

Abstract
BAD (Bcl-2 antagonist of cell death) belongs to the proapoptotic BH3-only subfamily of Bcl-2 proteins. Physiological activity of BAD is highly controlled by phosphorylation. To further analyze the regulation of BAD function, we investigated the role of recently identified phosphorylation sites on BAD-mediated apoptosis. We found that in contrast to the N-terminal phosphorylation sites, the serines 124 and 134 act in an antiapoptotic manner because the replacement by alanine led to enhanced cell death. Our results further indicate that RAF kinases represent, besides PAK1, BAD serine 134 phosphorylating kinases. Importantly, in the presence of wild type BAD, co-expression of survival kinases, such as RAF and PAK1, leads to a strongly increased proliferation, whereas substitution of serine 134 by alanine abolishes this process. Furthermore, we identified BAD serine 134 to be strongly involved in survival signaling of B-RAF-V600E-containing tumor cells and found that phosphorylation of BAD at this residue is critical for efficient proliferation in these cells. Collectively, our findings provide new insights into the regulation of BAD function by phosphorylation and its role in cancer signaling.
Keywords: MAP Kinases (MAPKs), Protein Kinases, Protein Phosphorylation, Raf, Signal Transduction, Bcl-2 Proteins
Introduction
Apoptosis is a programmed cell death mechanism that regulates the destruction of cells. This is crucial for multicellular organisms in processes such as development and tissue homeostasis (1–3). Mitochondria constitute a convergence point in the process of apoptosis (3, 4). Crucial regulators of apoptosis at the level of mitochondria are the proteins of the Bcl-2 family that are characterized by the presence of Bcl-2 homology domains (BH1–BH4).2 The Bcl-2 family of proteins is divided into three subfamilies, including proteins that either inhibit (e.g. Bcl-XL, Bcl-2, or Bcl-w) or promote (e.g. Bak, Bax, or Bok) programmed cell death (4–6). A second subclass of proapoptotic Bcl-2 family members consists of the BH3-only proteins that share sequence homology only at the BH3 domain. This subclass comprises BAD, Bmf, Bik, Noxa, Hrk, Bim, Bid, and Puma (4). A complex interplay between anti- and proapoptotic proteins of this family mediates induction and execution of programmed cell death. Surprisingly, several proteins of the Bcl-2 family shaped up as regulators of various physiological processes other than apoptosis, such as autophagy, mitochondrial respiration, and the glucose metabolism (7).
BAD (Bcl-2-associated death promoter, Bcl-2 antagonist of cell death) is a BH3-only protein that promotes apoptosis by forming heterodimers with the prosurvival proteins Bcl-2 and Bcl-XL (8). Phosphorylation of specific serine residues, Ser-112 and Ser-136 of murine BAD (mBAD) or the corresponding phosphorylation sites Ser-75 and Ser-99 of human BAD (hBAD) leads to complex formation with 14-3-3 proteins and subsequent relocation of BAD (9, 10). Phosphorylation of mBAD at Ser-155 (Ser-118 of hBAD) disrupts the association with Bcl-XL or Bcl-2, provoking cell survival (11). Therefore, the phosphorylation status of BAD at specific serine residues reflects a checkpoint for cell death or survival. Numerous kinases were shown to phosphorylate BAD (12–21), including RAF kinases (14, 22–25). Regarding RAF kinases, we demonstrated that B-RAF phosphorylates BAD in a direct manner, leading to inhibition of BAD-induced cell death (25). This finding is of particular importance because highly active B-RAF has been demonstrated to represent one of the crucial players in cancer development (26). In total, five serine phosphorylation sites (at positions 112, 128, 136, 155, and 170) and two threonine phosphorylation sites (117 and 201) have been reported so far for murine BAD. In human BAD, we recently identified several novel in vivo phosphorylation sites (serines 25, 32/34, 97, 124, and 134) besides the established phosphorylation sites at Ser-75, Ser-99, and Ser-118 (25).
In this study, we investigated the putative role of hBAD phosphorylation sites located at the N and C terminus and demonstrate that, contrary to the N-terminal part of hBAD, the residues Ser-124 and Ser-134 are directly involved in regulation of apoptosis. Additionally, our results indicate that RAF kinases represent, besides PAK1, in vivo BAD Ser-134-phosphorylating kinases. Furthermore, we demonstrated that phosphorylation of BAD Ser-134 by RAF kinases and PAK1 triggers cell proliferation and disclose that BAD cooperates with RAF in promoting proliferation in B-RAF mutant cancer cells.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Benzamidine, leupeptin, aprotinin, and Nonidet P-40 were obtained from Sigma. Glutathione-Sepharose was purchased from GE Healthcare, and Ni2+-nitrilotriacetic acid-agarose was from Qiagen. Monoclonal anti-phospho-ERK antibody (sc-7383), polyclonal anti-B-RAF antibody (sc-166), polyclonal anti-BAD antibodies (sc-943 and sc-8044), anti-Myc antibody (sc-40), and polyclonal anti-actin antibody (sc-1616) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and polyclonal anti-Akt/PKB antibody was from Cell Signaling Technology. Phosphospecific antibody directed against phosphoserine 134 of human BAD (or corresponding phosphoserine 170 in murine BAD) was from Abnova. Phosphospecific antibodies directed against phosphoserines 75 and 118 of human BAD (or corresponding phosphoserines 112 and 155 in murine BAD) were obtained from Cell Signaling Technology (catalog numbers 9296 and 9297, respectively). Horseradish peroxidase-conjugated polyclonal anti-rabbit and anti-mouse IgG were obtained from GE Healthcare.
Cell Culture, Transfection, and Immunoblotting
HEK-293 (ATCC CRL-1573), HeLa 229 (ATCC CCL-2.1), A375, SK-MEL-28, DX3, and MEL-Juso cells (kindly provided by Daniela Haug, University of Würzburg, Germany) were cultivated in DMEM supplemented with 10% fetal bovine serum (Biochrom), 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin at 37 °C in humidified air with 5% CO2. The cell lines PC3 and HCT 116 (kindly provided by Joachim Fensterle (Æterna Zentaris, Frankfurt, Germany) were cultivated in RPMI and McCoy's 5a medium, respectively, that were supplemented with 10% fetal bovine serum (Biochrom), 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin. Prior to transfection, cells were seeded at 3 × 105 cells/well of a 6-well plate and grown for 24 h before transfection by jetPEI (Polyplus). 16 h post-transfection, cells were washed twice with phosphate-buffered saline (PBS) and cultivated for 22 h in medium supplemented with 0.1–0.3% serum. To investigate the putative involvement of different survival kinases, the following kinase inhibitors were used: U0126 (Promega), PD0325901, wortmannin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and sorafenib (Bayer, Leverkusen). All inhibitors were dissolved in DMSO and used at the indicated concentrations. After harvesting, the cells were washed once with PBS and then lysed in Nonidet P-40 buffer (50 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2 mm EDTA, 2 mm EGTA, 10% (v/v) glycerol, 0.1% (v/v) β-mercaptoethanol, 25 mm β-glycerophosphate, 10 mm sodium pyrophosphate, 1 mm Na3VO4, 25 mm NaF, 1% Nonidet P-40, and a mixture of standard proteinase inhibitors). Protein concentration was determined by the Bradford method. SDS-PAGE and immunoblotting were performed as described previously (10).
siRNA Transfection
One day prior to transfection, cells were seeded into 12-well plates to a confluence of 70%. Transfection of siRNAs targeting the coding sequence (5′-ACGAGTTTGTGGACTCCTTTA-3′) or the 3′-UTR (5′-TCACTACCAAATGTTAATAAA-3′) of BAD or luciferase as control (5′-AACUUACGCUGAGUACUUCGA-3′) was performed with Lipofectamine 2000 reagent (Invitrogen) and Optimem medium (Invitrogen) with a final siRNA concentration of 100 nm. After 48 h, cells were harvested and used for Western blotting and monitoring of the number of viable cells by trypan blue (Sigma) exclusion.
DNA Expression Plasmids and Purification of Proteins
The hBAD expression plasmid was generated as described (25). Site-specific mutations of hBAD were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The accuracy of the BAD mutants was confirmed by DNA sequencing. PAK1 constructs were from Jonathan Chernoff (Philadelphia, PA). Akt/PKB plasmids were kindly provided by Jakob Troppmair (Innsbruck, Austria). Expression and purification of RAF kinases, PAK1, and Akt/PKB from Sf9 insect cells were performed as described previously (27, 28). GST-tagged hBAD was expressed in Escherichia coli using pGEX-2T vector (GE Healthcare) and isolated by glutathione-Sepharose affinity chromatography. Further purification was achieved by ion exchange chromatography using an ÄKTA system (GE Healthcare). The purity of the proteins was assessed by SDS-PAGE and Coomassie Blue staining (see also supplemental Fig. S1).
Cell Survival Assay
For the analysis of cell survival, cells were transiently transfected in triplicates. After 16 h, cells were washed twice with PBS and grown for 30 h in medium supplemented with 0.1% serum. Cell viability was assessed by staining cells with trypan blue.
Analyses of Cell Proliferation and Growth Inhibition
To monitor cell proliferation, HEK-293 and HeLa cells were transiently transfected in triplicates. 16 h post-transfection, cells were washed twice with PBS and grown in medium supplemented with 0.3% serum. SK-MEL-28, A375, PC-3, HCT 116, MEL-Juso, and DX3 cells were incubated with and without the indicated kinase inhibitors or transfected with the named siRNAs for 48 h. Cell proliferation and growth inhibition were analyzed by photographing and counting the cells as well as by counting living cells by use of trypan blue in a hemicytometer at different time points (2, 3, and 6 days following transfection). To visualize cell proliferation, mitochondria of viable cells were stained by MitoTracker Deep Red FM (catalog no. M22425, Invitrogen) according to the manufacturer's instructions, and fluorescence intensity was measured at 633-nm excitation and 670/30-nm emission by a Typhoon 9200 imager (GE Healthcare).
Kinase Activity Measurements
Kinase activity of RAF-containing samples was monitored using recombinant MEK and ERK-2 as substrates. For that purpose, HeLa cell lysates (5 μg) or purified proteins (1 μg) were mixed with 0.5 mm ATP in 25 mm Hepes, pH 7.6, 150 mm NaCl, 25 mm β-glycerophosphate, 10 mm MgCl2, 1 mm dithiothreitol, and 1 mm sodium vanadate buffer (50-μl final volume). Following the addition of MEK and ERK-2, the reaction mixture was incubated for 30 min at 30 °C. The incubation was terminated by the addition of Laemmli sample buffer, and the proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. The extent of ERK phosphorylation was determined with anti-phospho-ERK antibodies.
RESULTS
In Contrast to the N-terminal Phosphorylation Sites, Ser-124 and Ser-134 of hBAD Contribute to Apoptosis Control
BAD function is regulated by phosphorylation at several residues in response to survival factors. Phosphorylation of mBAD at serines 112, 136, and 155 (corresponding to serines 75, 99, and 118 in hBAD) was a subject of numerous studies. In contrast, little is known about the function of the recently published phosphorylation sites (25). Because the function of the internal phosphoserines, regulating the interactions of BAD with 14-3-3 proteins, has been thoroughly investigated (9–11, 29–31), we examined in this study the role of phosphorylation of serines located at the N- and C-terminal parts of hBAD. For that purpose, we analyzed the role of phosphorylation of serines 25, 32, and 34, located at the N terminus as well as phosphorylation sites located at the C-terminal part of the hBAD protein (i.e. serines 124 and 134 with respect to survival regulation) (see Fig. 1A). The functional role of these novel phosphorylation sites was assessed by site-directed mutagenesis, where the serines of interest were replaced by alanine. The analysis of the proapoptotic function of hBAD and hBAD mutants was carried out in HeLa cells. The cells were transiently transfected with the indicated expression plasmids and starved for 30 h in medium supplemented with 0.1% serum. The number of apoptotic cells was determined by trypan blue.
FIGURE 1.

Amino acid sequence of human BAD protein (A) and of the BAD fragment surrounding the BH3 domain in human and mouse BAD (B). Amino acid sequences were aligned using the ClustalW algorithm (available at the European Bioinformatics Institute Web site). All published phosphorylation sites are highlighted in magenta, and their positions within the sequence are indicated by numbers. The BH3 domain is shown in blue, and the putative lipid-binding domains (LBD1 and LBD2) are indicated by orange and green rectangles, respectively. In B, the FKK/FK regions vicinal to the BH3 domain are indicated in yellow.
As displayed in Fig. 2A, the substitution of the N-terminal Ser-25, Ser-32, and Ser-34 by alanine did not result in significant changes of the proapoptotic activity of BAD. In contrast, the substitution of Ser-124 or Ser-134 by alanine led to enhanced cell death, indicating that phosphorylation at these positions plays a pivotal role in the regulation of cell survival. In these experiments, BAD was expressed to the same levels (see Fig. 2B). These results were also verified by poly(ADP-ribose) polymerase cleavage (data not shown).
FIGURE 2.

Serines 124 and 134 located at the C-terminal part of hBAD contribute to apoptosis control. HeLa cells were transiently transfected with the indicated expression vectors. 16 h post-transfection, cells were cultivated for 30 h in medium supplemented with 0.1% serum. A, the extent of apoptotic cells was detected by trypan blue staining. B, expression levels of BAD wild type and BAD variants in transfected cell lysates were detected with an antibody directed against BAD. The experiments were repeated three times with the same results. Error bars, S.D.
Characterization of hBAD Ser-134 Phosphorylation
The proapoptotic protein BAD has been reported to be a substrate for a wide spectrum of kinases (see references within Ref. 25). Recently, we and others demonstrated that mammalian RAF isoforms represent direct BAD-phosphorylating kinases (14, 22–25). However, in our previous study (25), we restricted our efforts to the regulatory serines 75, 99, and 118. In this study, we focused our efforts on characterization of human BAD serine 134 phosphorylation. To monitor the in vivo phosphorylation at this critical serine residue, we co-expressed different RAF isoforms (B- and C-RAF) and performed a comparative analysis involving other BAD-phosphorylating kinases, such as PAK1 or Akt/PKB. By analyzing co-transfected HeLa cells, we found that Ser-134 of hBAD becomes efficiently phosphorylated by B- and C-RAF and PAK1 (Fig. 3A). Because the co-expression of active Akt/PKB caused relative low extents of Ser-134 phosphorylation (in the range of about 25% of the value obtained by RAF), we concluded that Akt/PKB is less involved in this process. To investigate whether the replacement of Ser-134 by alanine influences the phosphorylation of the survival sites Ser-75 and Ser-118 by B-RAF, we monitored also the extent of phosphorylation at these sites. As demonstrated in Fig. 3A (bottom), no difference was observed between S134A mutant and hBAD wild type. RAF kinases appear to be able to phosphorylate BAD serine 134 directly because two different MEK inhibitors (U0126 and PD0325901) did not affect the phosphorylation signal by RAF (Fig. 3C).
FIGURE 3.
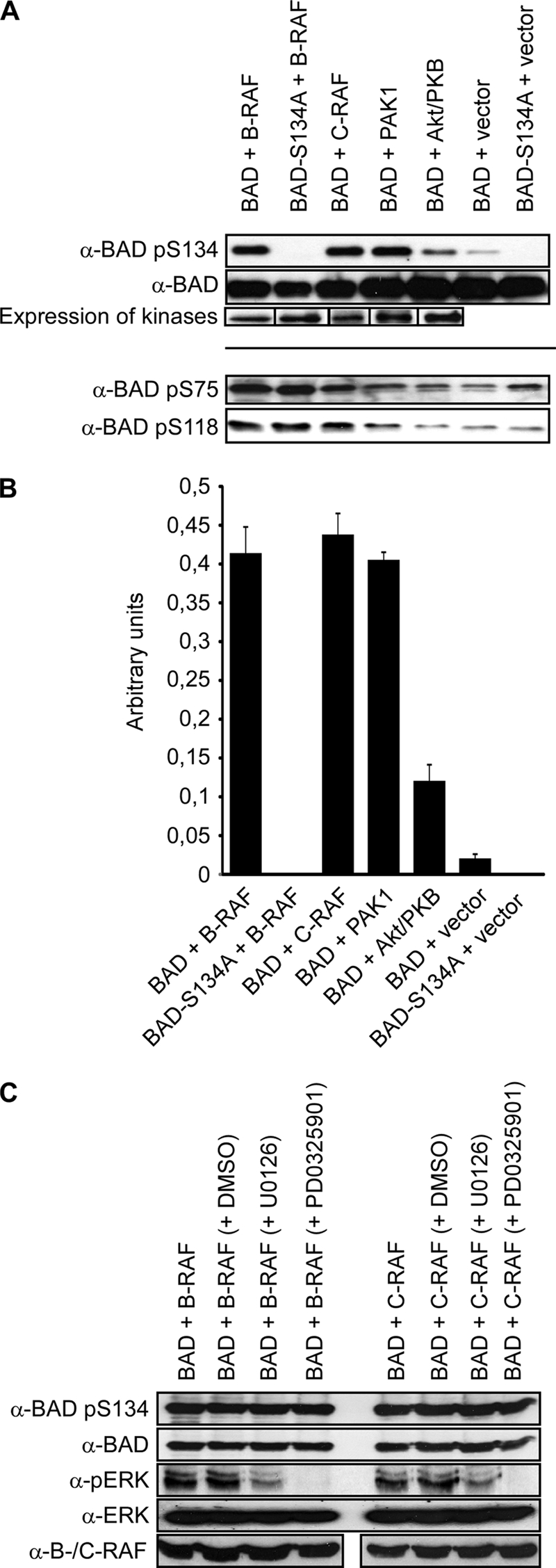
Comparative analysis of hBAD phosphorylation by Akt/PKB, PAK1, and RAF kinases using anti-BAD-pS134 antibody. HeLa cells were transiently transfected with the indicated expression vectors. In the case of Akt/PKB and PAK1, activating mutants (T308D/S473D and T423E, respectively) were used. 16 h post-transfection, cells were cultivated for 30 h in medium supplemented with 0.3% serum with (C) or without (A) of the indicated MEK inhibitors (10 μm). Total cell lysates were separated on a 15% SDS-polyacrylamide gel and blotted onto nitrocellulose membrane. Phosphorylation of human BAD at serine 134 as well as BAD expression was analyzed. Phosphorylation degrees of the survival serines 75 and 118 are also included (see lower lanes). The expression of different kinases was verified by use of specific antibodies. The phosphorylation degree of BAD at serine 134 by kinases shown in A was quantified by optical densitometry (B). In C, the efficiency of MEK inhibitors was analyzed by an antibody directed against phosphorylated ERK. These experiments were repeated three times with comparable results. Error bars, S.D.
To monitor the direct phosphorylation of Ser-134 by the indicated kinases (Fig. 3), we investigated also the in vitro phosphorylation of hBAD by purified kinases (supplemental Fig. S1). To this end, non-phosphorylated hBAD has been purified from E. coli, whereas RAF kinases, PAK1, and Akt/PKB were expressed and isolated from Sf9 insect cells. The purity of the isolated proteins was assessed by SDS-PAGE and Coomassie Blue staining (see supplemental Fig. S1). The phosphorylating kinases PAK1, Akt/PKB, and B-RAF provided similar results in vivo and in vitro (Fig. 3A and supplemental Fig. S1). In contrast, although C-RAF proved to be a potent in vivo Ser-134-phosphorylating kinase, in vitro it failed to phosphorylate this site significantly. The reason for this discrepancy could be explained by the ability of C-RAF to build an active heterodimeric complex with B-RAF (32–34) in vivo.
As previously reported (25), we demonstrated that co-expression of B-RAF (or activated C-RAF) inhibits BAD-mediated apoptosis following growth factor removal. These effects were ascribed to RAF-mediated phosphorylation of serine 75 and 118 in human BAD because the substitution of these residues by alanine led to increased apoptotic levels even in the presence of B-RAF (25). In this study, we investigated the putative impact of serine 134 phosphorylation on apoptosis degree in the presence of active RAF, such as B-RAF. As exhibited in Fig. 4A, the single mutation of serine 134 to alanine does not influence the proapoptotic activity of BAD as much as observed in Fig. 2A. The reason for this effect might be that BAD-induced apoptosis is regulated through an interplay between the serine residues 75, 118, and 134 that is controlled by B-RAF. In the presence of B-RAF, single mutation of serine 134 to alanine barely affects the proapoptotic activity of BAD because co-expression of B-RAF leads to increased phosphorylation of BAD serines 75 and 118 (see also Fig. 3A). In a previous report (25), we demonstrated that B-RAF-mediated phosphorylation of BAD serines 75 and 118 leads to inhibition of BAD-induced apoptosis. Without co-expression of B-RAF, the phosphorylation levels of the regulatory serines 75 and 118 are low, resulting in strong induction of apoptosis. The degree of apoptosis induction can even be strengthened by mutation of serine 134. This situation was simulated by using a triple BAD mutant (BAD-S75A/S118A/S134A) that resulted in intense induction of apoptosis, even in the presence of B-RAF (Fig. 4A). These results, together, indicate that residues 75 and 118 are crucial for regulation of BAD proapoptotic activity, whereas serine 134 mediates the fine tuning of BAD-induced apoptosis. These observations, together, indicate that residues 75 and 118 are crucial for the regulation of BAD proapoptotic activity, whereas serine 134 mediates the fine tuning of BAD-induced apoptosis.
FIGURE 4.

BAD-induced apoptosis is regulated by interplay between serines 75, 118, and 134. HeLa cells were transiently transfected in triplicates with the expression vectors as indicated. 16 h post-transfection, cells were washed and grown in medium supplemented with 0.1% serum. A, the extent of apoptotic cells was detected by trypan blue staining. B, expression levels of BAD constructs and B-RAF in transfected cell lysates were detected via specific antibodies. Error bars, S.D.
Phosphorylation of hBAD Ser-134 Enhances Cell Proliferation
Data shown in Fig. 3 reveal that RAF kinases and PAK1 phosphorylate BAD at position serine 134. Because these survival kinases play a crucial role in several types of cancer, we analyzed here in more detail the effect of BAD phosphorylation on cell proliferation. To this end, we co-expressed RAF kinases and PAK1 with wild type BAD or BAD-S134A mutant in HeLa cells and induced apoptosis through starvation conditions. Cell proliferation was analyzed by counting of living cells (Fig. 5, A and C) as well as by staining of mitochondria of viable cells and detection of fluorescence intensity by a laser scanner (Fig. 5, B and C).
FIGURE 5.

BAD stimulates RAF-mediated cell proliferation. HEK-293 and HeLa cells were transiently transfected in triplicates with the expression vectors as indicated. 16 h post-transfection, cells were washed and grown in medium supplemented with 0.3% serum. A, cell proliferation was analyzed by counting living cells by trypan blue exclusion 2 days following transfection. B, to visualize cell proliferation, mitochondria of viable cells were stained by MitoTracker, and fluorescence intensity of the wells was measured by a Typhoon 9200 imager. C, combination of results obtained in A and B. To show that the measured fluorescence intensities correlate with counted cell numbers, the results presented in A and B were quantitatively compared. BAD constructs and kinases were expressed to comparable levels in each sample (data not shown). These experiments were repeated three times with comparable results. Error bars, S.E.
Results obtained by both methods support the finding that RAF and PAK1 serve as phosphorylating kinases for hBAD serine 134. Surprisingly, proliferation activity of cells expressing RAF and PAK1 was considerably elevated in the presence of wild type hBAD (Fig. 5). On the other hand, in the presence of BAD-S134A mutant, the degree of cell proliferation was reduced to control levels. The measured fluorescence intensities reflect the amount of viable cells because they correlate with counted cell numbers (Fig. 5C). The change of mitochondrial activity may play a minor role in this context.
Of note, the induction of cell proliferation by the co-expression of BAD with survival kinases is not only due to an inhibition of apoptosis because the percentage of apoptotic cells is similar to the vector control (Fig. 4A). These observations are not cell type-specific because comparable results were obtained using HEK-293 cells (data not shown).
Taken together, we show here that serine 134 has two functions during survival signaling. On the one hand, this site controls intensity of BAD-mediated apoptosis, and on the other hand, it is involved in the regulation of proliferation.
Phosphorylation of BAD Serine 134 Plays an Exclusive Role in B-RAF Mutant Tumor Cells
In experiments performed with cells containing wild type RAF or RAS, we showed by overexpression that RAF kinases effectively phosphorylate BAD at serine 134, whereas Akt/PKB caused a relatively low extent of Ser-134 phosphorylation (Fig. 3A). To test whether this observation is valid in naturally occurring tumor cells, we investigated cancer cell lines possessing endogenously mutated RAF and RAS proteins. Two melanoma cell lines used in this study (A375 and SK-MEL-28) carry a valine-to-glutamic acid mutation at residue 600 of B-RAF (B-RAF-V600E), which leads to elevated RAF signaling in these cell lines (26) (see also Fig. 9). In contrast, the cell lines HCT 116, DX3, and MEL-Juso are known to harbor activating mutations within the RAS genes. RAS has been reported to activate a variety of cellular targets besides RAF, such as RAS-GDS, Tiam-1, and PI3K (for a review, see Ref. 35). Concerning driving tumorigenesis, one of the most probable targets of RAS is PI3K. This is known to activate the prosurvival Akt/PKB pathway (36) (see also Fig. 9). As a control, we also used the tumor cell line PC3, which carries neither a RAF nor a RAS mutation. Surprisingly, the phosphorylation of BAD serine 134 was only observed in the cell lines containing B-RAF mutant (Fig. 6A). These results are in agreement with the experiments presented in Fig. 3A, where we overexpressed RAF kinases and Akt/PKB in HeLa cells.
FIGURE 9.

Schematic presentation of BAD serine 134 phosphorylation in RAF and RAS mutant tumor cells. Stimulation of receptor tyrosine kinase (TRK) at the plasma membrane (PM) leads to activation of RAS GTPases and RAF kinases. The cell lines HCT 116, DX3, and MEL-Juso carry mutated RAS genes, resulting in enhanced activation of the PI3K/Akt pathway. Although Akt was shown to be a BAD-targeting kinase, it has a minor contribution in phosphorylating BAD at serine 134. The cell lines A375 and SK-MEL-28 harbor an activating mutation within B-RAF (V600E) that leads to elevated phosphorylation of BAD serine 134, mainly through the MEK-ERK cascade. The specific inhibitors of RAF, MEK, and PI3K are highlighted in blue boxes. Phosphorylation of BAD at serine 134 affects the efficiency of apoptosis and proliferation. Thick arrows indicate enhanced activation or phosphorylation. For more details, see “Results” and “Discussion” sections.
FIGURE 6.

BAD serine 134 plays an exclusive role in B-RAF mutant tumor cells. A, to monitor the phosphorylation degree of BAD at serine 134 in naturally occurring cancer cells, two cell lines that carry B-RAF-V600E mutant (A375 and SK-MEL-28), three cell lines that harbor activating mutations within the RAS genes (HCT 116, DX3, and MEL-Juso), and a control tumor cell line that carries neither RAF nor RAS mutation (PC3) were investigated. The amounts of the endogenous BAD protein, phosphorylation of BAD at serine 134, and endogenous actin were detected by immunoblotting. B and C, A375 and SK-MEL-28 cells were treated with the indicated kinase inhibitors, and cell growth (B) and expression levels of endogenous BAD and actin as well as phosphorylation of BAD serine 134 (C) were analyzed. The efficiencies of the kinase inhibitors wortmannin, sorafenib, and PD0325901 have been verified by change of Akt serine 473 and ERK phosphorylation, respectively. Error bars, S.D.
The RAF inhibitor sorafenib (also known as BAY 43-9006 or Nexavar®) has been reported to decrease proliferation in B-RAF mutant tumor cells (37) (see also Fig. 6B). Interestingly, impaired proliferation goes along with a diminished phosphorylation of hBAD serine 134 in sorafenib-treated A375 and SK-MEL-28 cells (Fig. 6, B and C). In contrast, the PI3K inhibitor wortmannin had a minor effect on cell proliferation and phosphorylation of hBAD serine 134 (Fig. 6, B and C). To investigate the putative synergistic effects between sorafenib and wortmannin, we also incubated the cells with both inhibitors together. Because wortmannin barely impaired the effects of sorafenib alone, we could exclude the possibility that inhibition of PI3K leads to enhanced RAF signaling that would mask the effect of wortmannin (Fig. 6, B and C). Inhibition of MEK by PD0325901 affects proliferation and phosphorylation of hBAD serine 134 similar to inhibition of RAF by sorafenib (Fig. 6, B and C). This leads to the conclusion that although RAF kinases are potentially able to phosphorylate hBAD serine 134 directly (Fig. 3C), in naturally occurring tumor cell lines, proliferation and phosphorylation of BAD serine 134 is apparently mediated through the RAF-MEK-ERK cascade. In agreement with published data (38), inhibition of MEK or RAF leads to a slight increase of the whole amount of BAD protein (Fig. 6C), indicating a dual role of BAD in MAPK-mediated survival signaling. The efficiency of wortmannin, sorafenib, or PD0325901 has been verified by change of Akt serine 473 and ERK phosphorylation, respectively. In cell lines containing mutated RAS, proliferation was more inhibited by wortmannin than by sorafenib or PD0325901 (supplemental Fig. S2). All three inhibitors barely caused inhibition of proliferation in cells containing neither a RAF nor RAS mutation (supplemnetal Fig. S2).
To analyze whether BAD directly contributes to cell proliferation in B-RAF mutant melanoma cells we used BAD-specific siRNA to down-regulate the endogenous BAD protein (Fig. 7A). Compared with mock cells and control cells that were transfected with siRNA directed against luciferase, knockdown of BAD led to marked inhibition of proliferation in A375 and SK-MEL-28 cells (Fig. 7B). This growth inhibition could be abrogated by overexpression of wild type BAD (Fig. 8). Importantly, overexpression of BAD serine 134 to alanine mutant was not able to rescue efficient proliferation upon knockdown of endogenous BAD. In cell lines containing mutated RAS and in cells containing neither a RAF nor RAS mutation, knockdown of BAD did not affect cell proliferation (supplemental Fig. S3).
FIGURE 7.

BAD is required for efficient proliferation in B-RAF mutant melanoma cells. A375 and SK-MEL-28 cells were transfected with siRNAs against the coding sequence of BAD or luciferase (Luci) as control. Two days post-transfection, knockdown of BAD at the protein level (A) and the effect of BAD knockdown on cell growth (B) were analyzed. In A, actin levels show equal loading. Error bars, S.D.
FIGURE 8.

Phosphorylation of BAD at the position serine 134 is critical for B-RAF driven proliferation. A375 (A) and SK-MEL-28 (B) cells were co-transfected with siRNAs against the 3′-UTR of endogenous BAD (but not against exogenous BAD) or luciferase (Luci) and the indicated expression vectors. Two days post-transfection, knockdown of endogenous BAD and overexpression of BAD wild type as well as BAD-S134A mutant was monitored by immunoblotting. Actin levels show equal loading. The resulting growth inhibition is presented below (see bar graph). Error bars, S.D.
DISCUSSION
Post-translational modifications of BH3-only proteins, such as phosphorylation, proteolytic processing, and lipid modifications, emerged as regulatory elements that integrate extracellular survival signals with the apoptotic machinery. Regarding regulation of BAD function, phosphorylation plays perhaps the most important role among these post-translational events. Recently, using mass spectrometry, we identified 10 distinct phosphorylation sites within the human BAD protein (25) (see also Fig. 1A). Although some of these phosphoserines have been found to be located at the N terminus of hBAD (serines 25, 32, and 34), three other phosphorylation sites are positioned at the C-terminal part of the protein (i.e. serines 118, 124, and 134). The third group of hBAD phosphorylation sites (serines 75, 91, 97, and 99) are either directly or indirectly involved in binding of 14-3-3 proteins. Because the function of the phosphoserines that regulate the interactions of BAD with 14-3-3 proteins has been thoroughly investigated already (9–11, 29–31), we examined in this study the role of serine phosphorylation at the N- and C-terminal parts of hBAD in more detail. Substitution of Ser-124 and/or Ser-134 by alanine led to an increase of apoptotic activity, indicating that phosphorylation at these positions is actively involved in the control of apoptotic pathways (Fig. 2A). By contrast, substitution of N-terminal serines 25, 32, and 34 by alanine did not significantly change the proapoptotic activities of BAD, allowing the conclusion that the phosphorylation of the N terminus does not play a decisive role in the regulation of cell survival. Although mutation of Ser-134 to alanine led to an increased apoptotic activity (Fig. 2A), in the presence of B-RAF, this effect was less pronounced (Fig. 4A). This observation is in accordance with our previous results showing that B-RAF phosphorylates also serines 75 and 118 of human BAD, thereby inhibiting BAD-mediated apoptosis (25). Thus, the inhibition of hBAD-induced apoptosis by B-RAF could be realized mostly through phosphorylation of serines 75 and 118, whereas serine 134 mediates the fine tuning of BAD-induced apoptosis.
In our previous attempts to characterize the translocation of BAD to mitochondria, we identified two lipid-binding domains (termed LBD1 and LBD2) within the C-terminal region of human BAD (10). Whereas LBD2 overlaps with helix-5 localized at the very C terminus, LBD1 encompasses the C-terminal half of the BH3 helix and covers also the short 124SFKK127 region (Fig. 1A). Thus, due to its close proximity to the FKK motif, it is feasible that the phosphorylation of Ser-124 may regulate BAD function by modulating the interaction of BAD with membrane lipids. However, the proposed regulation of BAD function by phosphorylation of Ser-124 seems to be unique for the human BAD protein because most of the mammalian homologues (e.g. murine BAD) do not contain the complete FKK motif (Fig. 1B). On the other hand, the alignment of human and murine BAD reveals that both BAD proteins contain the conserved segment PRPKS134/170AG including either Ser-134 in human or Ser-170 in murine BAD (Fig. 1B). Ser-170 has previously been identified as a BAD phosphorylation site in murine BAD (39). The recent availability of the phosphospecific antibodies against phosphoserine 134/170 allows the search for potential kinases that can phosphorylate Ser-134. Although the consensus sequence RXXS makes Akt/PKB probable as a potential phosphorylating kinase, the two proline residues present in the motif PRPKSAG render this possibility unlikely. Indeed, co-expression of hBAD with Akt/PKB did not result in significant phosphorylation of Ser-134. Instead, as shown in Fig. 3A, we identified PAK1 and RAF as the most potent BAD-phosphorylating kinases under both in vivo and in vitro conditions. Concerning PAK1, one should take into consideration that this kinase was described to phosphorylate BAD in an indirect manner targeting RAF as a downstream effector kinase (14).
Intriguingly, our data indicate that co-expression of wild type BAD with RAF kinases and PAK1 strongly increases cell proliferation (Fig. 5), whereas the BAD-S134A mutant abolishes this effect. As generally accepted, the RAS-RAF-MEK-ERK pathway regulates cellular survival, differentiation, and proliferation (40–43). Enhanced activation of this cascade, often caused through activating mutations in the composite proteins, plays an important role during cancer development (44) and is found in many tumors (45–46). In human cancer, mutated RAF (mainly B-RAF) was identified in 60% of melanomas (46) and with lower incidence in papillary thyroid cancers (47), colorectal carcinomas (26, 46, 48), and lung cancers (46). RAS mutations were found in about 15–30% of human cancers overall (46, 49–50). Activating RAS and B-RAF mutations typically show mutual exclusivity in tumors (26, 46, 51). Notably, although either mutated RAS or B-RAF is required for tumor development, it was demonstrated that both mutations do not result in similar downstream effects (52–53). In this study, we disclose an additional parameter that is apparently involved in tumor progression. In this regard, we demonstrate that the phosphorylation of BAD serine 134 is increased in cell lines with elevated RAF activity but not in cells harboring a high RAS or Akt/PKB activity (Figs. 3A and 6A). Additionally, we found that in melanoma cells, the phosphorylation of this site is preferentially realized through the RAF-MEK-ERK cascade (Fig. 6C), although RAF kinases are potentially able to phosphorylate this site directly (Fig. 3C). This result underlines the former observation that mutation of B-RAF leads to an exquisite dependence on MEK activity (52, 53), where the authors demonstrated that B-RAF mutant tumor cells are considerably more sensitive to MEK inhibition than either RAS mutant or B-RAF/RAS WT cells. These studies showed that only in B-RAF mutant cells does MEK inhibition cause potent inhibition of proliferation (52, 53). The increased sensitivity to MEK inhibitors of B-RAF mutant cells was observed to be based on a different regulation of Bcl-2 proteins in these cell lines (53). The aberrant regulation of Bcl-2 proteins in B-RAF mutant tumor cells may be realized by a specific mitochondrial localization of B-RAF-V600E compared with wild type B-RAF, as recently reported (54). Concerning BAD phosphorylation, Eisenmann et al. (55) demonstrated that the enhanced sensitivity to MEK inhibition is based on a melanoma-specific MAPK-mediated survival signaling. In normal melanocytes, BAD was shown to be phosphorylated at serines 75, 99, and 118, leading to insensitivity to MEK inhibition (55). In contrast, in B-RAF mutant melanoma cells, BAD was only phosphorylated at serine 75. MEK inhibition resulted in dephosphorylation of this site and induction of apoptosis (55). Another study linked resistance to anoikis in melanoma cells to phosphorylation of BAD serine 75 (38). Accordingly, the MAPK-dependent phosphorylation of BAD serine 75 seems to represent an important mechanism to escape from apoptosis, especially in melanoma cells. However, all former studies considered BAD to be only an inducer of apoptosis that is inactivated through phosphorylation, especially at serine 75. We demonstrate here for the first time that phosphorylation of serine 134 of hBAD is required for efficient proliferation in B-RAF-V600E-containing tumor cells (Figs. 7 and 8). Serine 170 of murine BAD that corresponds to serine 134 of human BAD was previously connected to cell proliferation (39). However, one has to consider that the properties of murine BAD may differ from human BAD due to a large N-terminal extension. Additionally, in Dramsi et al. (39), phosphorylation of serine 170 was mimicked by mutation of this residue to aspartic acid. Notably, in this study, the kinases responsible for serine 134 phosphorylation as well as an involvement of this regulatory site in naturally occurring tumor cells has not been addressed. Here we disclose that BAD serine 134 is phosphorylated in a RAF-dependent manner and that BAD cooperates with RAF in promoting proliferation and therefore may play an active role during tumor development. The observation that MEK inhibition leads to a decrease in BAD serine 134 phosphorylation but to an increase in the amount of the whole BAD protein underlines the dual role of BAD. Under survival conditions, BAD is phosphorylated at serine 134 and promotes proliferation in cells with elevated RAF activity (Fig. 9). On the other hand, when entering apoptotic conditions, dephosphorylated BAD accumulates within the cell, leading to complex formation with Bcl-2/XL and induction of apoptosis. Thus, apoptosis and proliferation seem to be regulated through interplay between the phosphorylation sites serine 75, 118, and 134.
CONCLUSIONS
Results presented in this study open new insights regarding the function of BAD and are in accordance with studies pointing to roles for BAD other than apoptosis control (56). Furthermore, our findings showing that human BAD is actively involved in proliferation of B-RAF-V600E-containing tumor cells may provide a new link between survival signaling and cancer development.
Supplementary Material
Acknowledgments
We thank Daniela Haug (University of Würzburg, Germany) and Joachim Fensterle (Æterna Zentaris) for providing cell lines and for fruitful discussions as well as Laura Goldberg (Lafayette College, Easton, PA) for excellent assistance and proofreading.
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 487 (Projects C3) and Graduate College 1141 Grant.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- BH1–BH4
- Bcl-2 homology domains 1–4, respectively
- mBAD
- mouse BAD
- hBAD
- human BAD
- LBD
- lipid binding domain
- PKB
- protein kinase B.
REFERENCES
- 1. Reed J. C., Doctor K. S., Godzik A. (2004) Sci. STKE 2004, re9. [DOI] [PubMed] [Google Scholar]
- 2. Danial N. N., Korsmeyer S. J. (2004) Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 3. Wang C., Youle R. J. (2009) Annu. Rev. Genet. 43, 95–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Youle R. J., Strasser A. (2008) Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 5. Adams J. M., Cory S. (1998) Science 281, 1322–1326 [DOI] [PubMed] [Google Scholar]
- 6. Gross A., McDonnell J. M., Korsmeyer S. J. (1999) Genes Dev. 13, 1899–1911 [DOI] [PubMed] [Google Scholar]
- 7. Danial N. N. (2008) Oncogene 27, Suppl. 1, S53–S70 [DOI] [PubMed] [Google Scholar]
- 8. Yang E., Zha J., Jockel J., Boise L. H., Thompson C. B., Korsmeyer S. J. (1995) Cell 80, 285–291 [DOI] [PubMed] [Google Scholar]
- 9. Zha J., Harada H., Yang E., Jockel J., Korsmeyer S. J. (1996) Cell 87, 619–628 [DOI] [PubMed] [Google Scholar]
- 10. Hekman M., Albert S., Galmiche A., Rennefahrt U. E., Fueller J., Fischer A., Puehringer D., Wiese S., Rapp U. R. (2006) J. Biol. Chem. 281, 17321–17336 [DOI] [PubMed] [Google Scholar]
- 11. Datta S. R., Katsov A., Hu L., Petros A., Fesik S. W., Yaffe M. B., Greenberg M. E. (2000) Mol. Cell 6, 41–51 [PubMed] [Google Scholar]
- 12. Harada H., Becknell B., Wilm M., Mann M., Huang L. J., Taylor S. S., Scott J. D., Korsmeyer S. J. (1999) Mol. Cell 3, 413–422 [DOI] [PubMed] [Google Scholar]
- 13. Datta S. R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M. E. (1997) Cell 91, 231–241 [DOI] [PubMed] [Google Scholar]
- 14. Jin S., Zhuo Y., Guo W., Field J. (2005) J. Biol. Chem. 280, 24698–24705 [DOI] [PubMed] [Google Scholar]
- 15. Schürmann A., Mooney A. F., Sanders L. C., Sells M. A., Wang H. G., Reed J. C., Bokoch G. M. (2000) Mol. Cell. Biol. 20, 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gnesutta N., Qu J., Minden A. (2001) J. Biol. Chem. 276, 14414–14419 [DOI] [PubMed] [Google Scholar]
- 17. Konishi Y., Lehtinen M., Donovan N., Bonni A. (2002) Mol. Cell 9, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 18. Shimamura A., Ballif B. A., Richards S. A., Blenis J. (2000) Curr. Biol. 10, 127–135 [DOI] [PubMed] [Google Scholar]
- 19. She Q. B., Ma W. Y., Zhong S., Dong Z. (2002) J. Biol. Chem. 277, 24039–24048 [DOI] [PubMed] [Google Scholar]
- 20. Klumpp S., Mäurer A., Zhu Y., Aichele D., Pinna L. A., Krieglstein J. (2004) Neurochem. Int. 45, 747–752 [DOI] [PubMed] [Google Scholar]
- 21. Macdonald A., Campbell D. G., Toth R., McLauchlan H., Hastie C. J., Arthur J. S. (2006) BMC Cell Biol. 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H. G., Rapp U. R., Reed J. C. (1996) Cell 87, 629–638 [DOI] [PubMed] [Google Scholar]
- 23. Panka D. J., Wang W., Atkins M. B., Mier J. W. (2006) Cancer Res. 66, 1611–1619 [DOI] [PubMed] [Google Scholar]
- 24. Kebache S., Ash J., Annis M. G., Hagan J., Huber M., Hassard J., Stewart C. L., Whiteway M., Nantel A. (2007) J. Biol. Chem. 282, 21873–21883 [DOI] [PubMed] [Google Scholar]
- 25. Polzien L., Baljuls A., Rennefahrt U. E., Fischer A., Schmitz W., Zahedi R. P., Sickmann A., Metz R., Albert S., Benz R., Hekman M., Rapp U. R. (2009) J. Biol. Chem. 284, 28004–28020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A. (2002) Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 27. Hekman M., Hamm H., Villar A. V., Bader B., Kuhlmann J., Nickel J., Rapp U. R. (2002) J. Biol. Chem. 277, 24090–24102 [DOI] [PubMed] [Google Scholar]
- 28. Fischer A., Baljuls A., Reinders J., Nekhoroshkova E., Sibilski C., Metz R., Albert S., Rajalingam K., Hekman M., Rapp U. R. (2009) J. Biol. Chem. 284, 3183–3194 [DOI] [PubMed] [Google Scholar]
- 29. Subramanian R. R., Masters S. C., Zhang H., Fu H. (2001) Exp. Cell Res. 271, 142–151 [DOI] [PubMed] [Google Scholar]
- 30. Masters S. C., Yang H., Datta S. R., Greenberg M. E., Fu H. (2001) Mol. Pharmacol. 60, 1325–1331 [DOI] [PubMed] [Google Scholar]
- 31. Zhang J., Liu J., Yu C., Lin A. (2005) Cancer Res. 65, 8372–8378 [DOI] [PubMed] [Google Scholar]
- 32. Weber C. K., Slupsky J. R., Kalmes H. A., Rapp U. R. (2001) Cancer Res. 61, 3595–3598 [PubMed] [Google Scholar]
- 33. Garnett M. J., Rana S., Paterson H., Barford D., Marais R. (2005) Mol. Cell 20, 963–969 [DOI] [PubMed] [Google Scholar]
- 34. Rushworth L. K., Hindley A. D., O'Neill E., Kolch W. (2006) Mol. Cell. Biol. 26, 2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaw R. J., Cantley L. C. (2006) Nature 441, 424–430 [DOI] [PubMed] [Google Scholar]
- 36. Amaravadi R., Thompson C. B. (2005) J. Clin. Invest. 115, 2618–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karasarides M., Chiloeches A., Hayward R., Niculescu-Duvaz D., Scanlon I., Friedlos F., Ogilvie L., Hedley D., Martin J., Marshall C. J., Springer C. J., Marais R. (2004) Oncogene 23, 6292–6298 [DOI] [PubMed] [Google Scholar]
- 38. Boisvert-Adamo K., Aplin A. E. (2008) Oncogene 27, 3301–3312 [DOI] [PubMed] [Google Scholar]
- 39. Dramsi S., Scheid M. P., Maiti A., Hojabrpour P., Chen X., Schubert K., Goodlett D. R., Aebersold R., Duronio V. (2002) J. Biol. Chem. 277, 6399–6405 [DOI] [PubMed] [Google Scholar]
- 40. Peyssonnaux C., Eychène A. (2001) Biol. Cell 93, 53–62 [DOI] [PubMed] [Google Scholar]
- 41. Rajalingam K., Schreck R., Rapp U. R., Albert S. (2007) Biochim. Biophys. Acta 1773, 1177–1195 [DOI] [PubMed] [Google Scholar]
- 42. Schreck R., Rapp U. R. (2006) Int. J. Cancer 119, 2261–2271 [DOI] [PubMed] [Google Scholar]
- 43. Wellbrock C., Karasarides M., Marais R. (2004) Nat. Rev. Mol. Cell Biol. 5, 875–885 [DOI] [PubMed] [Google Scholar]
- 44. Rapp U. R. (2007) Nat. Precedings 2007-08-23 [Google Scholar]
- 45. Cohen C., Zavala-Pompa A., Sequeira J. H., Shoji M., Sexton D. G., Cotsonis G., Cerimele F., Govindarajan B., Macaron N., Arbiser J. L. (2002) Clin. Cancer Res. 8, 3728–3733 [PubMed] [Google Scholar]
- 46. Brose M. S., Volpe P., Feldman M., Kumar M., Rishi I., Gerrero R., Einhorn E., Herlyn M., Minna J., Nicholson A., Roth J. A., Albelda S. M., Davies H., Cox C., Brignell G., Stephens P., Futreal P. A., Wooster R., Stratton M. R., Weber B. L. (2002) Cancer Res. 62, 6997–7000 [PubMed] [Google Scholar]
- 47. Cohen Y., Xing M., Mambo E., Guo Z., Wu G., Trink B., Beller U., Westra W. H., Ladenson P. W., Sidransky D. (2003) J. Natl. Cancer Inst. 95, 625–627 [DOI] [PubMed] [Google Scholar]
- 48. Rajagopalan H., Bardelli A., Lengauer C., Kinzler K. W., Vogelstein B., Velculescu V. E. (2002) Nature 418, 934. [DOI] [PubMed] [Google Scholar]
- 49. Bos J. L. (1989) Cancer Res. 49, 4682–4689 [PubMed] [Google Scholar]
- 50. Pavey S., Johansson P., Packer L., Taylor J., Stark M., Pollock P. M., Walker G. J., Boyle G. M., Harper U., Cozzi S. J., Hansen K., Yudt L., Schmidt C., Hersey P., Ellem K. A., O'Rourke M. G., Parsons P. G., Meltzer P., Ringnér M., Hayward N. K. (2004) Oncogene 23, 4060–4067 [DOI] [PubMed] [Google Scholar]
- 51. Gorden A., Osman I., Gai W., He D., Huang W., Davidson A., Houghton A. N., Busam K., Polsky D. (2003) Cancer Res. 63, 3955–3957 [PubMed] [Google Scholar]
- 52. Solit D. B., Garraway L. A., Pratilas C. A., Sawai A., Getz G., Basso A., Ye Q., Lobo J. M., She Y., Osman I., Golub T. R., Sebolt-Leopold J., Sellers W. R., Rosen N. (2006) Nature 439, 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cragg M. S., Jansen E. S., Cook M., Harris C., Strasser A., Scott C. L. (2008) J. Clin. Invest. 118, 3651–3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee M. H., Lee S. E., Kim D. W., Ryu M. J., Kim S. J., Kim Y. K., Park J. H., Kweon G. R., Kim J. M., Lee J. U., De Falco V., Jo Y. S., Shong M. (2011) J. Clin. Endocrinol. Metab. 96, E19–E30 [DOI] [PubMed] [Google Scholar]
- 55. Eisenmann K. M., VanBrocklin M. W., Staffend N. A., Kitchen S. M., Koo H. M. (2003) Cancer Res. 63, 8330–8337 [PubMed] [Google Scholar]
- 56. Danial N. N., Gramm C. F., Scorrano L., Zhang C. Y., Krauss S., Ranger A. M., Datta S. R., Greenberg M. E., Licklider L. J., Lowell B. B., Gygi S. P., Korsmeyer S. J. (2003) Nature 424, 952–956 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.