

Abstract
The annexins, a family of Ca2+- and lipid-binding proteins, are involved in a range of intracellular processes. Recent findings have implicated annexin A1 in the resealing of plasmalemmal injuries. Here, we demonstrate that another member of the annexin protein family, annexin A6, is also involved in the repair of plasmalemmal lesions induced by a bacterial pore-forming toxin, streptolysin O. An injury-induced elevation in the intracellular concentration of Ca2+ ([Ca2+]i) triggers plasmalemmal repair. The highly Ca2+-sensitive annexin A6 responds faster than annexin A1 to [Ca2+]i elevation. Correspondingly, a limited plasmalemmal injury can be promptly countered by annexin A6 even without the participation of annexin A1. However, its high Ca2+ sensitivity makes annexin A6 highly amenable to an unproductive binding to the uninjured plasmalemma; during an extensive injury accompanied by a massive elevation in [Ca2+]i, its active pool is severely depleted. In contrast, annexin A1 with a much lower Ca2+ sensitivity is ineffective at the early stages of injury; however, it remains available for the repair even at high [Ca2+]i. Our findings highlight the role of the annexins in the process of plasmalemmal repair; a number of annexins with different Ca2+-sensitivities provide a cell with the means to react promptly to a limited injury in its early stages and, at the same time, to withstand a sustained injury accompanied by the continuous formation of plasmalemmal lesions.
Keywords: Annexin, Calcium, Membrane, Membrane Function, Plasma Membrane, Membrane Injury, Membrane Repair, Microparticle, Microvesicle, Blebbing
Introduction
The annexins are a family of Ca2+-binding proteins expressed in most phyla and species (1–4). Twelve annexins are present in vertebrates (A1–A11 and A13) with different splice variants (1). Annexins share a common folding motif, the “annexin core,” which harbors the Ca2+- and membrane-binding sites (2–4). In their Ca2+-bound form, the annexins translocate from the cytoplasm to the plasma membrane and associate with negatively charged phospholipids (2–4). The N-terminal region precedes the conserved core and is unique for a given member of the annexin family. It mediates interactions with protein ligands and regulates the annexin-membrane association (2–4). Different annexins have been shown to orchestrate a variety of intracellular processes, ranging from the regulation of membrane dynamics to cell migration, proliferation, and apoptosis (2–12). However, the intriguing question why the majority of cells express several annexins, which differ only slightly in their biochemical properties, remains unanswered.
Recent findings have implicated annexin A1 in the resealing of plasmalemmal lesions following cell injury (13, 14). An injury-induced rise in the local concentration of intracellular Ca2+ (15) is sensed by annexin A1 and triggers its binding to the plasma membrane at the site of the injury (13, 14). Subsequently, annexin A1 promotes fusion of the damaged membrane around the pore, forming sealed, lesion-containing structures: large, cytosol-containing blebs (14) or smaller, cytosol-free microvesicles (16). The microvesicles subsequently can be shed by the cell (16, 17).
Here, we show that, similar to annexin A1, annexin A6 is directly involved in the repair of plasmalemmal lesions induced by streptolysin O (SLO).2 The shedding of microvesicles appears to be predominant in the elimination of pores by annexin A6-dependent repair. Annexin A6 requires lower [Ca2+]i for its plasmalemmal binding and, thus, responds faster to an injury than annexin A1. Correspondingly, a plasmalemmal lesion can be repaired by annexin A6 even without involvement of annexin A1; however, the concerted action of both annexins is instrumental for the efficient repair of multiple, simultaneously occurring plasmalemmal lesions.
EXPERIMENTAL PROCEDURES
Reagents
Monoclonal anti-annexin A6 and anti-annexin A1 antibodies were from BD Biosciences; an antiserum against SLO was from Bioacademia. Restriction endonucleases, Taq polymerase, and T4 DNA ligase were from New England Biolabs. Living Colors Fluorescent protein vectors peCFP-N1, peYFP-N1, and pmCherry-N1 were from Clontech. SureSilencing shRNA plasmids were from SA Biosciences (Frederick, MD). Other reagents were from Sigma.
Cell Culture and Transfections
Human embryonic kidney cells (HEK 293), human astrocytoma cells (U373MG), and primary smooth muscle cells (human myometrium) were maintained and transfected as described previously (18). The coding sequence of annexin A1 and annexin A6 were cloned into the Living Colors Fluorescent protein vectors following the PCR amplification from human bladder smooth muscle cDNA (19). Annexin A1-YFP (yellow fluorescent protein), annexin A1-CFP (cyan fluorescent protein), annexin A6-YFP, or annexin A6-CFP were expressed transiently in target cells (19).
Imaging
Transfected cells seeded on glass coverslips were mounted in a perfusion chamber at 25° C in Tyrode's buffer (140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES; pH = 7.4) containing 2.5 mm CaCl2. At time point = 0, the cells were challenged with 100 ng/ml (if not stated otherwise) SLO from Streptococcus pyogenes preactivated with 20 mm DTT. The fluorescence was recorded in an Axiovert 200 m microscope with a laser scanning module LSM 510 META (Zeiss) using a ×63 oil immersion lens (16). Intracellular calcium was measured with a fluorescent calcium indicator Fluo-4FF as described previously (14). The images were analyzed using the “Physiology Evaluation” software package (Zeiss).
Cell Lysis
A loss of a cytoplasmic protein (CFP or YFP) from SLO-perforated cells was analyzed as described previously (14).
RNAi Knockdown of Annexin A6 Expression
Annexin A6 knockdown experiments were performed with shRNA targeting human annexin A6 (clone 4, Pos. 2010–2030; 5′-ATGGTATCCCGCAGTGAGATT-3′) cloned into SureSilencing shRNA plasmids. Cells were transfected with shRNA using electroporation, and stable cell lines were established using puromycin resistance followed by clonal selection (20). Levels of annexin A6 and annexin A1 were assessed by Western blotting as described (21).
Ca2+ Sensitivities of Annexin A1 and Annexin A6 Plasmalemmal Translocations
The plasmalemmal translocations of annexin A1-YFP or annexin A6-YFP were recorded in HEK 293 cells maintained in Tyrode's buffer containing varying concentrations of CaCl2 buffered with 5 mm HEDTA or EGTA. The concentration of free Ca2+ in the extracellular milieu ([Ca2+]e,free) was determined using MaxChelator software (22). Cells were challenged with SLO (400 ng/ml) at the time point t = 0. High SLO concentrations used in these experiments allowed efficient plasmalemmal permeabilization, which resulted in an equilibration between the limited intracellular compartment and the infinitely larger extracellular space ([Ca2+]i = [Ca2+]e).
FACS
Non-transfected HEK 293 cells, cells co-transfected with annexin A1-YFP and annexin A6-mCherry, cell transfected with annexin A1-YFP alone, or cells transfected with annexin A6-mCherry alone were grown to confluence. 1 × 107 cells were used per experiment. To generate microvesicles, cells were washed three times with Tyrode's buffer containing 2.5 mm CaCl2 and challenged for 20 min with SLO (100 ng/ml) preactivated with 20 mm DTT. The microvesicle-containing buffer was collected, centrifuged first for 5 min at 2000 rpm and then for 60 min at 40,000 rpm. The pellet containing microvesicles was resuspended in PBS and analyzed by FACS (SORB LSRII, Becton Dickinson). YFP and mCherry were exited at 488 and 561 nm, respectively. The emitted light was collected using 525/50 nm and 610/20 nm filters. Single-transfected cells were used for gating the negative signal. The Flowjo program suite (version 9.2) was used for data analysis.
Electron Microscopy and Immunogold Labeling
Microvesicles from nontransfected confluent HEK 293 cells were obtained as described above. The microvesicle-containing buffer was filtered through a Whatman syringe filter with a pore size of 0.2 μm. The polypropylene housing was forced open, and the filter, containing the microvesicles trapped in the mesh on its upper side, was retrieved and cut into segments of ∼3 × 3 mm. The pieces were either immediately subjected to high pressure freezing and freeze substitution or used for antibody immunogold-labeling before freezing. After incubation for 1 h with monoclonal antibodies against annexin A1, A6, an antiserum against SLO, or a nonbinding control antibody, the samples were washed in Tyrode's solution containing 2.5 mm Ca2+ and reacted with secondary antibodies (IgG goat anti-mouse, goat anti-rabbit 10 nm gold (Sigma) for 1 h and briefly washed in Tyrode's solution containing 2.5 mm Ca2+. Then, the filter pieces were frozen with a pressure of 210MPa (using a Leica EMPACT instrument (FEI Company, The Netherlands)) and simultaneously cooled down to −196 °C by a double jet of liquid nitrogen, followed by freeze substitution. In brief, the samples were dehydrated at −90 °C, treated with 2% osmium tetroxide at −70 °C, and embedded in Epon at temperatures between −50 °C and ambient temperature. Ultrathin sections were prepared and viewed, without contrasting, in a Philips CM12 transmission electron microscope. Images were recorded using Olympus analySIS software.
RESULTS
Annexins A1 and A6 Participate in Repair of Plasmalemmal Injuries
We have shown recently that the down-regulation of annexin A1 expression levels using annexin A1-specific siRNA significantly decreased the capability of HEK 293 cells to repair plasmalemmal lesions induced by SLO (14). To establish the role of other members of the annexin protein family in plasmalemmal repair, annexin A6-specific siRNA was stably expressed in HEK 293 cells. In these cells, a 2-fold reduction of annexin A6 protein levels (Fig. 1A) resulted in a significant acceleration of SLO-induced lysis (Fig. 1B), whereas scrambled siRNA failed to decrease annexin A6 levels (Fig. 1A) and had no effect on cell lysis (Fig. 1B). In cells expressing annexin A6-specific siRNA, the expression levels of annexin A1 remained unchanged (supplemental Fig. S1A); a similar result was observed for annexin A6 in cells expressing annexin A1-specific siRNA (supplemental Fig. S1B, see also Ref. 14).
FIGURE 1.

Down-regulation of annexin A6 enhances lysis in SLO-perforated cells. A, annexin A6 expression in wild type HEK 293 cells (WT) or in HEK 293 cells transfected with either annexin A6-specific siRNA (siRNA) or scrambled siRNA (Scr) was analyzed by Western blotting using a monoclonal annexin A6-specific antibody. Equal amounts of WT cells, siRNA cells, or scrambled siRNA cells were initially collected. To generate a calibration curve for the determination of annexin A6 expression in siRNA cells, the samples of WT cells were subjected to serial dilution (dilution factor = 3). An undiluted sample of siRNA or scrambled siRNA cells was run on the same gel. Experiments were performed in triplicate. The blots were analyzed using Image Quant software (version 3.3). The averaged intensity of the annexin A6 bands obtained for individual siRNA cell samples (square) was fitted manually to the averaged WT calibration curve (circles). A 2.35-fold (57 ± 5.9%) reduction of annexin A6 protein levels was observed in siRNA-expressing cells, whereas transfection with scrambled siRNA had no effect. B, SLO (100 ng/ml) induced lysis was significantly accelerated in HEK 293 cells stably expressing annexin A6-specific siRNA compared either with wild type HEK 293 or HEK 293 cells transfected with scrambled siRNA (WT, 207 individual cells (n = 7); siRNA, 230 individual cells (n = 7); scrambled siRNA, 125 individual cells (n = 5); t test (p = 0.002)). Mean values (±S.E.) are presented.
Annexin A6 Responds Faster to Plasmalemmal Injury Than Annexin A1
An elevation in [Ca2+]i, presumably, the earliest event occurring in the cytoplasm of injured cells (15), leads to the translocation of cytoplasmic annexins to the plasmalemma (16, 19, 23). In SLO-permeabilized cells, annexin A6 was more sensitive to [Ca2+] elevation than annexin A1; the half-maximal [Ca2+] required for the translocation of annexin A6 was 8.12 ± 0.199 μm (Fig. 2A), whereas that of annexin A1 was 19.0 ± 0.64 μm (Fig. 2B). In case of annexin A1 (Fig. 2B), the cytoplasmic protein initially translocated to the plasmalemma and was subsequently internalized (18). High SLO concentrations (400 ng/ml) were used in these experiments to prevent plasmalemmal resealing (18) and, thus, to allow efficient [Ca2+] equilibration between intra- and extracellular compartments.
FIGURE 2.

Ca2+ sensitivities of annexin A1 and annexin A6 plasmalemmal translocations. A and B, HEK 293 cells were transfected with annexin (Anx) A1-YFP or annexin A6-YFP. SLO (400 ng/ml) was added to the cells maintained in sodium-Tyrode buffer containing varying concentrations of free Ca2+. SLO forms pores in the plasma membrane, which are large enough to allow a passage of cytoplasmic proteins (32). Correspondingly, annexin A6 (A) and annexin A1 (B) leaked out of the cells, provided [Ca2+]e was below the concentration that allows their binding to the plasma membrane (see [Ca2+]e < 100 nm). However, they were retained by cells at a [Ca2+]e high enough to support their membrane association. In the case of annexin A1, the cytoplasmic protein initially translocated to the plasmalemma and was subsequently internalized, whereas the intranuclear protein was bound to the membrane of the nuclear envelope (18). To obtain a dose-response curve, the percentage of cells, in which annexins were retained, was established for every calcium concentration tested (for each point at least 79 cells were recorded in five separate experiments). The data points were fitted with a sigmoid curve to estimate the half maximal [Ca2+] of annexin-membrane translocations. Mean values (±S.E.) are presented. Scale bar, 10 μm.
Due to its higher Ca2+ sensitivity, the plasmalemmal translocation of annexin A6 precedes that of annexin A1 in SLO-permeabilized cells (16, 18), suggesting that annexin A6 might be more efficient in recognizing and eliminating plasmalemmal injuries. This was obvious in those cells in which the pore formation and their subsequent resealing, accompanied by the reversible, “on-off” translocation of annexin A6, occurred without any noticeable translocation of annexin A1 (Fig. 3 and supplemental Movie 1). In this set of experiments, the efficient plasmalemmal resealing was assisted by using relatively low concentrations of the toxin (50 ng/ml). In all other experiments, SLO was used at an intermediate concentration (100 ng/ml). This particular concentration was sufficiently low to allow plasmalemmal resealing in a substantial number of cells (see Fig. 1B) and, at the same time, was high enough to induce [Ca2+]i elevation required for annexin A1 translocation.
FIGURE 3.

Plasmalemmal repair accompanied by annexin A6 translocation occurs without annexin A1 involvement. HEK 293 cells were double-transfected with annexin (Anx) A6-YFP and annexin A1-CFP. SLO (50 ng/ml) was added to the cells at time point = 0. The formation of SLO-pores resulted in the elevation of [Ca2+]i accompanied by plasmalemmal translocation of annexin A6. After successful repair, the excess of Ca2+ was pumped out from the cytoplasm by intracellular Ca2+-sequestrating mechanisms. Following the decrease in [Ca2+]i, annexin A6 back-translocated to the cytoplasm. Note that [Ca2+]i never reached levels required for the plasmalemmal translocation of annexin A1. Scale bar, 10 μm.
Microvesicles Containing SLO and Either Annexin A1 or Annexin A6 Are Shed by SLO-perforated Cells
The mechanism underlying the annexin A1-dependent repair of SLO-pores is based on its ability to cross-link and fuse adjacent membranes (13, 14, 24); the regions of the plasmalemma surrounding the toxin pores are fused by annexin A1 and are shed in the form of microvesicles (16).
To characterize released microvesicles in more detail, we purified them by differential centrifugation. Flow cytometry analysis revealed that although small amounts of microvesicles were recovered from control, untreated cells (1522 ± 239, n = 3), their release from the SLO-damaged cells was greatly elevated (83,500 ± 20,615, n = 5, p = 0.003). Electron microscopy revealed that purified microvesicles varied in size from 100 nm to 1 μm (481 ± 310 nm; n = 70).
The presence of SLO and annexin A1 on the membranes of shed microvesicles was confirmed by immunogold staining (Fig. 4). Immunogold electron microscopy and laser-scanning confocal microscopy of SLO-perforated cells revealed that annexin A6 was likewise shed with the microvesicles (Fig. 4 and supplemental Fig. S2 and Movie 2). Recently, in similar experiments (see supplemental Fig. S2), we have reported the release of annexin A1-containing microvesicles but failed to report that of annexin A6-containing ones (16). This discrepancy was the result of the differences in the visibility of YFP and CFP used to label the two annexins (note a prevalence of YFP-containing microvesicles in Video 2). Careful re-examination of the published images revealed that both annexin A1-containing (YFP, yellow) and annexin A6-containing (CFP, blue) microvesicles were shed from SLO-perforated cells (see supplemental Fig. S2).
FIGURE 4.
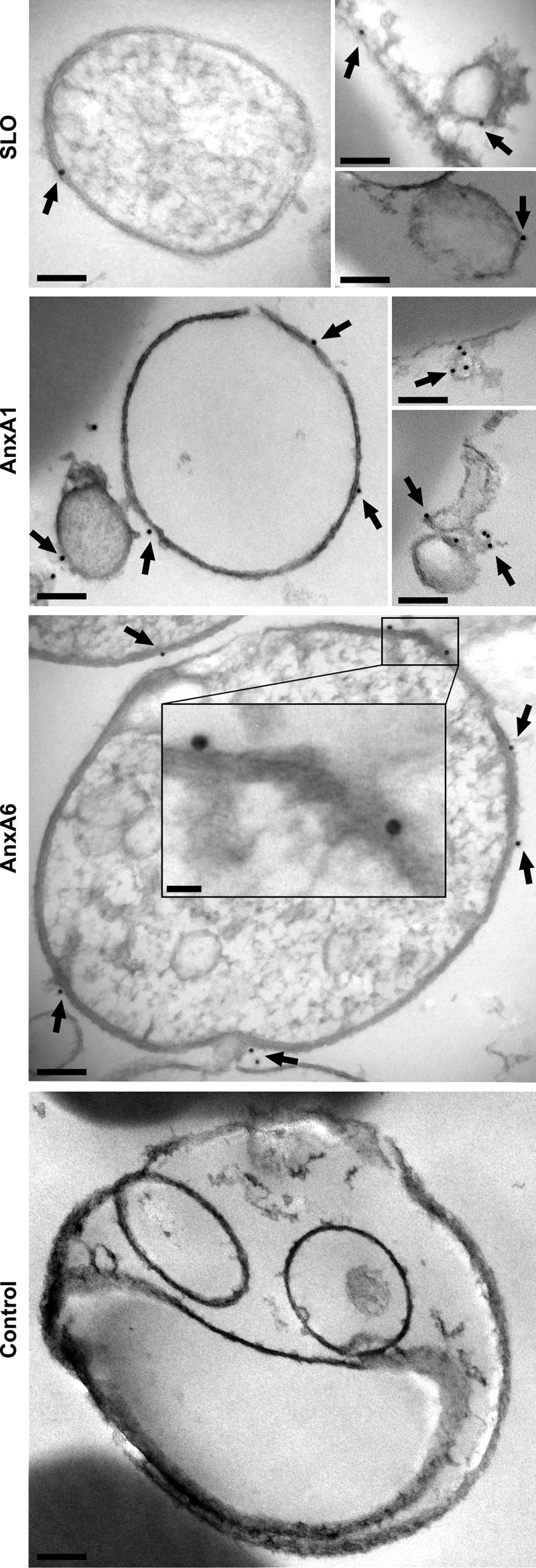
Released microvesicles contain SLO, annexin A1, and annexin A6. Electron micrographs of isolated microvesicles reacted with an antiserum against SLO (SLO) or monoclonal antibodies against annexin A1 (AnxA1) or annexin A6 (AnxA6). Immunogold labeling (arrows) is visible on the membranes of the microvesicles. Microvesicles reacted with a nonbinding primary antibody (Control) were devoid of a gold label. Scale bars, 100 nm; insert, 20 nm.
An analysis of individual microvesicles discarded by cells revealed that microvesicles containing both proteins as well as those containing individual annexins were generated (Fig. 5 and supplemental Movie 3). Flow cytometric analysis revealed that the majority of shed microvesicles contained both annexin A1 and annexin A6 (57 ± 1%; n = 3); 22 ± 0.7% of microvesicles contained only annexin A6, whereas annexin A1 alone was present in 21 ± 0.5% of microvesicles (Fig. 6). Because the relative amount of cells expressing either annexin A1 or annexin A6 in the population of double-transfected cells was <2%, the majority of microvesicles containing single annexins were shed from cells that simultaneously expressed both annexins.
FIGURE 5.

Microvesicles containing different annexins are shed by SLO-perforated cells. HEK 293 cells double-transfected with annexin A6 (AnxA6)-YFP and annexin A1 (AnxA1)-CFP were treated with SLO (100 ng/ml) at time point = 0. As SLO permeabilization and ensuing plasmalemmal repair progress, the microvesicles containing either both annexins (Region of interest (ROI) 1) or individual annexins (Regions of interest (ROI) 2 and 3; white arrows, annexin A1, yellow arrows, annexin A6) appear in the extracellular milieu. Scale bar, 10 μm.
FIGURE 6.

Microvesicles containing different annexins are shed by SLO-perforated cells: FACS analysis. Nontransfected HEK 293 cells or cells transfected with either annexin A6 (AnxA6)-YFP or annexin A1 (AnxA1)-mCherry or double-transfected with annexin A6-YFP and annexin A1-mCherry were treated with SLO (100 ng/ml) at time point = 0. Microvesicles released from the SLO-treated cells were purified by differential centrifugation and subjected to FACS analysis. A, microvesicles released from nontransfected HEK 293 cells, cells transfected with annexin A6-YFP, or annexin A1-mCherry were used for gating; quadrant gates were set to segregate fluorophores (≤1%) as well as the negative signal from nontransfected cells (≤5%). B, cells double-transfected with annexin A6-YFP and annexin A1-mCherry were used to estimate the amount of microvesicles, which contained either individual annexins (annexin A1, upper-left quadrant or annexin A6, lower-right quadrant) or both annexin A1 and annexin A6 (upper-right quadrant). C, the graph shows the relative amounts of different microvesicles released by double-transfected cells (n = 3); the microvesicles that contained neither annexin (26 ± 2.3%, lower-left quadrant) were excluded from the analysis. Mean values (±S.E.) are present.
Annexin A6 and Annexin A1 Translocate to Spatially Distinct Regions of Plasmalemma
Shedding of microvesicles containing either annexin A6 or annexin A1 suggests that the annexins bind to spatially distinct regions of the plasmalemma. To establish whether the intracellular dynamics of annexins A1 and A6 differ spatially, we simultaneously monitored the translocations of both annexins in HEK 293 cells double-transfected with annexin A1-YFP and annexin A6-CFP. The spatial segregation between the two annexins is, most likely, governed by the intracellular gradients of [Ca2+] that direct the plasmalemmal binding of a particular annexin in accord with its Ca2+ sensitivity. Due to the restricted diffusion within elongated thin cellular protrusions, significant [Ca2+]i gradients are likely to develop between the protrusions and a voluminous, spherical cell body. A preferential accumulation of Ca2+ within cellular protrusions was indeed observed in cells loaded with a calcium-sensitive dye Fluo-4FF (Fig. 7A and supplemental Movie 4). The localized intraprotrusion [Ca2+]i elevation led to spatially restricted translocation of annexin A6 (Fig. 7, B and C).
FIGURE 7.

Localized intraprotrusion [Ca2+]i elevation accompanied by spatially restricted translocation of annexin A6. A, HEK 293 cells loaded with Ca2+-sensitive dye Fluo-4-FF were challenged with SLO (50 ng/ml) at time point = 0. Arrows denote cellular protrusions with elevated [Ca2+]i. B and C, U373MG cells transfected with annexin A6 (AnxA6)-mCherry and loaded with the Ca2+-sensitive dye Fluo-4-FF were challenged with SLO (50 ng/ml) at time point = 0. Arrows denote cellular protrusions with elevated (relative to the cell body) [Ca2+]i. Note the simultaneous translocation of annexin A6 within the protrusions. The graph in C shows changes in Fluo-4-FF fluorescence (Fluo4-FF, red and black diamonds) and annexin A6-mCherry fluorescence (Anx6, red and black circles) in the cytoplasm of the protrusion (red symbols) and in the cytoplasm of the cell body (black symbols). The transient drop in the annexin A6 fluorescence within the protrusion (red circles) is due to its transient translocation to the plasmalemma in response to the spatially and temporary restricted [Ca2+]I elevation (red diamonds). a.u., fluorescence intensity units.
Therefore, our analysis of the spatial segregation between annexins A1 and A6 was directed toward the cells displaying well developed protrusions. In intact cells, the annexins co-localized within the cytoplasm (Fig. 8A, time after SLO addition = 479 s, and B, time after SLO addition = 186 s) (16). Annexin A1 was additionally localized within the nucleus (Fig. 8) (16). An efficient membrane permeabilization resulted in an increase in [Ca2+]i (Fig. 7) (18), which led to the translocation of annexin A6 from the cytoplasm to the plasma membrane (Fig. 8, A, time after SLO addition = 647 s and B, time after SLO addition = 239 s). At these time points, annexin A1 was still diffusely distributed within the cytoplasm. Later, a localized translocation of annexin A1 took place within the cellular protrusions (Fig. 8, A, time after SLO addition = 745 s, and B, time after SLO addition = 630 s). Subsequently, most of the cellular annexin A1 concentrated within the protrusions moving away from the cell body (note the conspicuous intranuclear annexin A1 staining). In the cell body, annexin A1 was associated with spatially restricted plasmalemmal puncta (Fig. 8, see arrows). In contrast, annexin A6 decorated the plasmalemma of the cell body more homogeneously and was absent from the plasma membrane of the protrusions (Fig. 8). The opposite movements of the annexins resulted in their almost complete segregation in the plane of the plasma membrane (Fig. 8 and supplemental Movie 5).
FIGURE 8.

Temporal and spatial segregation of plasmalemmal translocations of annexins A1 and A6. A and B, HEK 293 cells double-transfected with annexin A6 (AnxA6)-CFP and annexin A1 (AnxA1)-YFP were treated with SLO (100 ng/ml) at time point = 0. The intracellular dynamics of two annexins were analyzed by confocal microscopy. Two individual cells are shown. Arrows denote plasmalemmal puncta of annexin A1 within the cell body. Scale bar, 10 μm.
Within thin protrusions, the plasmalemmal association of the annexins and their segregation often advanced in the form of dynamic waves (Fig. 9 and supplemental Movie 6). The cell, in the figure and movie, responded to a SLO attack by a global translocation of annexin A6 occurring within the cell body and within the initial segment of the cell protrusion (Fig. 9, time after SLO addition = 896 s). Simultaneously, a local translocation of annexin A1 occurred, which initially was confined to the base of the cellular protrusion. Subsequently, annexin A1 moved along the protrusion and concentrated at its very tip (Fig. 9, times after SLO addition = 922–984 s, and supplemental Movie 6). The wave of annexin A6 within the protrusion followed that of annexin A1 and occurred only after annexin A6 had dissociated from the plasma membrane of the cell body (Fig. 9, times after SLO addition = 922–984 s, and supplemental Movie 6).
FIGURE 9.

Wave-like propagation of annexins A1 and A6 along the plasmalemma of a cellular protrusion. HEK 293 cells double-transfected with annexin A6 (AnxA6)-CFP and annexin A1 (AnxA1)-YFP were treated with SLO (100 ng/ml) at time point = 0. The intracellular dynamics of two annexins were analyzed by confocal microscopy. Scale bar, 10 μm.
In contrast to spherical cells, the SLO-induced segregation between the two annexins in flat cells manifested itself in the form of transient local accumulations of either annexin A1 or annexin A6 at the plane of the plasma membrane (supplemental Movie 7). The segregation of the annexins was observed not only in immortalized cell lines but also in primary cultures of human smooth muscle cells (Fig. 10 and supplemental Movie 8).
FIGURE 10.

Spatial segregation of annexins A1 and A6 plasmalemmal translocations in primary smooth muscle cells. Primary smooth muscle cells derived from human myometrium were double-transfected with annexin A6 (AnxA6)-CFP and annexin A1 (AnxA1)-YFP and treated with SLO (100 ng/ml) at time point = 0. The intracellular dynamics of two annexins were analyzed by confocal microscopy. Two different focal planes of the same cell are shown: (− SLO), before SLO was added to the cells; (+ SLO), 10 min after SLO addition. Arrows point to the regions of the plasmalemma at which the segregation of the annexins occurs. Scale bar, 10 μm.
DISCUSSION
Plasmalemmal injury is a common event in the life of cells that often leads to necrosis or apoptosis. Nucleated cells survive the disruption of their plasma membrane by a process of resealing (25–31). Recent data suggest that annexins might play a leading role in this process, and the role of annexin A1 in this event has been confirmed (13, 14). Here, we demonstrate that annexin A6, another member of the annexin protein family, contributes to plasmalemmal resealing.
Our findings suggest a cooperative role of annexins A1 and A6 in the repair of plasmalemmal injury (Fig. 11A). In an uninjured cell, [Ca2+]i is uniformly low, and the annexins are distributed diffusely within the cytoplasm (Fig. 11A, step 1). After binding to the plasma membrane, the toxin oligomerizes and forms active pores (Fig. 11A, step 2, magnified view), and the number of active pores is dependent on the toxin concentration and on the ability of the cell to neutralize the pores, i.e. on the activity of cellular repair mechanisms. The formation of an active SLO pore leads to an elevation in [Ca2+]i, which enters the cell from the extracellular milieu (Fig. 11A, step 2). Provided the injury is not extensive, such an elevation is localized within the cytoplasmic region adjacent to the injured plasmalemma. Within the rest of the cytoplasm, the endoplasmic reticulum and the mitochondria actively sequestrate excessive Ca2+. Thus, a Ca2+ hot-spot forms in the cytoplasm surrounding the SLO pore, where it is recognized promptly by the highly Ca2+-sensitive annexin A6. In the presence of elevated [Ca2+], cytoplasmic annexin A6 binds to the injured plasmalemma and seals off its damaged portions, which are subsequently released in the form of annexin A6-containing microvesicles (Fig. 11A, step 2a). Following a complete sequestration of the excessive intracellular Ca2+, the cell returns to its native, uninjured state (Fig. 11, step 1).
FIGURE 11.

A cooperative action of annexins A1 and A6 in plasmalemmal repair. A, the scheme depicts the repair of a single SLO pore (steps 2 and 2a) or that of three SLO pores assembled simultaneously (steps 3 and 3a). B, the scheme depicts the repair of a single SLO pore assembled within the plasmalemma of the cell body (step 2) or that of two pores assembled within the plasmalemma of a thin cellular protrusion (steps 3–5). Two annexins, which differ in their Ca2+ sensitivity of plasmalemmal association, are shown by blue crosses (the annexin with a higher Ca2+ sensitivity, annexin A6) and orange circles (the annexin with a lower Ca2+-sensitivity, annexin A1). The green lines depict spatiotemporal variations in [Ca2+]i corresponding to each step. Ca2+-sequestrating organelles (endoplasmic reticulum, mitochondria, not shown) are preferentially localized in the perinuclear region (n = nucleus).
However, should several toxin pores assemble in rapid succession, an ensuing globalized elevation in [Ca2+]i drives an undirected binding of annexin A6 even to the uninjured plasmalemma (Fig 11A, step 3). A rapid depletion of the free cytoplasmic pool of this protein makes it no longer available for the repair of the already existing or newly formed pores. In the presence of active, unrepaired SLO pores, the [Ca2+]i continues to rise. In the vicinity of the pores the local [Ca2+]i increases beyond the concentration required for the plasmalemmal translocation of annexin A1. A second round of plasmalemmal repair, driven by a less Ca2+-sensitive member of the annexin family ensues; it is accompanied by the release of annexin A1-containing microvesicles (Fig 11A, steps 3 and 3a).
Our data also reveal that the geometry of a cell plays an important role in plasmalemmal repair. As shown in Fig. 11B, the formation of a single SLO pore within the plasmalemma of the cell body results in a limited elevation in [Ca2+]i (Fig. 11B, step 2). The [Ca2+] elevation is restricted by its diffusion within the cytoplasm of the bulky cell body where it is actively sequestrated within the endoplasmic reticulum and mitochondria. Thus, within the cell body, the limited [Ca2+]i elevation favors the highly Ca2+-sensitive annexin A6-dependent repair. However, even a single pore induces a much higher increase in [Ca2+]i within a thin cellular protrusion (Fig. 11B, step 3). The reasons are 2-fold; first, such protrusions do not contain Ca2+-sequestrating organelles, and second, a much smaller volume favors faster diffusion of the inflowing Ca2+ in the cytoplasm of the protrusion. Thus, within the protrusion, an explosion-like elevation of [Ca2+]i would favor an instant, ineffective binding of annexin A6 to the plasmalemma. Moreover, a depleted intraprotrusion annexin A6 cannot be replenished from its vast pool of the cell body cytoplasm. Small Ca2+ ions, entering a cell body at the site of the protrusion, diffuse much faster than bulky protein molecules. Thus, a [Ca2+]i elevation reaches the cytoplasm of the cell body before any substantial amount of annexin A6 is able to diffuse into the protrusion from the cell body. Therefore, the inflowing annexin A6 is arrested, due to its binding to the uninjured sarcolemma, at the site of the protrusion-cell body contact and, thus, unable to reach the site of the injury (Fig. 11B, step 3).
In contrast, the depleted annexin A1 of the protrusion can be replenished from its cell body pool due to the low Ca2+ sensitivity of the protein. The directed movement of free annexin A1 from the cell body toward the lesion allows the protein to reach concentrations required for an efficient plasmalemmal repair (Fig. 11B, step 4). Successful repair leads to a decrease in [Ca2+]i that is followed by the dissociation of the annexins from the plasma membrane. Annexin A1 now becomes available for an additional round of repair and is recruited to another lesion (Fig. 11B, step 5). The directed movements of the annexins between the already repaired and still active lesions manifest themselves in the form of intracellular annexin waves (see Fig. 9).
By expressing several annexins with different Ca2+ sensitivities, a cell is able to react promptly to a limited injury in its early stages and, at the same time, to withstand a sustained injury accompanied by the continuous formation of plasmalemmal lesions. The first task is assigned to the most Ca2+-sensitive annexins (here, annexin A6) which are able to react to initially small increases in [Ca2+]i but are depleted quickly due to an unproductive binding to the uninjured plasmalemma. In contrast, the annexins with low Ca2+ sensitivity (here, annexin A1) are ineffective during the early stages of injury, an efficient plasmalemmal repair can occur even without them. However, they are indispensable when a cell faces a sustained injury. Serving as “strategic reserve,” they are deployed when their highly Ca2+-sensitive kin fails. Within a single cell, a number of annexins with overlapping Ca2+ sensitivities allow a continuous fine tuning of the repair responses. Moreover, by varying the expression levels of different annexins, a cell is able to set its own Ca2+ tolerance limits to fit precisely its physiological requirements.
Supplementary Material
This work was supported by Swiss National Science Foundation Grants 3100A0-121980/1 (to E. B. B.) and 320030_128064/1 (to A. D.) and the National Research Programme Grant NRP 53 “Musculoskeletal Health-Chronic Pain” 405340-104679/1 (to A. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Movies 1–8.
- SLO
- streptolysin O
- CFP
- cyan fluorescent protein
- HEDTA
- N-(2-Hydroxyethyl)ethylenediamine-N,N′,N′-triacetic acid.
REFERENCES
- 1. Morgan R. O., Fernández M. P. (1997) Cell Mol. Life Sci. 53, 508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerke V., Moss S. E. (2002) Physiol. Rev. 82, 331–371 [DOI] [PubMed] [Google Scholar]
- 3. Raynal P., Pollard H. B. (1994) Biochim. Biophys. Acta 1197, 63–93 [DOI] [PubMed] [Google Scholar]
- 4. Gerke V., Creutz C. E., Moss S. E. (2005) Nat. Rev. Mol. Cell Biol. 6, 449–461 [DOI] [PubMed] [Google Scholar]
- 5. Monastyrskaya K., Babiychuk E. B., Draeger A. (2009) Cell Mol. Life Sci. 66, 2623–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grewal T., Enrich C. (2009) Cell Signal. 21, 847–858 [DOI] [PubMed] [Google Scholar]
- 7. Babiychuk E. B., Draeger A. (2000) J. Cell Biol. 150, 1113–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buckingham J. C., Flower R. J. (1997) Mol. Med. Today 3, 296–302 [DOI] [PubMed] [Google Scholar]
- 9. Perretti M., D'Acquisto F. (2009) Nat. Rev. Immunol. 9, 62–70 [DOI] [PubMed] [Google Scholar]
- 10. John C. D., Gavins F. N., Buss N. A., Cover P. O., Buckingham J. C. (2008) Curr. Opin. Pharmacol. 8, 765–776 [DOI] [PubMed] [Google Scholar]
- 11. Rescher U., Gerke V. (2008) Pflugers Arch. 455, 575–582 [DOI] [PubMed] [Google Scholar]
- 12. Chasserot-Golaz S., Vitale N., Umbrecht-Jenck E., Knight D., Gerke V., Bader M. F. (2005) Mol. Biol. Cell 16, 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McNeil A. K., Rescher U., Gerke V., McNeil P. L. (2006) J. Biol. Chem. 281, 35202–35207 [DOI] [PubMed] [Google Scholar]
- 14. Babiychuk E. B., Monastyrskaya K., Potez S., Draeger A. (2011) Cell Death. Differ. 18, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan B. P., Luzio J. P., Campbell A. K. (1986) Cell Calcium 7, 399–411 [DOI] [PubMed] [Google Scholar]
- 16. Babiychuk E. B., Monastyrskaya K., Potez S., Draeger A. (2009) Cell Death. Differ. 16, 1126–1134 [DOI] [PubMed] [Google Scholar]
- 17. Moskovich O., Fishelson Z. (2007) J. Biol. Chem. 282, 29977–29986 [DOI] [PubMed] [Google Scholar]
- 18. Babiychuk E. B., Monastyrskaya K., Draeger A. (2008) Traffic. 9, 1757–1775 [DOI] [PubMed] [Google Scholar]
- 19. Monastyrskaya K., Babiychuk E. B., Hostettler A., Rescher U., Draeger A. (2007) Cell Calcium 41, 207–219 [DOI] [PubMed] [Google Scholar]
- 20. Monastyrskaya K., Babiychuk E. B., Hostettler A., Wood P., Grewal T., Draeger A. (2009) J. Biol. Chem. 284, 17227–17242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Babiychuk E. B., Draeger A. (2006) Biochem. J. 397, 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patton C., Thompson S., Epel D. (2004) Cell Calcium 35, 427–431 [DOI] [PubMed] [Google Scholar]
- 23. Skrahina T., Piljić A., Schultz C. (2008) Exp. Cell Res. 314, 1039–1047 [DOI] [PubMed] [Google Scholar]
- 24. White I. J., Bailey L. M., Aghakhani M. R., Moss S. E., Futter C. E. (2006) EMBO J. 25, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McNeil P. L., Steinhardt R. A. (2003) Annu. Rev. Cell Dev. Biol. 19, 697–731 [DOI] [PubMed] [Google Scholar]
- 26. McNeil P. L., Kirchhausen T. (2005) Nat. Rev. Mol. Cell Biol. 6, 499–505 [DOI] [PubMed] [Google Scholar]
- 27. Idone V., Tam C., Andrews N. W. (2008) Trends Cell Biol. 18, 552–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schapire A. L., Valpuesta V., Botella M. A. (2009) Trends Plant Sci. 14, 645–652 [DOI] [PubMed] [Google Scholar]
- 29. Tam C., Idone V., Devlin C., Fernandes M. C., Flannery A., He X., Schuchman E., Tabas I., Andrews N. W. (2010) J. Cell Biol. 189, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thiery J., Keefe D., Saffarian S., Martinvalet D., Walch M., Boucrot E., Kirchhausen T., Lieberman J. (2010) Blood 115, 1582–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambert O., Gerke V., Bader M. F., Porte F., Brisson A. (1997) J. Mol. Biol. 272, 42–55 [DOI] [PubMed] [Google Scholar]
- 32. Walev I., Bhakdi S. C., Hofmann F., Djonder N., Valeva A., Aktories K., Bhakdi S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3185–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.