in virus-infected cells by dsRNA produced during the virus life cycle (Tan and Katze, 1999; Ung et al., 2001). PKR consists of 2 functionally distinct domains: a N-terminal dsRNA binding regulatory domain and a C-terminal catalytic domain. When PKR binds to dsRNA it causes PKR to form homodimers and to autophosphorylate on multiple serine/threonine residues including Thr446 and Thr451, resulting in PKR activation (Williams, 1999, 2001). In addition to dsRNA, PKR can be activated by cytokines, growth factors, serum deprivation, disruption of intracellular Ca2+ homeostasis, oxidative stress, and endoplasmic reticulum (ER) stress (Patel and Sen, 1998; Patel et al., 2000; Shimazawa and Hara, 2006; Wang et al., 2007a,b; Williams, 1999, 2001). Activation of PKR leads to an inhibition of protein synthesis, and sometimes apoptosis in the cells (Saelens et al., 2001; Williams, 1999, 2001). We have previously shown that TD activates PKR in vitro and in vivo (Wang et al., 2007a). In this study, we use an in vitro model, cultured cerebellar granule neurons (CGNs), to investigate the interaction between TD and ethanol and evaluate the contribution of their interaction to neuronal loss. We show that ethanol promotes TD-induced death of CGNs and that PKR plays an important role in the interaction.
MATERIAL AND METHODS
Animals and Reagents
Sprague–Dawley rats were obtained from Shanghai Laboratory Animals Co. Ltd (Shanghai, China). All animal experiments were performed in accordance with the Guideline of Animal Care and Use Committee of the Institute for Nutritional Sciences, Shanghai Institute for Biological Science (SIBS), Chinese Academy of Science (CAS). A specific inhibitor for PKR, 8-(imidazol-4-ylmethylene)-6H-azolidinol(5,4-g)benzothiazol-7-one, was purchased from Calbiochem (Cat. #527450; San Diego, CA). Chemicals were obtained from Sigma Chemical Co. (St Louis, MO) unless otherwise mentioned. Anti-α-tubulin antibody was purchased from Sigma Chemical Co. Other antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA).
Cultures of Cerebellar Granule Neurons
Cultures of CGNs were generated from 7-day-old rat pups using a previously described method (Wang et al., 2007b). Briefly, the rat pups were decapitated under deep anesthesia and cerebella removed. The cerebella were minced with a sterile razor blade and suspended in 10 ml of trypsin solution (0.025%) at 37°C. After incubation for 15 minutes, an equal volume of solution containing DNAse (130 Kunitz units/ml) and trypsin inhibitor (0.75 mg/ml) was added, and the tissue was sedimented by a brief (5 seconds) centrifugation. The tissue was dissociated by trituration, and the cell suspension was mixed with 4% bovine serum albumin and centrifuged. The cell pellet was resuspended in Neurobasal/B27 medium (Invitrogen, Carlsbad, CA) containing B27 (2%), KCl (25 mM), glutamine (1 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were plated into poly-D-lysine (50 μg/ml) coated cell culture wells or dishes, and maintained at 37°C in a humidified environment containing 5% CO2 for 7 days before the initiation of the experiment.
Induction of Thiamine Deficiency and Ethanol Exposure Protocol
Thiamine deficiency in CGNs was induced by the treatment of amprolium as described previously (Wang et al., 2007a). Briefly, after cultured in Neurobasal/B27 medium for 7 days, CGNs were treated with amprolium (0.5, 1, or 1.5 mM) for indicated times. Amprolium is a competitive inhibitor of thiamine transport and effectively depletes intracellular thiamine (Bettendorff et al., 1995; Park et al., 2000). We have verified that amprolium inhibited the activity of alpha ketoglutarate dehydrogenase complex and transketolase in cultured CGNs (Wang et al., 2007a).
To initiate ethanol exposure, the appropriate amount of ethanol (95% ethanol) was added directly to the culture medium in the multiwell cell culture trays. The trays were placed on a rack inside a sealed plastic container. A 200-ml bath containing the same ethanol concentration as the culture medium was present in the bottom of each container. The nonethanol control container only had a bath of water. A small volume of CO2 (60 ml) was injected into each container before sealing and all containers were incubated at 37°C. Although the ethanol containing bath was changed daily, no additional ethanol was added directly to the culture media. With this method, ethanol concentrations in the culture medium can be accurately maintained (Luo et al., 2001).
Determination of Cell Viability
Cell viability was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as previously described (Wang et al., 2007a). The assay is based on the cleavage of the yellow tetrazolium salt, MTT, to purple formazan crystals by metabolically active cells. Briefly, the cells were plated into 96-well microtiter plates and exposed to 6-hydroxydopamine (0 or 50 μM) for 48 hours. Following treatments, 10 μl of MTT-labeling reagent was added to each well and the plates were incubated at 37°C for 4 hours. The cultures were then solubilized and spectrophotometric absorbance of the samples was detected by a microtiter plate reader. The wavelength to measure absorbance of formazan product is 570 nm, with a reference wavelength of 750 nm.
Sample Preparation and Immunoblotting
Cytoplasmic and nuclear proteins were extracted and fractionated with a previously described method (Wang et al., 2007a). The procedure for immunoblotting has been previously described (Wang et al., 2007a). Aliquots of cytoplasmic or nuclear proteins (50 μg) were loaded into the lanes of a sodium dodecyl sulfate–polyacrylamide gel. The proteins were separated by electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk or 5% bovine serum albumin (for detection of phosphorylation) in 0.01 M phosphate-buffered saline (PBS) (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 hour. Subsequently, the membrane was incubated with primary antibodies directed against target proteins overnight at 4°C. The final dilutions for the primary antibodies were: PKR, 1:1000; p-PKR (Thr446), 1:1000; α-tubulin, 1:10000; Histone, 1:1000. After 2 quick washes in TPBS, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (Amersham, Arlington Heights, IL), diluted at 1:5000 in TPBS for 1 h. The immunocomplexes were detected by the enhanced chemiluminescence method (Amersham). The density of immunoblotting was quantified with the software of Quantity One (Bio-Rad Laboratories, Hercules, CA).
Immunofluorescence Staining
Immunocytofluorescent staining of p-PKR (Thr446) was performed as previously described (Wang et al., 2007a). CGNs cultured on cover slips were incubated with the primary antibody (1:500) overnight at 4°C, and the cells were then treated with a fluorescein isothiocyanate-labeled goat antirabbit IgG secondary antibody (1:500 dilution in PBS; Vector Laboratories Burlingame, CA). Nuclei were labeled with 4′, 6-dianidino-2-phenylindole dihydrochloride (1 μg/ml in PBS). Images of fluorescence were acquired using the Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss, Jena, Germany). The same settings for filters, pinhole size, stack size, and resolution were used for all captured images.
RESULTS
Ethanol Promotes TD-Induced Death of CGNs and PKR Phosphorylation
As shown in Fig. 1, TD reduced the viability of CGNs in a dose-dependent manner. At 1.5 mM, amprolium decreased the viability of CGNs by approximately 25%. This result was consistent with our previous finding (Wang et al., 2007a). TD-induced death of CGNs is in the form of apoptosis which has been previously confirmed by Annexin V expression and chromatin condensation/breakdown (Wang et al., 2007a,b). At 200 or 400 mg/dl, ethanol exposure alone did not cause significant cell death (Fig. 1). However, when TD was induced in CGNs in association with ethanol exposure, a much greater cell loss was observed. Treatment with amprolium (0.5 or 1 mM) in combination with ethanol (400 mg/dl) produced significantly more cell loss than amprolium treatment only. In the cultures that were treated with 1.5 mM amprolium, ethanol at 200 mg/dl significantly potentiated amprolium-induced cell death, and 400 mg/dl ethanol caused much more damage. Thus, the effect of ethanol was concentration-dependent.
Fig. 1.

Effect of thiamine deficiency (TD) and ethanol (EtOH) on the viability of cerebellar granule neurons (CGNs). CGNs were treated with amprolium (AMP; 0, 0.5, 1, or 1.5 mM) and ethanol (0, 200, or 400 mg/dl) for 5 days. The viability of cells was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) assay and expressed as a relative MTT reading. The experiment was replicated 3 times. *Denotes statistically significant difference from non-ethanol-treated control in the same TD group. #Denotes statistically significant difference from cultures treated with 200 mg/dl ethanol in the same TD group.
We have previously shown that TD caused PKR phosphorylation at Thr446 and Thr451, resulting in PKR activation (Wang et al., 2007a). We sought to determine whether ethanol affected TD-induced PKR phosphorylation. As the function of PKR is regulated by its activity as well as subcellular distribution, we examined the effect of TD and ethanol on the phosphorylation/expression of PKR in both the cytoplasm and nucleus. TD was induced by 1.5 mM amprolium treatment. As shown in Fig. 2, TD promoted p-PKR (Thr446) expression in the cytoplasm; the level of p-PKR (Thr446) increased after 6 hours of TD, and the high expression remained after 48 hours of TD. Ethanol exposure had little effect on PKR phosphorylation/expression (data not shown); however, it significantly potentiated TD-induced p-PKR (Thr446) expression. TD combined with ethanol exposure produced a stronger expression of p-PKR (Thr446), compared with TD alone (Fig. 2). TD also caused an increase in nuclear accumulation of p-PKR (Thr446), suggesting p-PKR (Thr446) translocated from the cytoplasm to the nucleus (Fig. 3). More importantly, ethanol significantly enhanced a TD-induced increase in nuclear p-PKR (Thr446) (Fig. 3). An immunocytochemical study confirmed ethanol’s potentiation of TD-induced nuclear localization of p-PKR (Thr446) (Fig. 4). In CGNs where TD and ethanol exposure were applied together, we observed more intense p-PKR (Thr446) expression in the nucleus compared with the cultures treated with TD only (Fig. 4B). We also examined the effect of TD and ethanol on p-PKR (Thr451), another PKR autophosphorylation site, and similar results were observed (data not shown).
Fig. 2.

Effect of thiamine deficiency (TD) and ethanol (EtOH) on the expression/phosphorylation of double-stranded RNA-activated protein kinase (PKR) in the cytoplasm. (A) Cerebellar granule neurons (CGNs) were treated with amprolium (0 or 1.5 mM) and ethanol (0 or 400 mg/dl) for indicated times. Cytoplasmic proteins were extracted, and the purity was confirmed by negative histone expression. The expression of PKR and p-PKR (Thr446) was determined with immunoblotting. The expression of tubulin served as an internal control. (B) The relative amounts of p-PKR (Thr446) were quantified microdensitometrically and normalized to the expression of tubulin. The experiment was replicated 3 times. *Denotes statistically significant difference from TD group at the same time point.
Fig. 3.
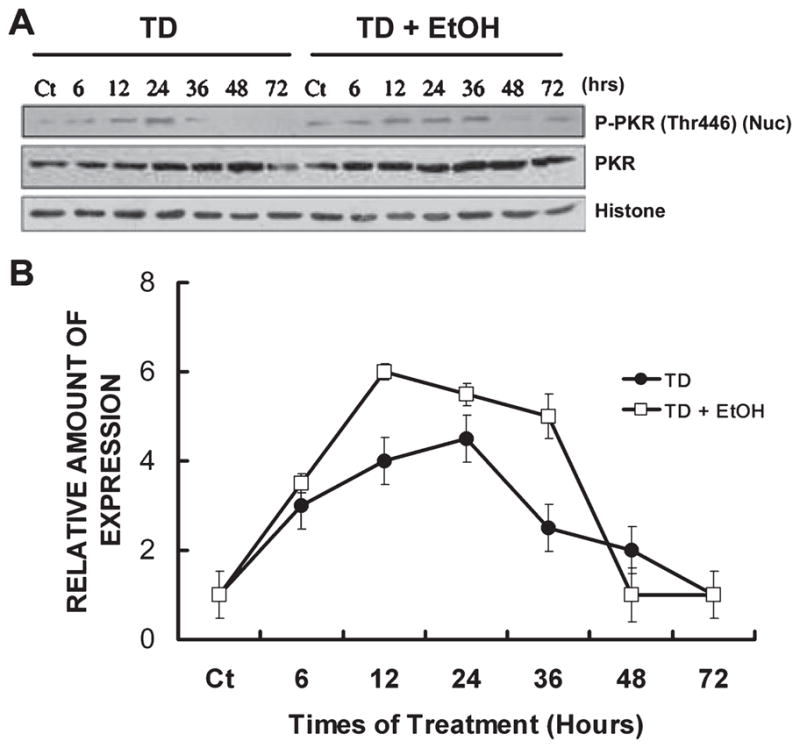
Effect of thiamine deficiency (TD) and ethanol (EtOH) on the expression/phosphorylation of double-stranded RNA-activated protein kinase (PKR) in the nucleus. Nuclear proteins were extracted, and the purity was confirmed by negative tubulin expression. The expression/phosphorylation of PKR in the nucleus was determined with immunoblotting. Notations are as in Fig. 2.
Fig. 4.

Immunocytochemical study of p-PKR (Thr446) expression/distribution. (A) Cerebellar granule neurons (CGNs) were treated with amprolium (0 or 1.5 mM) for 24 hours. (B) CGNs were treated with amprolium (1.5 mM) and ethanol (400 mg/dl) for indicated times. The expression of p-PKR (Thr446) (Green) was examined with immunocytochemistry as described under Materials and Methods. The nuclei (Blue) were visualized with 4′, 6-dianidino-2-phenylindole dihydrochloride (DAPI) staining. Bar = 5 μM. PKR, double-stranded RNA-activated protein kinase; TD, thiamine deficiency; EtOH, ethanol.
PKR Mediates the Effect of TD and Ethanol
Double-stranded RNA-activated protein kinase is an important modulator of apoptosis (Barber, 2005; Gil and Esteban, 2000). As TD activated PKR, we sought to determine the role of PKR in TD- and ethanol-induced death of CGNs by using a PKR inhibitor, 8-(imidazol-4-ylmethylene)- 6H-azolidinol[5,4-g]benzothiazol-7-one. This is an ATP-binding site-directed inhibitor of PKR that selectively blocks PKR activity (Jammi et al., 2003; Shimazawa and Hara, 2006). We have previously demonstrated that pretreatment with PKR inhibitor at 500 nM effectively blocked amprolium-induced PKR activation in cultured CGNs (Wang et al., 2007a). As shown in Fig. 5, treatment with PKR inhibitor not only protected CGNs against TD-induced cell death but also abolished ethanol’s potentiation of TD-mediated cell loss.
Fig. 5.

Effect of double-stranded RNA-activated protein kinase (PKR) inhibitor on thiamine deficiency (TD) and ethanol (Et)-induced death of cerebellar granule neurons (CGNs). CGNs were pretreated with a selective inhibitor of PKR (PKR-I, 0 or 500 nM) for 30 minutes and exposed to amprolium (0 or 1.5 mM) and ethanol (0 or 400 mg/dl) for 5 days. Cell viability was determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide assay as described under Material and Methods. The experiment was replicated 3 times. *Denotes statistically significant difference from untreated controls (Ct). #Denotes statistically significant difference from TD cultures.
DISCUSSION
Thiamine deficiency-mediated brain damage and ethanol-induced neurotoxicity may share some common mechanisms. For example, both insults cause mitochondrial malfunction, oxidative stress, and ER stress (Alexander-Kaufman et al., 2007; Chen et al., 2008; Fadda and Rossetti, 1998; Gibson et al., 2005; Gonthier et al., 1991; Ke and Gibson, 2004; Montoliu et al., 1994; Park et al., 2000, 2001; Singleton and Martin, 2001; Wang et al., 2007a,b). However, the interaction of TD and alcohol exposure and the contribution of this interaction to neurotoxicity remain to be elucidated.
We demonstrate that ethanol potentiates TD neurotoxicity on CGNs. Although ethanol alone did not produce significant cell loss, it dramatically enhanced TD-induced loss of CGNs. For example, in cultures treated with 1.5 mM amprolium, TD-induced loss of CGNs was potentiated by ethanol in a concentration-dependent manner (Fig. 1). A number of studies have investigated the interaction between TD and alcohol exposure. An animal study assessed the contribution of TD, associated or not with ethanol, in causing cortical and hippocampal cholinergic alterations and open-field behavioral impairment (Pires et al., 2001). TD decreased the acetylcholinesterase (AChE) activity both in the cortex and hippocampus of rats; ethanol treatment only reduced hippocampal enzyme activity. When rats were induced with brief TD associated with ethanol exposure, a greater reduction in cortical and hippocampal AChE activity was observed compared with ethanol treatment alone. Furthermore, TD and ethanol together resulted in more intense behavioral changes in some categories of open-field tests compared with treatment of either TD or ethanol alone (Pires et al., 2001). Similarly, Palencia and colleagues (1995) showed that both ethanol and TD alone caused similar and mild damage to the brain of rats, while the combination of these treatments led to much more pronounced neurological damage. Bâ and colleagues (1999) investigated the effect of developmental exposure to ethanol associated with TD. They demonstrated that ethanol and TD-induced cell atrophy (shrinkage of nucleus) and cell death in hippocampus CA3 pyramidal cells. Thiamine administration during developmental ethanol exposure partially restored the mean nuclear size. However, cell loss generated by ethanol treatment was not suppressed by thiamine administration. They therefore suggested that common and separate mechanisms underlined the effects of alcohol and TD on cell atrophy and cell death. Furthermore, a behavioral study revealed that TD yielded memory deficits in chicks and administration of thiamine reversed thememory deficit (Crowe and Kempton, 1997). A combination of TD and alcohol administration also resulted in memory deficits; in contrast to the effect of TD alone, the memory deficits induced by TD in association with alcohol cannot be reversed by thiamine resupplementation. Mulholland and colleagues (2005) used an in vitro model, organotypic cerebellar slice cultures to investigate the interaction of TD and ethanol withdrawal. They showed that TD modestly caused cerebellar cytotoxicity; neither ethanol exposure nor ethanol withdrawal induced cerebellar cytotoxicity. However, when TD and ethanol withdrawal were coapplied, a marked increase in cerebellar cytotoxicity was observed. In chronic alcoholics, a loss of cerebellar Purkinje cells and a decrease in molecular layer volume were more marked in patients suffering from Wernicke’s encephalopathy (Baker et al., 1999). Furthermore, atrophy of cerebellar vermis in alcoholics correlated with serum thiamine levels (Maschke et al., 2005). Together, these results suggest that TD and ethanol may act in a synergetic or additive pattern to produce more intense damage to the brain. The preexistent TD may make the subject susceptible to the neurotoxic effects of alcohol.
On the other hand, some results indicated that the interaction of ethanol consumption and TD was not always synergistic or additive. When rats were exposed to both ethanol and TD, some behavioral aspects appeared to be sensitive to the synergistic interaction between ethanol exposure and TD, whereas others were most affected by ethanol only; neurological symptoms were mostly associated with TD (Ciccia and Langlais, 2000). Therefore, common and separate mechanisms underlie the effects of alcohol intoxication and TD on the brain.
Double-stranded RNA-activated protein kinase has emerged as a potential mediator of neuronal death and is implicated in the neurodegeneration observed in Alzheimer’s disease (AD), Parkinson’s disease, and Huntington’s disease (Bando et al., 2005; Chang et al., 2002; Peel, 2004; Peel et al., 2001; Suen et al., 2003). We showed that ethanol promotes TD-induced PKR phosphorylation and its nuclear distribution. The activity of PKR was regulated by its phosphorylation on Thr446 and Thr451. In addition, the action of PKR depended on its subcellular distribution, and the nuclear localization of PKR was associated with neuronal apoptosis (Onuki et al., 2004). For example, phosphorylated PKR was detected in the nuclei of autopsied brain tissues in AD patients (Onuki et al., 2004). We demonstrated that blocking PKR activity by a selective inhibitor provided protection against TD neurotoxicity and abolished ethanol potentiation of TD-induced loss of CGNs. These results suggest that PKR may be a convergent protein and mediate TD and ethanol interaction.
It is likely that TD and ethanol activate PKR by triggering oxidative stress or ER stress in the cells because TD and ethanol are known to induce oxidative stress and ER stress (Calingasan et al., 1999; Chen et al., 2008; Pannunzio et al., 2000; Wang et al., 2007a,b). PKR can be activated by oxidative stress or ER stress through interaction with its protein activator PACT or RAX (Onuki et al., 2004; Shimazawa and Hara, 2006; Wang et al., 2007a). Regardless of how TD and ethanol activate PKR, our results provide 2 implications: first, preexistent TD may increase susceptibility to alcoholic cerebellar degeneration; second, PKR, as a convergent point of TD and ethanol’s action, is a potential therapeutic target for the treatment of TD and ethanol neurotoxicity.
Acknowledgments
We would like to thank Ms. Kimberly A. Bower for reading this manuscript. This research was supported by grants from the National Natural Science Foundation of China (30870812 and 30570580), the Ministry of Science and Technology of China (2007CB9471000), the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-YW-R-115), the Chief Scientist Program of Shanghai Institutes for Biological Sciences, the Chinese Academy of Sciences (SIBS2008006), and the Science and Technology Commission of Shanghai Municipality (07DJ14005). Dr. Z.-J.Ke was also supported by the One Hundred Talents Program of the Chinese Academy of Sciences and Shanghai Pujiang Program. Dr. J. Luo was supported by a grant from NIH/NIAAA(AA015407).
References
- Alexander-Kaufman K, Harper C, Wilce P, Matsumoto I. Cerebellar vermis proteome of chronic alcoholic individuals. Alcohol Clin Exp Res. 2007;31:1286–1296. doi: 10.1111/j.1530-0277.2007.00437.x. [DOI] [PubMed] [Google Scholar]
- Andersen BB. Reduction of Purkinje cell volume in cerebellum of alcoholics. Brain Res. 2004;1007:10–18. doi: 10.1016/j.brainres.2004.01.058. [DOI] [PubMed] [Google Scholar]
- Bâ A, Seri BV, Aka KJ, Glin L, Tako A. Comparative effects of developmental thiamine deficiencies and ethanol exposure on the morphometry of the CA3 pyramidal cells. Neurotoxicol Teratol. 1999;21:579–586. doi: 10.1016/s0892-0362(99)00014-8. [DOI] [PubMed] [Google Scholar]
- Baker KG, Harding AJ, Halliday GM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91:429–438. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem Int. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Barber GN. The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ. 2005;12:563–570. doi: 10.1038/sj.cdd.4401643. [DOI] [PubMed] [Google Scholar]
- Bettendorff L, Goessens G, Sluse F, Wins P, Bureau M, Laschet J, Grisar T. Thiamine deficiency in cultured neuroblastoma cells: effect on mitochondrial function and peripheral benzodiazepine receptors. J Neurochem. 1995;64:2013–2021. doi: 10.1046/j.1471-4159.1995.64052013.x. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Chun WJ, Park LC, Uchida K, Gibson GE. Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J Neuropathol Exp Neurol. 1999;58:946–958. doi: 10.1097/00005072-199909000-00005. [DOI] [PubMed] [Google Scholar]
- Chang RC, Suen KC, Ma CH, Elyaman W, Ng HK, Hugon J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J Neurochem. 2002;83:1215–1225. doi: 10.1046/j.1471-4159.2002.01237.x. [DOI] [PubMed] [Google Scholar]
- Charness ME. Brain lesions in alcoholics. Alcohol Clin Exp Res. 1993;17:2–11. doi: 10.1111/j.1530-0277.1993.tb00718.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res. 2008;86:937–946. doi: 10.1002/jnr.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia RM, Langlais PJ. An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcohol Clin Exp Res. 2000;24:622–634. [PubMed] [Google Scholar]
- Crowe SF, Kempton S. Both ethanol toxicity and thiamine deficiency are necessary to produce long-term memory deficits in the young chick. Pharmacol Biochem Behav. 1997;58:461–470. doi: 10.1016/s0091-3057(97)10001-6. [DOI] [PubMed] [Google Scholar]
- Diamond I, Messing RO. Neurologic effects of alcoholism. West J Med. 1994;161:279–287. [PMC free article] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;56:385–431. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Gastaldi G, Casirola D, Ferrari G, Rindi G. Effect of chronic ethanol administration on thiamine transport in microvillous vesicles of rat small intestine. Alcohol Alcohol. 1989;24:83–89. doi: 10.1093/oxfordjournals.alcalc.a044888. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5:107–114. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- Gonthier B, Jeunet A, Barret L. Electron spin resonance study of free radicals produced from ethanol and acetaldehyde after exposure to a Fenton system or to brain and liver microsomes. Alcohol. 1991;8:369–375. doi: 10.1016/0741-8329(91)90588-n. [DOI] [PubMed] [Google Scholar]
- Harper C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J Neuropathol Exp Neurol. 1998;57:101–110. doi: 10.1097/00005072-199802000-00001. [DOI] [PubMed] [Google Scholar]
- Harper C. Thiamine (vitamin B1) deficiency and associated brain damage is still common throughout the world and prevention is simple and safe. Eur J Neurol. 2006;13:1078–1082. doi: 10.1111/j.1468-1331.2006.01530.x. [DOI] [PubMed] [Google Scholar]
- Harper C, Kril JJ. The changing face of the Wernicke–Korsakoff syndrome. Drug Alcohol Rev. 1990;9:299–301. doi: 10.1080/09595239000185411. [DOI] [PubMed] [Google Scholar]
- Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5:73–78. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- He X, Sullivan EV, Stankovic RK, Harper CG, Pfefferbaum A. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology. 2007;32:2207–2216. doi: 10.1038/sj.npp.1301332. [DOI] [PubMed] [Google Scholar]
- Hoyumpa AM., Jr Mechanisms of thiamin deficiency in chronic alcoholism. Am J Clin Nutr. 1980;33:2750–2761. doi: 10.1093/ajcn/33.12.2750. [DOI] [PubMed] [Google Scholar]
- Jammi NV, Whitbv LR, Beal PA. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem Biophys Res Commun. 2003;308:50–57. doi: 10.1016/s0006-291x(03)01318-4. [DOI] [PubMed] [Google Scholar]
- Joyce EM. Aetiology of alcoholic brain damage: alcoholic neurotoxicity or thiamine malnutrition? Br Med Bull. 1994;50:99–114. doi: 10.1093/oxfordjournals.bmb.a072888. [DOI] [PubMed] [Google Scholar]
- Ke ZJ, Gibson GE. Selective response of various brain cell types during neurodegeneration induced by mild impairment of oxidative metabolism. Neurochem Int. 2004;45:361–369. doi: 10.1016/j.neuint.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX, Savage LM. Neuropathology of thiamine deficiency: an update on the comparative analysis of human disorders and experimental models. Metab Brain Dis. 1996;11:19–37. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- Luo J, Lindström CL, Donahue A, Miller MW. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J Neurochem. 2001;76:1354–1363. doi: 10.1046/j.1471-4159.2001.00129.x. [DOI] [PubMed] [Google Scholar]
- Martin PR, Singleton CK, Hiller-Sturmhöfel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27:134–142. [PMC free article] [PubMed] [Google Scholar]
- Maschke M, Weber J, Bonnet U, Dimitrova A, Bohrenkämper J, Sturm S, Müller BW, Gastpar M, Diener HC, Forsting M, Timmann D. Vermal atrophy of alcoholics correlate with serum thiamine levels but not with dentate iron concentrations as estimated by MRI. J Neurol. 2005;252:704–711. doi: 10.1007/s00415-005-0722-2. [DOI] [PubMed] [Google Scholar]
- Montoliu C, Vallés S, Renau-Piqueras J, Guerri C. Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: effect of chronic alcohol consumption. J Neurochem. 1994;63:1855–1862. doi: 10.1046/j.1471-4159.1994.63051855.x. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ. Susceptibility of the cerebellum to thiamine deficiency. Cerebellum. 2006;5:55–63. doi: 10.1080/14734220600551707. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Self RL, Stepanyan TD, Little HJ, Littleton JM, Prendergast MA. Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience. 2005;135:1129–1139. doi: 10.1016/j.neuroscience.2005.06.077. [DOI] [PubMed] [Google Scholar]
- Nicolás JM, Fernández-Solà J, Robert J, Antúnez E, Cofán M, Cardenal C, Sacanella E, Estruch R, Urbano-Márquez A. High ethanol intake and malnutrition in alcoholic cerebellar shrinkage. QJM. 2000;93:449–456. doi: 10.1093/qjmed/93.7.449. [DOI] [PubMed] [Google Scholar]
- Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K. RNA-dependent protein kinase is involved in tunicamycin- induced apoptosis and Alzheimer’s disease. EMBO J. 2004;23:959–968. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia G, Teixeira F, Ortiz A, Perez R, Sotelo J. Reversibility of the alterations induced by chronic alcoholism and malnutrition in rats after alcohol withdrawal and proper nutrition. J Stud Alcohol. 1995;56:140–146. doi: 10.15288/jsa.1995.56.140. [DOI] [PubMed] [Google Scholar]
- Pannunzio P, Hazell AS, Pannunzio M, Rao KV, Butterworth RF. Thiamine deficiency results in metabolic acidosis and energy failure in cerebellar granule cells: an in vitro model for the study of cell death mechanisms in Wernicke’s encephalopathy. J Neurosci Res. 2000;62:286–292. doi: 10.1002/1097-4547(20001015)62:2<286::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Park LC, Albers DS, Xu H, Lindsay JG, Beal MF, Gibson GE. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J Neurosci Res. 2001;66:1028–1034. doi: 10.1002/jnr.10062. [DOI] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Uchida K, Zhang H, Gibson GE. Metabolic impairment elicits brain cell type-selective changes in oxidative stress and cell death in culture. J Neurochem. 2000;74:114–124. doi: 10.1046/j.1471-4159.2000.0740114.x. [DOI] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL. PKR activation in neurodegenerative disease. J Neuropathol Exp Neurol. 2004;63:97–105. doi: 10.1093/jnen/63.2.97. [DOI] [PubMed] [Google Scholar]
- Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, Bredesen DE. Double-stranded RNA-dependent protein kinase, PKR binds preferentially to Huntington’s disease (HD) transcripts and its activated in HD tissue. Hum Mol Genet. 2001;10:1531–1538. doi: 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- Pires RG, Pereira SR, Pittella JE, Franco GC, Ferreira CL, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacol Biochem Behav. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Rajgopal Y, Chetty CS, Vemuri MC. Differential modulation of apoptosis- associated proteins by ethanol in rat cerebral cortex and cerebellum. Eur J Pharmacol. 2003;470:117–124. doi: 10.1016/s0014-2999(03)01795-3. [DOI] [PubMed] [Google Scholar]
- Ron MA. Syndromes of alcohol-related brain damage. Br Med Bull. 1982;38:91–94. doi: 10.1093/oxfordjournals.bmb.a071742. [DOI] [PubMed] [Google Scholar]
- Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem. 2001;276:41620–41628. doi: 10.1074/jbc.M103674200. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Hara H. Inhibitor of double stranded RNA-dependent protein kinase protects against cell damage induced by ER stress. Neurosci Lett. 2006;409:192–195. doi: 10.1016/j.neulet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- Singleton CK, Martin PR. Molecular mechanisms of thiamine utilization. Curr Mol Med. 2001;1:197–207. doi: 10.2174/1566524013363870. [DOI] [PubMed] [Google Scholar]
- Suen KC, Yu MS, So KF, Chang RC, Hugon J. Upstream signaling pathways leading to the activation of double-stranded RNA-dependent serine/threonine protein kinase in beta-amyloid peptide neurotoxicity. J Biol Chem. 2003;278:49819–49827. doi: 10.1074/jbc.M306503200. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Deshmukh A, Desmond JE, Lim KO, Pfefferbaum A. Cerebellar volume decline in normal aging, alcoholism, and Korsakoff’s syndrome: relation to ataxia. Neuropsychology. 2000;14:341–352. doi: 10.1037//0894-4105.14.3.341. [DOI] [PubMed] [Google Scholar]
- Tan SL, Katze MG. The emerging role of the interferon-induced PKR protein kinase as an apoptotic effector: a new face of death? J Interferon Cytokine Res. 1999;19:543–545. doi: 10.1089/107999099313677. [DOI] [PubMed] [Google Scholar]
- Tavares MA, Paula-Barbosa MM, Cadete-Leite A. Chronic alcohol consumption reduces the cortical layer volumes and the number of neurons of the rat cerebellar cortex. Alcohol Clin Exp Res. 1987;11:315–319. doi: 10.1111/j.1530-0277.1987.tb01315.x. [DOI] [PubMed] [Google Scholar]
- Ung TL, Cao C, Lu J, Ozato K, Dever TE. Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR. EMBO J. 2001;20:3728–3737. doi: 10.1093/emboj/20.14.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M, Davis RD, Collins GH. The Wernicke–Korsakoff Syndrome and Related Neurological Disorders Due to Alcoholism and Malnutrition. F. A. Davis Company; Philadelphia, PA: 1989. [Google Scholar]
- Wang X, Fan Z, Wang B, Luo J, Ke ZJ. Activation of double-stranded RNA-activated protein kinase by mild impairment of oxidative metabolism in neurons. J Neurochem. 2007a;103:2380–2390. doi: 10.1111/j.1471-4159.2007.04978.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang BW, Fan ZQ, Shi XL, Ke ZJ, Luo J. Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience. 2007b;144:1045–1056. doi: 10.1016/j.neuroscience.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR. PKR: a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- Williams BR. Signal Integration via PKR. Sci STKE. 2001;89:RE2. doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- Zimitat C, Kril J, Harper CG, Nixon PF. Progression of neurological disease in thiamin-deficient rats is enhanced by ethanol. Alcohol. 1990;7:493–501. doi: 10.1016/0741-8329(90)90038-e. [DOI] [PubMed] [Google Scholar]