

Abstract
Influx of calcium is an essential but insufficient signal in sustained nutrient-stimulated insulin secretion, and increased metabolic rate of the beta cell is also required. The aim of the study was to test the hypothesis that the reduced state of cytochrome c is a metabolic co-factor necessary for insulin secretion, over and above its participation in the ATP-generating function of electron transport/oxidative phosphorylation. We found that nutrient stimulation of insulin secretion by isolated rat islets was strongly correlated with reduced cytochrome c, and agents that acutely and specifically reduced cytochrome c led to increased insulin secretion, even in the face of decreased oxygen consumption and calcium influx. In contrast, neither sites 1 nor 4 of the electron transport chain were both necessary and essential for the stimulation of insulin secretion to occur. Importantly, stimulation of islets with glucose, α-ketoisocaproate, or glyceraldehyde resulted in the appearance of cytochrome c in the cytosol, suggesting a pathway for the regulation of exocytotic machinery by reduction of cytochrome c. The data suggest that the metabolic factor essential for sustained calcium-stimulated insulin secretion to occur is linked to reduction and translocation of cytochrome c.
Keywords: Calcium, Cytochrome c, Electron Transport, Insulin Secretion, Pancreatic Islet
Introduction
It is well established that a major determinant in the development of Type 2 diabetes is failure of pancreatic beta cells to compensate for increased demand for insulin secretion (1, 2). It follows that there is a critical need to understand the fundamental mechanisms regulating insulin secretion. An essential factor for sustained insulin secretory response to glucose is an increase in calcium (Ca2+) influx via L-type Ca2+ channels (3–6). This increased Ca2+ influx occurs when the metabolism of glucose generates ATP, leading to the closure of ATP-sensitive potassium (KATP) channels and the resultant opening of voltage-dependent L-type Ca2+ channels (7). However, it is recognized that increased Ca2+ in the absence of the elevated metabolic rate does not elicit second-phase insulin secretion (8, 9). For example, at low glucose, agents that increased Ca2+ influx such as potassium (K+) (10), tolbutamide, or arginine (11) elicited only a rapid transient response in insulin secretion that quickly waned. Thus, there are additional co-factors generated when substrates are metabolized that in concert with Ca2+ are essential for a physiologic response of insulin secretion.
These factors have been long sought after as reviewed (12–14) and a number of candidates have been investigated including products of pyruvate cycling (NADPH and α-ketoglutarate) (15), anaplerosis (16), as well as KATP channel-independent affects of ATP/ADP (17) and long chain acyl-CoA (12). Since we first began measuring reduced cytochrome c in rat islets (18), we noticed that sustained insulin secretion never occurred unless accompanied by an increase in this parameter. An increase in cytochrome c reduction occurred in response to changes in metabolic substrates that can independently increase insulin secretion such as glucose and α-ketoisocaproate (KIC),2 but not with potentiators of nutrient-stimulated insulin secretion such as GLP-1, arginine, and acetylcholine that do not have an affect on ISR in the absence of nutrients. Notably, cytochrome c reduction did not change in response to agents that altered the oxygen consumption rate (OCR) due to changes in workload (18, 19), an intuitively requisite attribute for a control element that would faithfully transduce the glucose signal. Thus, we reasoned that cytochrome c reduction behaves like an ideal signal transduction mechanism supporting the function of the beta cell as a glucose sensor that would be independent of metabolic need by intracellular processes. Cytochrome c is the only protein in the electron transport chain (ETC) that is mobile, and is a known regulatory protein representing a committed step in apoptosis by binding to APAF-1 (20). In addition, its reductive state is linked to the generation of reactive oxygen species (21, 22) and mitochondrial pH (23), factors thought to stimulate ISR. Based on these data we hypothesized that the reduction of cytochrome c is a metabolic co-factor necessary for Ca2+-mediated insulin secretion, over and above its participation in the ATP-generating function of ETC/oxidative phosphorylation.
In addition to testing this hypothesis, in this study, we endeavored to elucidate where in the stimulus-secretion coupling pathway reduced cytochrome c might act. Our predictions of how the essential metabolic factor could facilitate Ca2+-stimulated insulin secretion were based on a conceptual model we developed over that past 5 years. The primary data supporting the model includes the findings that glucose-stimulated Ca2+ influx is associated with about 35% of total glucose-stimulated OCR, a measurement that was tightly correlated with ISR (19). These findings suggested that Ca2+-sensitive OCR reflects a highly energetic process that couples Ca2+ to the secretion of insulin (6). In lieu of the identification of the proteins involved with this process, we termed this process the Ca2+/metabolic coupling process (CMCP) (24). Further studies revealed that potentiation of the insulin secretion rate (ISR) by protein kinase C (PKC) and cAMP-dependent pathways occur with only very small changes in OCR, demonstrating that activation of insulin secretion by these mechanisms is mediated by steps that are downstream of the CMCP (6). Based on the findings that increased Ca2+ influx did not stimulate ISR at subthreshold concentrations of glucose (25), we predicted that the CMCP and ISR would be dually regulated upstream by both Ca2+ and the metabolic co-factor reduced cytochrome c as shown in Fig. 1.
FIGURE 1.

Hypothesized role of reduced cytochrome c in mediating glucose-stimulated insulin secretion. We previously defined a highly energetic, Ca2+-sensitive process that couples the influx of Ca2+ to the release of insulin secretion and denoted it as the calcium-metabolic coupling process or CMCP (24). We hypothesize that the CMCP is dually controlled by Ca2+ influx through L-type Ca2+ channels and a factor related to reduced cytochrome c. The model predicts that Ca2+-sensitive OCR and ISR are only activated when both calcium influx is stimulated and cytochrome c is reduced.
To test our predictions, we compared responses by isolated rat islets to the stimulation of various secretagogues, metabolic poisons, and other agents selected to separate the proposed signaling function of reduced cytochrome c from its established role in the mitochondrial generation of ATP. Measured parameters included reduced cytochrome c and cytochrome c oxidase, OCR, NAD(P)H, Ca2+ influx, and ISR assessed in real time. We subsequently tested the possibility that reduced cytochrome c interacts with cytosolic processes mediating exocytosis by its translocation from the mitochondria to the cytosol.
EXPERIMENTAL PROCEDURES
Chemicals
KRB was used for the perifusion analyses, prepared as described previously (19). Potassium cyanide (KCN), KIC, N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD), S(−)-BayK 8644, sodium ascorbate, nimodipine, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and glyceraldehyde were purchased from Sigma.
Rat Islet Isolation and Culture
Islets were harvested from Sprague-Dawley rats (≈250 g, Charles River) anesthetized by intraperitoneal injection of sodium pentobarbital (35 mg/230 g of rat). All procedures were approved by the University of Washington Institutional Animal Care and Use Committee. Islets were prepared and purified as described (18, 26), and then cultured at 37 °C in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) for 18 h prior to the experiments.
Measurement of OCR, Cytochromes, and ISR
A flow culture system was used that concomitantly measures OCR, cytochrome c, and cytochrome c oxidase reduction, while collecting outflow fractions for subsequent measurement of ISR (described previously (18, 27, 28)). OCR was calculated as the flow rate (∼80 μl/min) times the difference between in-flow and out-flow oxygen tension and measured as described previously (28) except that a different system was used to detect oxygen tension. In the present study, oxygen tension was measured by detecting the phosphorescence lifetime of an oxygen-sensitive dye painted on the inside of the perifusion chamber using a MFPF-100 multifrequency phase fluorometer lifetime measurement system made by TauTheta Instruments (Boulder, CO). The sensor was excited and detected by a single 2-mm light fiber (TauTheta number SFO-026) that bifurcated with one fiber illuminated by a 405-nm light emitting diode and the other connected to the phosphorometer. The detection/excitation end of the light guide was positioned so it was just touching the outside of the glass perifusion chamber opposite where the dye was painted. Because TMPD/ascorbate interfered with the oxygen-sensitive dye use in the flow system, a Seahorse XF24 was used to measure its affect as per the manufacturer's instructions (Seahorse Bioscience Inc., North Billerica, MA).
Reduction of cytochrome c was measured as absorbance at 550 nm by the layer of intact islets in the perifusion chamber as previously described (18). Reduced cytochrome c oxidase in islets was measured similarly to cytochrome c reduction by light transmission through the column of islets using a Tungsten/Halogen 780 Lamp (NRC; Newport, Irvine, CA), a spectrometer (Ocean Optics USB2000, Dunedin, FL), and two 600-μm jacketed optical fibers. Integration times were 25−150 ms, and 55 signals were averaged per spectrum and ported into a laptop computer (Dell Latitude D610, Round Rock TX; Excel software, Microsoft, Redmond, WA). After each experiment, calibration spectra for fully oxidized and reduced cytochromes were acquired with 12 μg/ml of antimycin A followed by 3 mm KCN. Baseline drift was virtually eliminated by using the second derivative of absorbance with respect to wavelength (29) at 605 nm calculated as Equation 1,
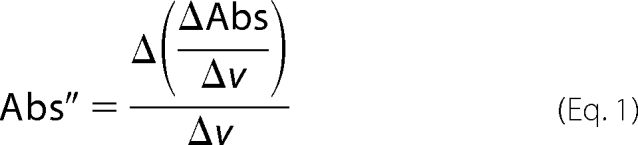 |
where Abs = log (intensity − intensitybkg)/(intensityref − intensitybkg), v = wavelength in nanometers, and Δ = change in the variable over the integration interval. Calculations were made using Excel (Microsoft). Background intensity (intensitybkg) was determined with the light source off, and the reference intensity (intensityref) was that obtained when cytochrome c was fully oxidized by antimycin A. Absorbance spectra were smoothed by a moving average in 10-nm intervals (15 points before and after a particular wavelength before calculating the second derivative). Percent reduction of cytochrome c oxidase (cytcoxred) was calculated following Kashiwagura et al. (30) as Equation 2,
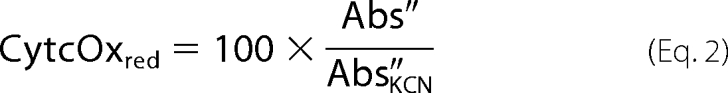 |
where Abs″ and Abs″KCN are values at 605 nm, and Abs″KCN is obtained in the presence of KCN when cytochrome c oxidase is fully reduced. Insulin was measured using an RIA kit (Linco Research Inc.).
Imaging and Quantification of Cytosolic Ca2+ and NAD(P)H
Cytosolic Ca2+ was measured by fluorescence imaging of islets after loading them with Fura-2/AM (Invitrogen) as previously described (24). Dyed islets were pipetted into a temperature-controlled, 250-μl perifusion dish (Bioptechs, Butler, PA) mounted onto the stage of a Nikon Eclipse TE-200 inverted microscope, and KRB (containing 5 mm NaHCO3) was pumped through the dish at a flow rate of 150 μl/min. Fluorescent emission was detected at 510 nm by a Photometrics Cool Snap EZ camera (Tucson, AZ) during alternating excitation at either 340 or 380 nm. Results are displayed as the ratio of the fluorescent intensities during excitation at these two wavelengths (F340/F380). NAD(P)H autofluorescence was measured similarly to Ca2+, except there was no need for loading with dye and the excitation and emission wavelengths were 360 and 460 nm, respectively. To calibrate the relative fluorescence units (RFU), at the end of the experiments the steady-state RFU in the presence of KCN and subsequently FCCP was measured. The normalized fluorescence of NAD(P)H was then calculated relative to RFUFCCP set to 0, and RFUKCN set to 100% (24).
Measurement of Ca2+ Influx
Ca2+ influx was measured by first preincubating islets in KRB containing 3 mm glucose for 60 min in a 5% CO2 incubator at 37 °C. Subsequently, islets were picked into 12 × 75 test tubes containing 90 μl of KRB (with 0.5 mm Ca2+) and the indicated additions (except 20 mm glucose) and incubated for 30 min, and then 10 μl of 45Ca2+ (1 μCi) was added by use of a repeater pipette. For islets being exposed to 20 mm glucose, the supplemental glucose was added immediately (within 1 min) after the 45Ca2+ so that the Ca2+ influx measurement would include the initial stimulation by high glucose. At about 14 min after addition of 45Ca2+, the islet suspension was transferred to 0.4-ml centrifuge tubes containing 100 μl of an oil mixture, and then the free and the cell-associated radioactivity were separated at exactly 15 min by spinning the islets through the oil layer and counting, as described previously (31).
Cytochrome c Appearance in Cytosol by Western Analysis
Cytochrome c and cytochrome c oxidase in the cytosol and mitochondria were detected using the ApoAlert Cell Fractionation Kit (Clontech, Mountain View, CA) per the manufacturer's instructions. Islets were incubated in 24-well plates (100/well) in 500 μl of KRB containing 3 mm glucose for 4 h (to allow the effects of high glucose to decay away), and then test compounds were added to wells as indicated. After specified times (typically 15 min), 480 μl of supernatant was removed (leaving 20 μl), and 1 ml of ice-cold wash buffer was added and after the islets were settled, the wash buffer was replaced with 250 μl of ice-cold homogenization buffer. After 10 min on ice, the content of each tube was transferred to a Dounce tissue homogenizer, and homogenized with 1 pass of the pestle. The homogenates were transferred to microcentrifuge tubes and spun down at 700 × g for 10 min to remove plasma membranes and nucleus. Supernatant was removed (230 μl), placed into another microcentrifuge tube, and spun at 10,000 × g for 25 min. The remaining material, representing the mitochondrial fraction, was resuspended in 100 μl of buffer. The supernatant representing the cytosolic fraction was again transferred to new tubes (200 μl). The protein was measured in both fractions using a Pierce BCA protein kit (ThermoFisher Scientific, Rockford, IL). Samples were then boiled in SDS and 30 μg of protein was loaded into 4–20% gel lanes (Bio-Rad). Western blots were analyzed for cytochrome c and cytochrome c oxidase subunit IV using the antibodies supplied with the kit. Bands were quantified using ImageJ (NIH). Lack of appearance of cytochrome c oxidase in the cytosolic fraction demonstrated that the cytosol was purified without contamination by mitochondria.
Data Analysis
All data were displayed as the average ± S.E. of at least 3 separate perifusions or experiments. Statistical significance was determined using paired t tests calculated with Excel (Microsoft). For perifusion data, statistical significance was determined on changes in steady-state values calculated as the average of the parameter for the final 15 min at that condition.
RESULTS
Pharmacologic Activation of Ca2+ Influx-stimulated ISR and OCR at High but Not Low Glucose
To demonstrate the essentiality of a metabolic co-factor for Ca2+ to stimulate ISR, we compared the effect of an activator of L-type Ca2+ channels (10 μm BayK 8644) at 20 versus 3 mm glucose on isolated rat islets (Fig. 2). In the presence of 20 mm glucose, exposure to BayK 8644 increased cytosolic Ca2+, OCR, and ISR by 83 ± 18, 18 ± 4, and 180 ± 14%, respectively, compared with 20 mm glucose alone (Fig. 2A and Table 1), consistent with previous observations that ATP turnover and ISR are tightly coupled to increased influx of Ca2+ (6, 19). In the presence of subthreshold concentrations of glucose (3 mm), BayK 8644 also potently increased cytosolic Ca2+ (by 92% compared with 20 mm glucose effect) but, in contrast to 20 mm glucose where cytochrome c was highly reduced, it had no effect on either OCR or ISR (Fig. 2B). Thus, increased Ca2+ influx alone is insufficient to stimulate ISR or OCR, and demonstrates that the metabolic factor needed for Ca2+ to stimulate ISR is acting at a step preceding Ca2+-sensitive OCR (i.e. the CMCP). Note that no first phase response of ISR was seen when glucose was changed from 3 to 20 mm. This is due to the use of low flow rates that are needed to resolve OCR. As a result, glucose changes resemble a ramp, a waveform that does not induce first phase insulin secretion and leads to longer times before a plateau is reached (32).
FIGURE 2.

Effect of increasing Ca2+ influx at 20 mm (A) and 3 mm (B) glucose on cytochrome c reduction, OCR, Ca2+, and ISR. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 μm BayK 8644 was added to the in-flow after (A) or before (B) glucose (glc) concentration was raised to 20 mm for 45 min. Third from the top, detection of cytosolic Ca2+ by fluorescence imaging (measured in separate experiments). Top, second from top and bottom, OCR, cytochrome c reduction, and ISR were measured concomitantly using the flow culture system. Data are displayed as the change in signal relative to the steady-state value obtained at 3 mm glucose (determined by averaging data obtained in the final 15 min prior to the increase in glucose). Steady-state values of cytochrome c, OCR, and ISR at 3 mm glucose were 28 ± 2.7% (n = 7), 0.31 ± 0.057 nmol/min/100 islets (n = 8), and 0.12 ± 0.038 ng/min/100 islets (n = 10), respectively. Statistical analysis was carried out by comparing steady-state values (determined by averaging data obtained in the final 15 min of each experimental condition) before and after each change in medium composition using a paired t test.
TABLE 1.
The effect of substrates and agents on changes in steady-state values of OCR, ISR, cytochrome c reduction, and NAD(P)H in rat pancreatic islets relative to the baseline (in the presence of 3 mm glucose)
Data were taken from Figs. 2–6, and each steady-state value shown was the average of the final 15 min of the indicated condition. Statistical analysis was carried out by comparing steady-state values before and after each change in medium composition using a paired t test. Abbreviations not defined in text: Ant A, antimycin A; Asc, ascorbate; Cyt c ox, cytochrome c oxidase; Glyc, glyceraldehyde.
| Process affected | Conditions | ΔOCR | ΔISR | ΔCytochrome c reduction | ΔNAD(P)H or Δcyt c ox reductiona |
|---|---|---|---|---|---|
| nmol/min/100 islets | ng/min/100 islets | % | |||
| Effect of increased Ca2+ influx in high glucose (Fig. 2A) | Condition 1: 20 mm Glc | +0.41 ± 0.032 (n = 4, p < 0.005) | +2.3 ± 0.35 (n = 6, p < 0.005) | +35 ± 4.3 (n = 4, p < 0.005) | NDb |
| Condition 2: 20 mm Glc +10 μm BayK 8644 | +0.074 ± 0.017 (n = 4, p < 0.05) | +3.1 ± 0.72 (n = 6, p < 0.01) | +5.2 ± 3 (n = 4) | ND | |
| Effect of increased Ca2+ influx in low glucose (Fig. 2B) | Condition 1: 3 mm Glc + 10 μm BayK 8644 | +0.025 ± 0.007 (n = 4, p < 0.05) | −0.02 ± 0.07 (n = 4) | −1.2 ± 0.31 (n = 3) | ND |
| Condition 2: 20 mm Glc + 10 μm BayK 8644 | +0.49 ± 0.038 (n = 4, p < 0.001) | +5.0 ± 1.0 (n = 4, p < 0.05) | +43 ± 1.9 (n = 3, p < 0.005) | ND | |
| Effect of increased TCA cycle activity in low glucose (Fig. 3A) | Conditon 1: 10 mm KIC | +0.33 ± 0.074 (n = 6, p < 0.005) | +2.3 ± 0.47 (n = 6, p < 0.005) | +28 ± 4.1 (n = 5, p < 0.005) | +25 ± 1.7 (n = 3, p < 0.005) |
| Condition 2: 10 mm KIC + 5 μm nimodipine | −0.097 ± 0.031 (n = 6, p < 0.005) | −2.5 ± 0.57 (n = 6, p < 0.01) | −6.4 ± 4.2 (n = 5) | −7.0 ± 1.0 (n = 3, p < 0.05) | |
| Effect of increased ETC in low glucose (Fig. 3B) | Cond 1: 10 mm Glyc | +0.13 ± 0.018 (n = 8, p < 0.005) | +0.9 ± 0.19 (n = 5, p < 0.005) | +15 ± 7.6 (n = 6, p < 0.05) | −7.0 ± 3.2 (n = 5) |
| Condition 2:10 mm Glyc + 5 μm nimodipine | −0.13 ± 0.011 (n = 8, p < 0.005) | −1.0 ± 0.17 (n = 5, p < 0.005) | −20 ± 7.2 (n = 6, p < 0.005) | +11 ± 4.8 (n = 5) | |
| Effect of reducing cytochrome c oxidase in low Glc (Fig. 4A) | Condition 1: 0.1 mm TMPD, 5 mm Asc | +0.40 ± 0.063c (n = 13, p < 0.005) | +0.39 ± 0.12 (n = 4) | +7.7 ± 7.0 (n = 4) | +94 ± 1.5a (n = 4, p < 0.0001) |
| Condition 2: 0.1 mm TMPD /5 mm Asc + 5 μm nimodipine | +0.094 ± 0.049c (n = 13) | +0.073 ± 0.031 (n = 4) | −4.7 ± 3.0 (n = 4) | −18 ± 12a (n = 4) | |
| Effect of reducing cytochrome c oxidase in high Glc (Fig. 4C) | Condition 1: 20 mm glucose | +0.26 ± 0.04 (n = 16, p < 0.0001) | +1.6 ± 0.1 (n = 5, p < 0.0001) | +37 ± 9.6 (n = 6, p < 0.01) | 7.5 ± 2.1 (n = 6, p < 0.01) |
| Condition 2: 20 mm Glc + TMPD/Asc (at 90 min) | +0.51 ± 0.06 (n = 16, p < 0.0001) | +0.96 ± 0.18 (n = 5, p < 0.01) | +4.0 ± 3.8 (n = 6) | 41.9 ± 8.7 (n = 6, p < 0.005) | |
| Effect of reducing cytochrome c with KCN (Fig. 5A, inset) | Condition 1: 20 mm Glc | +0.43 ± 0.016 (n = 6, p < 0.005) | +1.3 ± 0.13 (n = 6, p < 0.005) | +38 ± 6.4 (n = 6, p < 0.005) | ND |
| Condition 2: 20 mm Glc +1 μm KCN | −0.034 ± 0.009 (n = 6, p < 0.05) | +0.70 ± 0.24 (n = 6, p < 0.05) | +6.9 ± 1.0 (n = 6, p < 0.005) | ND | |
| Effect of oxidizing cytochrome c with antimycin A (Fig. 5A, inset) | Condition 1: 20 mm Glc | +0.50 ± 0.062 (n = 3, p < 0.05) | +2.2 ± 0.42 (n = 3, p < 0.05) | +41 ± 2.6 (n = 3, p < 0.005) | ND |
| Condition 2: 20 mm Glc + 0.6 ng/ml Ant A | −0.024 ± 0.002 (n = 3, p < 0.01) | +0.13 ± 0.2 (n = 3) | −1.4 ± 1.8 (n = 3) | ND | |
| Effect of glucose stimulation and no further additions (Fig. 5A) | Condition 1: 20 mm glucose | +0.36 ± 0.02 (n = 12, p < 0.0001) | +2.0 ± 0.2 (n = 6, p < 0.0005) | +42 ± 2.5 (n = 12, p < 0.0001) | ND |
| Condition 2: 20 mm Glc (at time = 90) | −0.006 ± 0.007 (n = 12) | +0.31 ± 0.23 (n = 6) | +2.1 ± 1.4 (n = 12) | ND | |
| Effect of reducing cytochrome c with KCl in high Glc/diazoxide (Fig. 6A) | Condition 1: 20 mm Glc | +0.28 ± 0.024 (n = 8, p < 0.005) | +0.25 ± 0.075 (n = 5, p < 0.05) | +28 ± 3.9 (n = 6, p < 0.005) | ND |
| Condition 2: 30 mm KCl + 20 mm Glc | +0.12 ± 0.014 (n = 8, p < 0.005) | +4.2 ± 0.66 (n = 5, p < 0.01) | +15 ± 3.2 (n = 6, p < 0.05) | ND | |
| Effect of increasing cytochrome c reduction with KCl at low Glc/diazoxide (Fig. 6D) | Cond 1: 30 mm KCl + 3 mm glucose | +0.051 ± 0.004 (n = 6, p < 0.0001) | +0.48 ± 0.14 (n = 4, p < 0.05) | +8.7 ± 4.4 (n = 6, p < 0.05) | ND |
| Condition 2: 20 mm Glc + 30 mm KCl | +0.27 ± 0.018 (n = 6, p < 0.0001) | +1.9 ± 0.4 (n = 4, p < 0.05) | +24.8 ± 9.0 (n = 6, p < 0.005) | ND | |
a Cytochrome c oxidase reduction was measured and not NAD(P)H.
b ND, not determined.
c OCR measured by the Seahorse was normalized by dividing by the OCR obtained at 3 mm glucose and is unitless.
Reduction of Cytochrome c Common to All Agents Able to Independently Stimulate ISR
In addition to glucose, nutrient secretagogues independently able to elicit sustained ISR at below 5 mm glucose are fairly limited and include KIC, glyceraldehyde, and leucine, agents that enter metabolic pathways at different points. To identify characteristics in beta cell response common to all the nutrient secretagogues, each agent or combination of agents was tested for its ability to stimulate OCR, NAD(P)H, cytochrome c reduction, Ca2+ influx, and ISR. The response of these parameters to 10 mm KIC, a fuel that enters the TCA cycle without traversing glycolysis, was very similar to the responses to 20 mm glucose (Fig. 3A, Table 1). The subsequent Ca2+-sensitive component of OCR (i.e. the decrease in OCR elicited by the L-type Ca2+-channel blocker nimodipine, about 30% of glucose-stimulated OCR) was also similar to that seen in previous studies of glucose-stimulated islets (19, 24). Notably, reduced cytochrome c was insensitive to changes in OCR induced by blocking Ca2+ influx, demonstrating that regulation of dehydrogenases by Ca2+ observed in some tissues (33) was not driving changes in Ca2+-sensitive OCR. Because KIC is a mitochondrial fuel that does not increase glycolytic rate (34), glycolysis cannot be generating metabolic factors coupling Ca2+ influx to ISR. Note that the responses of Ca2+ and NADH were more rapid than OCR and cytochrome c reduction, but this was due to the faster response time of the imaging system compared with our flow culture system. In general, the resolution of the flow culture system is limited to about 5 min. The responses of OCR, cytochrome c reduction, and ISR to 10 mm leucine in the presence of 2 mm glutamine were similar to the responses to KIC (data not shown). Like glucose and KIC, glyceraldehyde (Fig. 3B) also increased cytochrome c reduction and OCR, reflecting the well established requirement for mitochondrial generation of ATP. The major difference between responses to the four agents was the lack of an increase in NAD(P)H elicited by glyceraldehyde, suggesting that the metabolic co-factor, if linked to the ETC, would be downstream of site 1.
FIGURE 3.

Effect of nutrient secretogogues (KIC and glyceraldehyde) and a blocker of l-type Ca2+ channels on NAD(P)H, cytochrome c reduction, OCR, cytosolic Ca2+, and ISR. Islets were perifused in the presence of 3 mm glucose (glc) for 90 min; at time = 0, 10 mm KIC (A) or 10 mm glyceraldehyde (B) was added for 45 min; subsequently, L-type Ca2+ channel activity was inhibited by nimodipine (5 μm) for 45 min. Top and fourth panel from top, detection of NAD(P)H and Ca2+ by fluorescence imaging. Second and third panel from the top and bottom panels: cytochrome c reduction, OCR, and ISR were measured concomitantly using the flow culture system. Data are displayed and analyzed as described in the legend of Fig. 2. For KIC studies, steady-state values of NAD(P)H, cytochrome c reduction, OCR, and ISR at 3 mm glucose were 8.6 ± 2.1 (n = 3), 23 ± 3.9 (n = 5), 0.35 ± 0.056 nmol/min/100 islets (n = 6), and 0.31 ± 0.061 ng/min/100 islets (n = 6), respectively. For the glyceraldehyde experiments, steady-state values of NAD(P)H, cytochrome c reduction, OCR, and ISR at 3 mm glucose were 17 ± 2.7 (n = 5), 22 ± 4.6 (n = 6), 0.39 ± 0.064 nmol/min/100 islets (n = 5), and 0.19 ± 0.095 ng/min/100 islets (n = 8), respectively.
Thus, it appeared that all agents that independently stimulated ISR also increased cytochrome c reduction. To test whether cytochrome c reduction had any functional significance over and above its role in supplying electrons to cytochrome c oxidase, we measured the response to an artificial electron donor TMPD/ascorbate. Following exposure of islets to 0.1 mm TMPD and 5 mm ascorbate, cytochrome c oxidase but not cytochrome c was reduced (Fig. 4A). Thus, TMPD/ascorbate appeared to represent an appropriate tool for investigating the affect of activation of the ETC downstream of cytochrome c. Despite an increase in cytochrome c oxidase reduction accompanied by increases in OCR (Fig. 4A) and Ca2+ influx (Fig. 4B) that were similar to or greater than those seen in response to glucose, ISR was only slightly increased (less than 25 and 10% of the responses to glyceraldehyde and glucose, respectively). In the presence of 20 mm glucose, TMPD/ascorbate also lead to increased OCR and cytochrome c oxidase, and in contrast to the results at 3 mm glucose, ISR was significantly increased (Fig. 4C, Table 1). The stimulatory effect of TMPD/ascorbate on ISR rules out the possibility that TMPD/ascorbate inhibited ISR at 3 mm glucose by an off-target effect or toxicity. Consistent with this, no changes in ISR or reduced cytochrome c were seen in response to ascorbate alone (data not shown). Unfortunately TMPD, being an optically active molecule, was not easily evaluated. In the absence of ascorbate, TMPD absorbs at both 550 and 605 nm and eclipsed the cytochrome signal. It did not increase OCR or ISR but without the cytochrome measurements, it is not possible to evaluate the hypothetical role of reduced cytochrome c on ISR. In fact, in vitro cytochrome c is reduced by the presence of TMPD/ascorbate (data not shown), so it is not clear why only cytochrome c oxidase is reduced in intact islets (Fig. 4A). So the mechanism for the affect of TMPD/ascorbate on cytochrome c oxidase but not cytochrome c is not clear.
FIGURE 4.

Effect of stimulation of the ETC at the level of cytochrome c oxidase and subsequent blockade of l-type Ca2+ channels. A, cytochrome c reduction, cytochrome c oxidase reduction, OCR, and ISR were measured in response to exposure to TMPD/ascorbate (0.1/5 mm) applied for 90 min at 3 mm glucose (glc), followed by the addition of 5 mm nimodipine. Because TMPD/ascorbate interfered with the oxygen-sensitive dye used in the flow experiments, for these OCR measurements, a Seahorse XF 24 was used. For reference, the OCR responses to an increase in glucose and subsequently nimodipine are shown (n = 6). Steady-state values of cytochrome c reduction and ISR at 3 mm glucose were 21 ± 10% (n = 4) and 0.17 ± 0.017 ng/min/100 islets (n = 4), respectively; cytochrome c oxidase was set to 0 at 3 mm glucose due to the difficulty of measuring a difference between this value and that in the presence of the antimycin A that is used for calibration. Due to non-uniform distribution of islets around the oxygen-sensitive dye in the well, well-to-well variation in the OCR data were normalized by dividing it by that obtained at 3 mm glucose. 70 islets were used for each well. 13 wells are included for average (filled circles). B, Ca2+ influx was measured in response to activation by 20 mm glucose, TMPD/ascorbate (0.1 mm/5 mm), 10 mm KIC, and 10 mm glyceraldehyde, in the absence or presence of nimodipine (5 mm) using radioactive 45Ca2+ and normalized with the 3 mm glucose (glc) effect in each experiment. Statistical significance compared with control or 20 mm glucose or TMPD, is indicated by either a single asterisk for p < 0.05 or a double asterisk for p < 0.01. C, cytochrome c reduction, cytochrome c oxidase reduction, OCR, and ISR were measured as described for A, except that TMPD/ascorbate was added after an increase of glucose as indicated. Steady-state values of cytochrome c reduction and ISR at 3 mm glucose were 18.4 ± 3.4% (n = 6) and 0.23 ± 0.02 ng/min/100 islets (n = 5), respectively.
Nonetheless, because reduced cytochrome c oxidase, OCR, and calcium influx all increased, it does appear that the classical regulatory arm of the metabolism-induced Ca2+ influx is operational in response to TMPD/ascorbate (similar to that of glucose). However, at low glucose the effects of these agents on cytochrome c oxidase and oxidative phosphorylation did not generate a metabolic co-factor capable of appreciably stimulating ISR, supporting the assertion that cytochrome c reduction is essential for the sustained elevation of ISR. Moreover, neither the increase in OCR nor the small increase in ISR were inhibited by nimodipine, despite complete inhibition of the affect of TMPD/ascorbate on Ca2+ influx (Fig. 4B), indicating that these agents do not stimulate the CMCP or the normal mechanism mediating Ca2+-sensitive secretion of insulin. It is noteworthy that in contrast to reduced cytochrome c, the change in cytochrome c oxidase reduction was only 7.5% in response to 20 mm glucose (Fig. 4C, Table 1), suggesting that cytochrome c is uniquely poised as a barometer of nutrient levels.
Elevated Cytochrome c Reduction by Low Levels of KCN or Mild Hypoxia Enhanced ISR
To further establish the role of reduced cytochrome c, we sought to test its relationship to ISR more broadly under a variety of conditions. The next approach was to test the effects of slight inhibition of ETC downstream of cytochrome c, at the level of cytochrome c oxidase. We postulated that additional reduction of cytochrome c by exposure of islets in 20 mm glucose to low levels of KCN due to inhibition of complex 4 would result in increased ISR even in the face of decreased Ca2+ influx and OCR. As a control, results were compared with the affects of antimycin A, an inhibitor of ETC at a site upstream of cytochrome c, at the level of complex 3. As expected, KCN increased, and antimycin A decreased, cytochrome c reduction, in a dose-dependent manner (Fig. 5A). Remarkably, in response to 1 μm KCN, we observed a 50% increase in ISR that coincided with increases in cytochrome c reduction (Fig. 5A, inset). In contrast, antimycin A (0.6 ng/ml) at a concentration that had similar decreases in OCR to 1 μm KCN, had only a small impact on ISR, which was not significantly different from glucose-stimulated ISR in the absence of added agent (Table 1 and Fig. 5A). These low concentrations of the two inhibitors resulted in only small (but significant) differences in steady-state OCR relative to the pre-addition baseline (96 ± 0.9% (p < 0.01, n = 6) for KCN, 97 ± 0.6% (p < 0.05, n = 3) for antimycin A, compared with 99 ± 1.1% (n = 6) for the control) and Ca2+ influx was slightly decreased (Fig. 5B). As such, the only difference in the state of the islets in the presence of the each inhibitor was the reductive state of cytochromes c1, c, and cytochrome c oxidase.
FIGURE 5.

Effect of KCN-induced increase in the reductive state of cytochrome c on ISR and OCR (A) and Ca2+ influx (B). A, islets were perifused in the presence of 3 mm glucose (glc) for 90 min; at time = 0, glucose was raised to 20 mm. After 45 min, islets were exposed to incrementally increasing levels of either KCN (1, 3 or 10 μm) or antimycin A (0.6, 1.8, or 6 ng/ml). Cytochrome c reduction, OCR, and ISR were measured concomitantly using the flow culture system. Steady-state values of cytochrome c reduction, OCR, and ISR at 3 mm glucose with KCN were 22 ± 3.2% (n = 6), 0.37 ± 0.035 nmol/min/100 islets (n = 6), and 0.25 ± 0.054 ng/min/100 islets (n = 6), respectively. Steady-state values of cytochrome c reduction, OCR, and ISR at 3 mm glucose with antimycin A were 25 ± 6.5% (n = 3), 0.42 ± 0.095 nmol/min/100 islets (n = 3), and 0.31 ± 0.036 ng/min/100 islets (n = 3), respectively. Steady-state values of cytochrome c reduction, OCR, and ISR at 3 mm glucose alone were 44.8 ± 3.8% (n = 3), 0.23 ± 0.03 nmol/min/100 islets (n = 6), and 0.12 ± 0.04 ng/min/100 islets (n = 6), respectively. As inclusion of two-sided error bars on individual data points made it difficult to distinguish the individual curves, we have shown just one-sided error bars. Inset, statistical comparison of effects of KCN relative to antimycin A at the lowest concentrations on cytochrome c and ISR. B, Ca2+ influx was measured in response to activation by 20 mm glucose, in the absence or presence of either 1 μm KCN or 0.6 ng/ml of antimycin A by using radioactive 45Ca2+ and normalized with the 3 mm glucose effect in each experiment. If statistically significant compared with 20 mm glucose then *, p < 0.05; **, p < 0.01.
Quantitatively similar results (ISR increased by about 50%) were obtained in response to a decrease in oxygen tension in the medium from 140 to 105 mm Hg that should mimic the action of KCN to block complex 4 (data not shown). Thus, both KCN and mild hypoxia resulted in a concurrent increase in reduced cytochrome c and insulin secretion, making it unlikely that the increase in ISR was a result of an off-target effect.
Increased Cytochrome c Reduction by K+ Occurred Concomitantly with OCR and ISR
As a final test of the correlation between reduction of cytochrome c and ISR, we assessed the effect of K+. Previous experiments done while maintaining a constant level of Ca2+ influx (in the presence of diazoxide and KCl) revealed a KATP-independent effect of glucose (10, 35). It has always been assumed that the sole affect of K+ on beta cells is electrophysiologic, which occurs in the absence of changes in the metabolism (36–38). However, we found that K+ increased cytochrome c reduction and OCR above the effects of 20 mm glucose (Fig. 6A) while having no effect on NAD(P)H (data not shown). This is unlikely to be mediated by the effect of K+ on Ca2+ influx because the sulfonylurea glibenclamide, which elicits Ca2+ influx by activating L-type Ca2+ channels via closing KATP channels, had no effect on cytochrome c reduction (18). Therefore, there must be additional effects of K+ over and above the effect on depolarization of the plasma membrane, but it does not appear to be related to an increase in glycolysis or the TCA cycle because no increase in NAD(P)H was seen, a factor typically increased by these two pathways. To test whether experiments done under these conditions would support a role of reduced cytochrome c as a critical mediator of KATP-independent enhancement of ISR, the affect of K+ in the presence of diazoxide and 20 mm glucose on ISR was compared with 20 mm glucose alone. These two conditions lead to a similar increase in Ca2+ influx (Fig. 6B), whereas K+ had about 3 times greater affect on ISR (Fig. 6C). Like BayK 8644, at low glucose, K+ increased Ca2+ (Fig. 6D). However, in contrast to BayK 8644, in the presence of 3 mm glucose, K+ had a small but significant effect on ISR, cytochrome c reduction, and OCR (Fig. 6D). Subsequent exposure to 20 mm glucose increased cytochrome c reduction and ISR in the absence of a significant increase in cytosolic Ca2+. Thus, it appears that the potent affect of K+ on ISR is partly due to its ability to reduce cytochrome c.
FIGURE 6.

Effect of K+ (30 mm) on cytochrome c reduction, OCR, and ISR and Ca2+ influx, in the presence of 50 μm diazoxide and 20 mm glucose. A, islets were perifused in the presence of 3 mm glucose (glc) and 50 μm diazoxide for 90 min. Diazoxide was present for the duration of the protocol. At time = 0, glucose was raised to 20 mm for 45 min; subsequently, K+ was raised from 5 to 30 mm (at the same time Na+ was lowered by 25 mm). Cytochrome c reduction, OCR, and ISR were measured concomitantly using the flow culture system. Data are displayed and analyzed as described in the legend of Fig. 2. Steady-state values of cytochrome c reduction, OCR, and ISR at 3 mm glucose were 25 ± 4.2% (n = 6), 0.45 ± 0.080 nmol/min/100 islets (n = 8), and 0.12 ± 0.045 ng/min/100 islets (n = 5), respectively. Normalized 45Ca2+ influx (B) and static ISR (C) were measured in response to activation by 20 mm glucose, or 30 mm KCl, in the absence or presence of diazoxide (50 μm). Statistical significance compared with 20 mm glucose or 20 mm glucose plus diazoxide were denoted with: *, p < 0.05, and **, p < 0.01. D, islets were perifused as described in A except that K+ was raised from 5 to 30 mm prior to switching to 20 mm glucose. Steady-state values of cytochrome c reduction, OCR, and ISR at 3 mm glucose were 26.6 ± 2.1% (n = 6), 0.20 ± 0.02 nmol/min/100 islets (n = 6), and 0.03 ± 0.02 ng/min/100 islets (n = 4), respectively.
Increased Nutrients Led to an Increase in Cytosolic Cytochrome c
Having demonstrated that nutrient secretagogues lead to the reduction of cytochrome c, we hypothesized that for cytochrome c to interact with exocytotic machinery residing near or at the plasma membrane, cytochrome c might translocate from the mitochondria to the cytosol. The appearance of cytochrome c in the cytosol was measured in response to nutrient secretagogues glucose, KIC, and glyceraldehyde by subcellular fractionation and subsequent analysis by Western blot. All three agents induced the appearance of cytochrome c in the cytosol within 15 min of adding the agents (Fig. 7). Remarkably, the glucose-stimulated appearance of cytosolic cytochrome c was inhibited by blocking Ca2+ influx (Fig. 7). Density of bands representing cytosolic cytochrome c were less than 20% of mitochondrial cytochrome c, and no changes in mitochondrial cytochrome c were detectable in response to nutrients (data not shown). Thus, ISR, reduction, and translocation of cytochrome c are highly correlated, but only translocation and insulin secretion are dependent on Ca2+ influx.
FIGURE 7.

Effect of nutrient secretagogues on appearance of cytochrome c in cytosol. Islets (100 per condition) were incubated in the presence of 3 mm glucose in KRB for 4 h and 45 min, before adding the indicated agents and incubating further for 15 min. Islets were then lysed and 30 mg of cytosolic protein were loaded into SDS gels as described under “Experimental Procedures.” Cytochrome c and cytochrome c oxidase subunit IV were detected using antibodies supplied in the kit and densitometry was used to quantify the intensity of the bands. Statistics were carried out comparing the effects of nutrient additions relative to control conditions, or the effect of nimodipine relative to 20 mm glucose, using paired t tests where, * denotes p < 0.05, and ** denotes p < 0.01. No cytochrome c oxidase bands were seen in the cytosolic fractions validating the fractionation process (data not shown).
DISCUSSION
Ca2+ Influx Does Not Independently Stimulate ISR but Requires the Action of a Factor Generated in the Metabolism of Glucose
It is commonly stated and depicted schematically that a rise in cytosolic Ca2+ levels leads to secretion of insulin. However, as recognized by others (38), and as we have recapitulated here in this study using an activator of L-type Ca2+ channels (BayK 8644), additional factors generated from the metabolism of nutrients are required for Ca2+ to induce sustained insulin secretion. Because glucose-stimulated ISR does not occur appreciably in the absence of Ca2+ influx through L-type Ca2+ channels, the mechanisms mediating it must be under dual control by both Ca2+-sensitive processes and factors generated by increased metabolic activity. It is well established that generation of ATP by the ETC and oxidative phosphorylation is a key process leading to closure of KATP channels and opening of L-type Ca2+ channels (7). Data obtained in the present study using substrates and inhibitors of various steps in the metabolic machinery suggest that reduced cytochrome c is an essential regulatory signal allowing Ca2+ to stimulate the secretion of insulin, over and above its participation in the ETC. This was initially suggested by measuring reduction of cytochrome c in response to all nutrients that independently stimulate ISR, although increasing the ETC downstream of cytochrome c (by the artificial electron donors TMPD/ascorbate) resulted in increased OCR and Ca2+ influx, but had little effect on ISR. The regulation of ISR by reduced cytochrome c was further supported by demonstrating increased ISR in response to reducing cytochrome c either by inhibiting steps downstream of cytochrome c (with KCN or hypoxia) or by action on the reductive state of cytochrome c by K+ at similarly elevated levels of Ca2+ influx. To summarize, under all conditions carried out in this study where Ca2+ influx was stimulated, the steady-state values of reduced cytochrome c and ISR were strongly correlated (Fig. 8). Remarkably, we found that nutrient secretagogues (glucose, KIC, and glyceraldehyde) elevated cytosolic levels of cytochrome c via a Ca2+-dependent pathway, providing an explanation of how cytochrome c could interact with exocytotic machinery located in the cytosol.
FIGURE 8.

Summary of steady-state levels of reduced cytochrome c versus ISR under conditions where Ca2+ influx is elevated. Legend for conditions: 1, BayK 8644/3 mm Glc (Fig. 2B); 2, 3 mm Glc, 0.1 mm TMPD, 5 mm ascorbate (Fig. 4A); 3, 30 mm K+, 3 mm Glc (Fig. 6D); 4, 10 mm glyceraldehyde, 3 mm Glc (Fig. 3B); 5, 10 mm KIC, 3 mm Glc (Fig. 3A); 6, 20 mm Glc (Fig. 2A); 7, 30 mm K+, 20 mm Glc/diazoxide (Fig. 6A). Note that these are absolute values not changes as are shown in the graphs.
Consideration of Approach
A major challenge in testing whether cytochrome c reduction has a signaling role in addition to its role as an electron donor to cytochrome c oxidase is altering its state while controlling or assessing the other regulatory parameters such as ATP production and Ca2+ influx. This situation makes it difficult to harness molecular approaches because any change in cytochrome c would strongly influence both roles of cytochrome c, making it difficult to separate their effects. Consequently, we chose to test a variety of agents that acutely affected different steps in metabolism and ETC to investigate the relationship between reduced cytochrome c and ISR. The most compelling of these agents were the presence of low levels of KCN and mild hypoxia, where ISR was increased even in the face of decreased Ca2+ influx and OCR. These experimental results meet the classical criteria of a control step where a specific and physiological change in the candidate variable (reduced cytochrome c) led to time-dependent changes in cell function (ISR). It is true that no inhibitors are without off-target effects, but the corroboration of increased ISR in response to reduced cytochrome c with both KCN and hypoxia strongly suggests it is caused by the common mechanisms of action of these agents.
Two of the agents used to increase cytochrome c reduction (K+ and glyceraldehyde) acted by mechanisms that are not clear from the data obtained. Nonetheless, they were still useful tools to demonstrate that cytochrome c reduction always accompanied stimulated ISR under conditions of widely different rates of glycolysis, the TCA cycle, and levels of NAD(P)H. The question arises whether other metabolic correlates thought to play a role in KATP-independent stimulation of ISR could equally well explain this data. A number of candidates were considered including the ATP/ADP ratio (38), and factors associated with anaplerosis such as NADPH (16, 39–41). Although ATP/ADP was not measured, it is highly unlikely that the increase in ISR and reduced cytochrome c observed in response to low levels of KCN were mediated by an increase in this ratio, especially in light of the decrease in OCR and Ca2+ influx that occurred. Likewise for anaplerosis, it seems that it would be unlikely to explain the KCN induced increase in ISR (relative to antimycin A) unless cytochrome c (or cytochrome c oxidase) directly regulated anaplerosis.
In the presence of glyceraldehyde, reduction of cytochrome c occurred without a measurable increase in autofluorescent NAD(P)H. Because levels of NADPH and NADH are on the same order of magnitude (42, 43), the lack of change in autofluorescent NAD(P)H suggests that NADPH does not correlate with the stimulation of ISR by glyceraldehyde.
Thus, the metabolic factors most commonly reported to contribute to KATP-independent ISR do not appear to explain the strong link between reduced cytochrome c and ISR. Eventually, a systematic comparison of reduced cytochrome c to other mediators will be important to carry out. However, the goal of this study was to show the critical importance of reduced cytochrome c as a KATP channel-independent signal. Future studies will need to be carried out affirming that relationships between cytochrome c reduction and ISR occurring in rat islets are also observed in human islets.
Potential Mechanisms of Action by Cytosolic Cytochrome c
It has been previously well established that cytochrome c functions as a signaling molecule in addition to its role of donating electrons to cytochrome c oxidase. During apoptosis, it binds to other regulatory proteins after translocating from the mitochondria to the cytosol (44). Under a wide variety of conditions tested, translocation of cytochrome c reflects the irreversible transition to an apoptotic state (45). That cytochrome c can appear in the cytosol under conditions that do not lead to beta cell death is interesting. Given the precedent of how cytochrome c functions to regulate apoptosis, it is plausible that it could translocate and bind to a different set of proteins involved with secretion. Evidence supports a scenario where much of the control of Ca2+-stimulated ISR may occur in a microdomain in the vicinity of L-type Ca2+ channels (4, 46), and it might be that cytosolic cytochrome c might translocate into this cellular region. There have been other reports that suggest that binding and translocation may be dependent on the redox state of the cytochrome c (47, 48). Supporting this notion, high Ca2+ (49), K+ (50), and ATP (51) have all been found to prevent cytosolic cytochrome c from activating caspase-9, conditions that are likely to be met in nutrient-stimulated beta cells. A major determinant of cytochrome c binding could just be the amount of this protein in the cytosol, because high levels overcame inhibition of ATP binding to APAF-1 (51). At this point, we have only shown a correlation between ISR and cytosolic cytochrome c, but if ISR were dependent on translocated cytochrome c, it might provide an explanation for previous findings that pro- and anti-apoptotic proteins that decrease or increase the translocation of cytochrome c have a reciprocal affect on ISR. For example, beta cell-specific overexpression of Bcl-xL (an anti-apoptotic protein that suppresses translocation of cytochrome c) in mice leads to a diabetic phenotype characterized by decreased ISR despite increased beta cell survival (52). The finding that the translocation is dependent on Ca2+ influx lends more credence to the involvement of cytochrome c translocation in mediating ISR. Indeed, the release of cytochrome c from mitochondria through the mitochondrial permeability transition pore is sensitive to Ca2+ (53) and is therefore possible that the requirements for both Ca2+ and reduced cytochrome c for stimulation of ISR could be mediated at this site. This theory is particularly attractive as it would explain the Ca2+-sensitive component of glucose-stimulated OCR (reflected by the CMCP) by mitochondrial permeability transition pore-mediated uncoupling.
Integration of Cytochrome c into a Conceptual Model of Insulin Secretion
Past studies conducted in our laboratory have focused on correlations between five fundamental islet parameters: Ca2+ influx, cytochrome c reduction, Ca2+-sensitive OCR (reflecting the operation of the CMCP), cytochrome c translocation, and ISR. Our data indicates that the first two parameters both have to be elevated for the final three to occur, suggesting signals from cytochrome c reduction and Ca2+ interact upstream to activate the latter processes. It is typically stated that an increase in Ca2+ in the cytosol leads to stimulation of ISR (38, 54). However, our model places equal emphasis on the metabolic factor (i.e. cytochrome c reduction) and Ca2+ influx, because both are essential for an increase in ISR to occur. An important feature of this model is the stability of reduced cytochrome c except in response to nutrient secretagogues. We have done experiments with many different preparations of islets and the reduction of cytochrome c in the presence of a given concentration of glucose is remarkably invariable (55), intuitively a requirement for it to play a role in the transduction of extracellular nutrient signals. Indeed, no changes in cytochrome c reduction occur in response to changes in Ca2+ turnover, fatty acids, protein synthesis, and various activators of protein kinases including acetylcholine, GLP-1, and arginine, even when these agents result in large changes in OCR (6, 19).3 That flux of electrons through the ETC can vary without a change in the number of electrons bound to cytochrome c suggests that mechanisms operate in the beta cell to actively maintain constant levels of the reductive state of cytochrome c. How or if the translocation of cytochrome c regulates ISR is not clear from our data, and we cannot at this point be sure that translocated (reduced) cytochrome c does not become oxidized once it gets into the cytosol, but we favor a mechanism where cytochrome c translocation interacts with downstream steps, possibly protein kinases, that determines the rate of insulin secretion.
This work was supported, in whole or in part, by National Institutes of Health Grant DK17047 (a DERC Islet Core and an R24 seed grant supplement) and National Science Foundation Grant IIP-0750508.
S.-R. Jung, I. T. D. Kuok, and I. R. Sweet, unpublished data.
- KIC
- α-ketoisocaproate
- CMCP
- Ca2+/metabolic coupling process
- ETC
- electron transport chain
- ISR
- insulin secretion rate
- K+
- potassium
- KATP
- ATP-sensitive potassium channel
- KCN
- potassium cyanide
- OCR
- oxygen consumption rate
- TMPD
- N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- RFU
- relative fluorescence unit.
REFERENCES
- 1. Prentki M., Nolan C. J. (2006) J. Clin. Invest. 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leahy J. L. (2005) Arch. Med. Res. 36, 197–209 [DOI] [PubMed] [Google Scholar]
- 3. Grodsky G. M., Bennett L. L. (1966) Diabetes 15, 910–913 [DOI] [PubMed] [Google Scholar]
- 4. Bokvist K., Eliasson L., Ammälä C., Renström E., Rorsman P. (1995) EMBO J. 14, 50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Satin L. S., Tavalin S. J., Kinard T. A., Teague J. (1995) Endocrinology 136, 4589–4601 [DOI] [PubMed] [Google Scholar]
- 6. Gilbert M., Jung S. R., Reed B. J., Sweet I. R. (2008) J. Biol. Chem. 283, 24334–24342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cook D. L., Hales C. N. (1984) Nature 311, 271–273 [DOI] [PubMed] [Google Scholar]
- 8. Henquin J. C. (2009) Diabetologia 52, 739–751 [DOI] [PubMed] [Google Scholar]
- 9. Prentki M. (1996) Eur. J. Endocrinol. 134, 272–286 [DOI] [PubMed] [Google Scholar]
- 10. Gembal M., Detimary P., Gilon P., Gao Z. Y., Henquin J. C. (1993) J. Clin. Invest. 91, 871–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishiyama N., Ravier M. A., Henquin J. C. (2006) Am. J. Physiol. Endocrinol. Metab. 290, E540–549 [DOI] [PubMed] [Google Scholar]
- 12. Deeney J. T., Prentki M., Corkey B. E. (2000) Semin. Cell Dev. Biol. 11, 267–275 [DOI] [PubMed] [Google Scholar]
- 13. Ishihara H., Wollheim C. B. (2000) IUBMB Life. 49, 391–395 [DOI] [PubMed] [Google Scholar]
- 14. Bratanova-Tochkova T. K., Cheng H., Daniel S., Gunawardana S., Liu Y. J., Mulvaney-Musa J., Schermerhorn T., Straub S. G., Yajima H., Sharp G. W. (2002) Diabetes 51, Suppl. 1, S83–90 [DOI] [PubMed] [Google Scholar]
- 15. Jensen M. V., Joseph J. W., Ronnebaum S. M., Burgess S. C., Sherry A. D., Newgard C. B. (2008) Am. J. Physiol. Endocrinol. Metab. 295, E1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacDonald M. J., Fahien L. A., Brown L. J., Hasan N. M., Buss J. D., Kendrick M. A. (2005) Am. J. Physiol. Endocrinol. Metab. 288, E1–15 [DOI] [PubMed] [Google Scholar]
- 17. Henquin J. C., Gembal M., Detimary P., Gao Z. Y., Warnotte C., Gilon P. (1994) Diabetes Metab. 20, 132–137 [PubMed] [Google Scholar]
- 18. Sweet I. R., Cook D. L., DeJulio E., Wallen A. R., Khalil G., Callis J., Reems J. (2004) Diabetes 53, 401–409 [DOI] [PubMed] [Google Scholar]
- 19. Sweet I. R., Gilbert M. (2006) Diabetes 55, 3509–3519 [DOI] [PubMed] [Google Scholar]
- 20. Schafer Z. T., Kornbluth S. (2006) Dev. Cell. 10, 549–561 [DOI] [PubMed] [Google Scholar]
- 21. Leloup C., Tourrel-Cuzin C., Magnan C., Karaca M., Castel J., Carneiro L., Colombani A. L., Ktorza A., Casteilla L., Pénicaud L. (2009) Diabetes 58, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pi J., Zhang Q., Fu J., Woods C. G., Hou Y., Corkey B. E., Collins S., Andersen M. E. (2010) Toxicol. Appl. Pharmacol. 244, 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wiederkehr A., Park K. S., Dupont O., Demaurex N., Pozzan T., Cline G. W., Wollheim C. B. (2009) EMBO J. 28, 417–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung S. R., Reed B. J., Sweet I. R. (2009) Am. J. Physiol. Endocrinol. Metab. 297, E717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malaisse-Lagae F., Mathias P. C., Malaisse W. J. (1984) Biochem. Biophys. Res. Commun. 123, 1062–1068 [DOI] [PubMed] [Google Scholar]
- 26. Matsumoto S., Shibata S., Kirchhof N. (1999) Transplantation 67, S220 [Google Scholar]
- 27. Sweet I. R., Cook D. L., Wiseman R. W., Greenbaum C. J., Lernmark A., Matsumoto S., Teague J. C., Krohn K. A. (2002) Diabetes Technol. Ther. 4, 67–76 [DOI] [PubMed] [Google Scholar]
- 28. Sweet I. R., Khalil G., Wallen A. R., Steedman M., Schenkman K. A., Reems J. A., Kahn S. E., Callis J. B. (2002) Diabetes Technol. Ther. 4, 661–672 [DOI] [PubMed] [Google Scholar]
- 29. Cavinato A. G., Mayes D. M., Ge Z. H., Callis J. B. (1990) Anal. Chem. 62, 1977–1982 [DOI] [PubMed] [Google Scholar]
- 30. Kashiwagura T., Wilson D. F., Erecińska M. (1984) J. Cell Physiol. 120, 13–18 [DOI] [PubMed] [Google Scholar]
- 31. Sweet I. R., Cook D. L., Lernmark A., Greenbaum C. J., Wallen A. R., Marcum E. S., Stekhova S. A., Krohn K. A. (2004) Biochem. Biophys. Res. Commun. 314, 976–983 [DOI] [PubMed] [Google Scholar]
- 32. Sweet I. R., Li G., Najafi H., Berner D., Matschinsky F. M. (1996) Am. J. Physiol. Endocrinol. Metab. 271, E606–E625 [DOI] [PubMed] [Google Scholar]
- 33. Denton R. M., McCormack J. G. (1985) Am. J. Physiol. Endocrinol. Metab. 249, E543–E554 [DOI] [PubMed] [Google Scholar]
- 34. Gao Z., Young R. A., Li G., Najafi H., Buettger C., Sukumvanich S. S., Wong R. K., Wolf B. A., Matschinsky F. M. (2003) Endocrinology 144, 1949–1957 [DOI] [PubMed] [Google Scholar]
- 35. Aizawa T., Sato Y., Komatsu M., Hashizume K. (1992) Endocr. Regul. 26, 159–162 [PubMed] [Google Scholar]
- 36. Malaisse W. J., Picton S., Malaisse-Lagae F., Sener A. (1999) Biochim. Biophys. Acta. 1451, 255–262 [DOI] [PubMed] [Google Scholar]
- 37. Ravier M. A., Gilon P., Henquin J. C. (1999) Diabetes 48, 2374–2382 [DOI] [PubMed] [Google Scholar]
- 38. Henquin J. C. (2000) Diabetes 49, 1751–1760 [DOI] [PubMed] [Google Scholar]
- 39. Farfari S., Schulz V., Corkey B., Prentki M. (2000) Diabetes 49, 718–726 [DOI] [PubMed] [Google Scholar]
- 40. Fransson U., Rosengren A. H., Schuit F. C., Renstrom E., Mulder H. (2006) Diabetologia 49, 1578–1586 [DOI] [PubMed] [Google Scholar]
- 41. Jensen M. V., Joseph J. W., Ilkayeva O., Burgess S., Lu D., Ronnebaum S. M., Odegaard M., Becker T. C., Sherry A. D., Newgard C. B. (2006) J. Biol. Chem. 281, 22342–22351 [DOI] [PubMed] [Google Scholar]
- 42. Ivarsson R., Quintens R., Dejonghe S., Tsukamoto K., in't Veld P., Renström E., Schuit F. C. (2005) Diabetes 54, 2132–2142 [DOI] [PubMed] [Google Scholar]
- 43. Matschinsky F. M., Ghosh A. K., Meglasson M. D., Prentki M., June V., von Allman D. (1986) J. Biol. Chem. 261, 14057–14061 [PubMed] [Google Scholar]
- 44. Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996) Cell 86, 147–157 [DOI] [PubMed] [Google Scholar]
- 45. Garrido C., Galluzzi L., Brunet M., Puig P. E., Didelot C., Kroemer G. (2006) Cell Death Differ. 13, 1423–1433 [DOI] [PubMed] [Google Scholar]
- 46. Satin L. S. (2000) Endocrine 13, 251–262 [DOI] [PubMed] [Google Scholar]
- 47. Brown G. C., Borutaite V. (2008) Biochim. Biophys. Acta 1777, 877–881 [DOI] [PubMed] [Google Scholar]
- 48. Pan Z., Voehringer D. W., Meyn R. E. (1999) Cell Death Differ. 6, 683–688 [DOI] [PubMed] [Google Scholar]
- 49. Bao Q., Lu W., Rabinowitz J. D., Shi Y. (2007) Mol. Cell. 25, 181–192 [DOI] [PubMed] [Google Scholar]
- 50. Cain K., Langlais C., Sun X. M., Brown D. G., Cohen G. M. (2001) J. Biol. Chem. 276, 41985–41990 [DOI] [PubMed] [Google Scholar]
- 51. Chandra D., Bratton S. B., Person M. D., Tian Y., Martin A. G., Ayres M., Fearnhead H. O., Gandhi V., Tang D. G. (2006) Cell 125, 1333–1346 [DOI] [PubMed] [Google Scholar]
- 52. Zhou Y. P., Pena J. C., Roe M. W., Mittal A., Levisetti M., Baldwin A. C., Pugh W., Ostrega D., Ahmed N., Bindokas V. P., Philipson L. H., Hanahan D., Thompson C. B., Polonsky K. S. (2000) Am. J. Physiol. Endocrinol. Metab. 278, E340–351 [DOI] [PubMed] [Google Scholar]
- 53. Halestrap A. P., McStay G. P., Clarke S. J. (2002) Biochimie 84, 153–166 [DOI] [PubMed] [Google Scholar]
- 54. Straub S. G., Sharp G. W. (2002) Diabetes Metab. Res. Rev. 18, 451–463 [DOI] [PubMed] [Google Scholar]
- 55. Sweet I. R., Gilbert M., Jensen R., Sabek O., Fraga D. W., Gaber A. O., Reems J. (2005) Transplantation 80, 1003–1011 [DOI] [PubMed] [Google Scholar]