

Abstract
The multimodular guanine nucleotide exchange factors (GEFs) of the Dbl family mostly share a tandem Dbl homology (DH) and pleckstrin homology (PH) domain organization. The function of these and other domains in the DH-mediated regulation of the GDP/GTP exchange reaction of the Rho proteins is the subject of intensive investigations. This comparative study presents detailed kinetic data on specificity, activity, and regulation of the catalytic DH domains of four GEFs, namely p115, p190, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG). We demonstrate that (i) these GEFs are specific guanine nucleotide exchange factors for the Rho isoforms (RhoA, RhoB, and RhoC) and inactive toward other members of the Rho family, including Rac1, Cdc42, and TC10. (ii) The DH domain of LARG exhibits the highest catalytic activity reported for a Dbl protein till now with a maximal acceleration of the nucleotide exchange by 107-fold, which is at least as efficient as reported for GEFs specific for Ran or the bacterial toxin SopE. (iii) A novel regulatory region at the N terminus of the DH domain is involved in its association with GDP-bound RhoA monitored by a fluorescently labeled RhoA. (iv) The tandem PH domains of p115 and PRG efficiently contribute to the DH-mediated nucleotide exchange reaction. (v) In contrast to the isolated DH or DH-PH domains, a p115 fragment encompassing both the regulator of G-protein signaling and the DH domains revealed a significantly reduced GEF activity, supporting the proposed models of an intramolecular autoinhibitory mechanism for p115-like RhoGEFs.
Keywords: Enzyme Catalysis, G Proteins, Protein Domains, Protein Motifs, Protein Structure, Dbl Homology, Fluorescence Spectroscopy, GEFs, Rho GTPases, Exchange Factors
Introduction
The small GDP/GTP-binding proteins (GTPases)4 of the Rho family are key regulators in a multitude of cellular processes (1, 2). Like almost all GTPases, the Rho proteins function as binary switches, cycling between an inactive GDP-bound state and an active GTP-bound state (3, 4). In response to diverse extracellular stimuli, guanine nucleotide exchange factors (GEFs) catalyze the exchange of bound GDP for cellularly abundant GTP in their cognate GTPase substrates and thereby initiate Rho signaling cascades (5–12).
The common structural module of Dbl family GEFs (69 members known), which is responsible for the nucleotide exchange activity, consists of a Dbl homology (DH) domain and an adjacent pleckstrin homology (PH) domain C-terminal to the DH domain (9, 11). The DH domain makes extensive contacts with switch I and II regions of RhoGTPases and contains virtually all the residues required for substrate recognition, binding, and guanine nucleotide exchange (9, 11)). The nearly invariant domain organization of the DH-PH tandem in all members of the Dbl family presumes a conserved function for the PH domain. However, a clear role of the tandem PH domain has not yet been established. In some cases, the PH domain seems to facilitate the catalytic activity of the DH domain. For example, residues within the PH domain of Dbs interact directly with the bound RhoGTPase (13, 14) and enhance nucleotide exchange on Cdc42 and RhoA (14, 15). A similar scenario has been reported for the PH domains of PDZ-RhoGEF/GTRAP48 (hereafter called PRG) and leukemia-associated RhoGEF (LARG) (16, 17). Conversely, the PH domains of son of sevenless homolog 1 (Sos1) and Trio-N appear to inhibit nucleotide exchange on Rac1 and RhoG, respectively (18, 19). The tandem PH domain of other GEFs, including Tiam1, Intersectin1 (ITSN1), and Pem-2/Collybistin II, has been shown not to contact the respective GTPase at all (13, 20, 21).
In contrast to the conservation of the DH-PH tandem, the GEFs of the Dbl family also exhibit a variety of functional domain compositions and domain organizations (7, 8, 11, 22), which link their GEF activity to specific signaling events. Interesting examples in this regard are regulator of G-protein signaling (RGS) domain-containing RhoGEFs, such as p115, PRG, and LARG. The RGS domain at the N terminus of p115 directly links the heterotrimeric G proteins Gα12/13 to RhoA regulation and acts as a GTPase-activating protein for Gα12/13 (23, 24). The association of p115 with Gα12 and Gα13 has been suggested to activate its GEF function toward Rho proteins (25–28). A similar regulatory model has been proposed for PRG and LARG (29–36). Recently, Zheng et al. (37) provided direct biochemical evidence for an autoinhibitory RGS-mediated regulation of the DH domain.
Despite intensive research, there is little comparative analysis of these RGS-containing GEFs available. Thus, we purified different protein domains of p190, p115, PRG, and LARG (Fig. 1) and characterized them functionally regarding their specificity, activity, and regulation with respect to each other. We measured their effects on the DH-catalyzed nucleotide exchange of RhoGTPases by means of fluorescence spectroscopy utilizing both GTPases loaded with fluorescently labeled guanine nucleotides (38) and a fluorescent RhoA itself (RhoA(V33C)-AEDANS) (this study). p190, a Rho-specific GEF (39), was used as a control. Our results suggest that the PH domains of PRG and p115 participate in the DH-catalyzed exchange but not in the association reaction and that the N-terminal regions of p115 possibly represent an autoregulatory module. In addition, we demonstrate that these four GEFs are specific for the Rho isoforms (RhoA, RhoB, and RhoC) and are able to catalyze their very slow intrinsic nucleotide dissociation up to 7 orders of magnitude beyond the capability of any other GEF reported so far.
FIGURE 1.

Schematic representation of domain organization and different constructs of p115, p190, PRG, and LARG used in this study. The numbers indicate the N- and C-terminal amino acids of the respective constructs. DH-PHn, DH-PHc, and DH-PHcn are shorter variants at the N and C termini of p115 DH-PH that are equivalent to LARG DH-PH (see supplemental Fig. S2A). DH-PHcnΔN and DH-PHΔN are N-terminally deleted variants of p115 and LARG and equivalent to each other. DH-PHΔN2m contains two point mutations at positions Asn946 and Lys949 that are substituted by Ser and Gln, the corresponding residues in p115. C1, cysteine-rich region; cc, coiled coil; L, leucine-rich; P, proline-rich.
EXPERIMENTAL PROCEDURES
Constructs
Constructs of human p115 DH (residues 382–645), DH-PH (residues 382–786), DH-PHc (residues 382–766), DH-PHcn (residues 396–766), DH-PHcnΔN (residues 411–766), RGS (residues 1–252), RGS-Linker (residues 40–400), Linker (residues 234–400), RGS-Linker-DH (residues 40–645), and Linker-DH (residues 234–645); murine p190 DH (residues 811–1081) and DH-PH (residues 811–1210); human PRG DH (residues 712–963), DHs (residues 729–939), and DH-PH (residues 712–1081); human LARG DH (residues 766–986), DH-PH (residues 766–1138), and DH-PHΔN (residues 782–1138); and C-terminal truncated human RhoA (residues 1–181), RhoB (residues 1–181), RhoC (residues 1–181), Cdc42 (residues 1–178), Rac1 (residues 1–184), and TC10 (residues 1–193) were amplified by standard PCR and cloned in pGEX-4T1 and pGEX-4T1-Ntev vector, respectively. Point mutations in RhoA (residues 1–181) at position Val33 to Cys and in LARG DH-PHΔN at positions Asn946 to Ser and Lys949 to Gln were generated using the QuikChangeTM site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing.
Proteins
All proteins were expressed as glutathione S-transferase (GST) fusion proteins in Escherichia coli BL21(DE3)pLyS or alternatively CodonPlusRIL, isolated in a first step by affinity chromatography on a glutathione-Sepharose column, and purified after proteolytic cleavage of GST in a second step by size exclusion chromatography (Superdex S200) as described (38). GTPases either in complex with non-labeled nucleotides (GDP or GppNHp), with fluorescent nucleotides (methylanthraniloyl-GDP (mantGDP) or mantGppNHp), or without nucleotide (the nucleotide-free form) were prepared as described (38). Purified proteins were snap frozen in liquid nitrogen and stored at −80 °C.
Fluorescence Labeling of RhoA with AEDANS
For coupling the AEDANS fluorescence reporter group, purified GDP-bound RhoA(V33C) was transferred to buffer containing 50 mm Tris/HCl, pH 7.5, 5 mm MgCl2, 2 mm ascorbate by repeated dilution and ultrafiltration steps. Protein was then incubated overnight with a 10-fold excess of 1,5-I-AEDANS (Sigma). It should be mentioned that other fluorescence reporter groups, such as fluorescein, Alexa Fluor, and pyrene, were not tested because they are proven to be less environmentally sensitive or result in precipitation of the proteins after labeling (40). The reaction was stopped by adding of dithioerythritol in excess. Unbound AEDANS was removed by sequential dilution and ultrafiltration steps. The efficiency of the labeling reaction was analyzed by mass spectrometry.
Fluorescence Measurements
All fluorescence measurements were performed in 30 mm Tris/HCl, pH 7.5, 10 mm K2HPO4/KH2PO4, pH 7.5, 5 mm MgCl2, 3 mm DTT at 25 °C. The mantGDP dissociation rates from RhoA (0.1 μm) were measured in the absence and presence of different amounts of respective DH proteins as described previously for Rac proteins (38, 41, 42). Fast kinetics (<1000 s) were performed with an Applied Photophysics (SX18MV) or with a Hi-Tech Scientific (SF-61) stopped-flow instrument, respectively. The excitation wavelengths were 366 nm for mant and 350 nm for AEDANS. Emission was detected through a cutoff filter of 408 nm for both mant and AEDANS. Slow kinetics (>1000 s) were measured on a PerkinElmer Life Sciences spectrofluorometer (LS50B) or on a FluoroMax spectrofluorometer (SPEX Instruments, Edison, NJ), respectively, using an excitation wavelength of 366 nm for mant and 350 nm for AEDANS and an emission wavelength of 450 nm for mant and 490 nm for AEDANS. Data were processed as described before (38, 40).
RESULTS
DH-PH Tandem Determines GEF Specificity
Considerable advantages in the investigation of the GEF-accelerated nucleotide exchange reaction are provided by fluorescence spectroscopy (38, 41, 43). In this method, the displacement of fluorescent mantGDP from RhoA in the presence of an excess amount of non-fluorescent GDP resulted in a significant change in fluorescence intensity over the time course of the reaction (Fig. 2A). The very slow intrinsic nucleotide dissociation rate (1.7 × 10−5 s−1) was efficiently accelerated 3.8 × 104-fold (with an observed rate constant or kobs of 0.65 s−1) when 2 μm of the DH-PH domain of LARG was mixed in the stopped-flow apparatus with 0.1 μm RhoA-mantGDP and 20 μm GDP (Fig. 2B). For comparison, the GEF activities of p115, p190, and PRG were measured under the same condition (Fig. 2C). All three GEFs were less efficient in the acceleration of nucleotide dissociation compared with LARG. Although PRG DH-PH accelerated the nucleotide exchange 9.6 × 103-fold, which was only 4 times slower than LARG, p115 and p190 only showed a 0.5 × 103- and 0.3 × 103-fold acceleration, which was 82- and 110-fold slower than LARG, respectively. It has been shown that PRG is not only a Rho-specific GEF but to a certain extent also active on Cdc42 (16). Therefore, we measured the rate of mantGDP dissociation from Rac1, Cdc42, and TC10 in the absence and in the presence of the DH-PH domain of p115, p190, PRG, or LARG, respectively. As shown in Fig. 2C, we did not observe any significant changes in the nucleotide dissociation rates of these GTPases by p115, p190, PRG, or LARG. The RacGEF Tiam1 (41, 42) and the Cdc42GEF Asef (44) were used as positive controls (supplemental Fig. S1). This clearly emphasizes the specificity of these GEFs for RhoA. To complete the scenario, we next determined the efficiency of p115, p190, PRG, and LARG on the exchange of mantGDP bound to RhoB and RhoC GTPases, which share 84 and 92% sequence identity with RhoA, respectively. We found that these two Rho isoforms together with RhoA are specific substrates for the investigated GEFs (Fig. 2C). Their intrinsic nucleotide dissociation rates (1.3 × 10−5 s−1 for RhoB and 4.7 × 10−5 s−1 for RhoC) were also accelerated up to 4 orders of magnitude in the presence of a 2 μm concentration of the respective DH-PH (Fig. 2C).
FIGURE 2.

Rho specificity of p115, p190, PRG, and LARG. A and B, DH-PH catalyzes the very slow intrinsic nucleotide exchange reaction by several orders of magnitude. The mantGDP dissociation from 0.1 μm RhoA was monitored after addition of 20 μm unlabeled GDP in the absence (A) and in the presence of 2 μm LARG DH-PH (B). Note the dimension of the x axis, which is in hours in A and in seconds in B, visualizing a rate acceleration of more than 38,000-fold. C, Rho isoform specificity of p115, p190, PRG, and LARG. The observed rate constants (kobs) of both intrinsic and DH-PH-catalyzed reactions of different GTPases were obtained by single exponential fitting of the data. The kobs values were determined using 0.1 μm mantGDP-bound GTPases (RhoA, RhoB, RhoC, Rac1, Cdc42, and TC10) and 20 μm non-fluorescent GDP in the mantGDP dissociation catalyzed by four different DH-PH proteins (2 μm each). D, DH-PH-catalyzed nucleotide exchange is independent of the type of bound nucleotide. GEF-catalyzed mantGDP and mantGppNHp dissociation from RhoA was monitored using 0.1 μm mant-nucleotide-loaded RhoA (RhoA-mantGDP or RhoA-mantGppNHp) and 20 μm non-fluorescent nucleotide (GDP or GppNHp) in the presence of 10 μm DH-PH domain of p115 or p190 or 2 μm DH-PH domain of PRG or LARG. Note that a 5-fold lower concentration of LARG and PRG has been used compared with the experiments with p190 and p115. Moreover, the LARG-catalyzed mant-nucleotide dissociation was measured in the presence of excess amounts of both GDP and GppNHp (white bar). The observed rate constants (kobs) were obtained by single exponential fitting of the data. For convenience, the exact kobs values are given as numbers above the bars in C and D.
RhoGEF-catalyzed Exchange Reaction Is Independent of Type of Incoming Nucleotide
The activation process of small GTPases is an intensively studied issue. However, it remains unclear how GEFs approach the inactive GDP-bound GTPase and which role the switch regions have in this context. To address this question, we measured the GEF-catalyzed mantGDP and mantGppNHp dissociation from RhoA in the presence of an excess amount of GDP or GppNHp (a non-hydrolyzable analog of GTP), respectively. The Dbl proteins p115, p190, PRG, and LARG were able to catalyze the mantGppNHp dissociation from RhoA, but the efficiency was 3–4-fold lower than that for mantGDP (Fig. 2D). Moreover, there was no difference whether the incoming nucleotide was GDP or GppNHp as shown for the LARG-catalyzed reaction (Fig. 2D). These data suggest that the conformation of the switch regions of RhoA plays a rather marginal role in GEF recognition, and the exchange machinery functions regardless of the type of nucleotide.
DH Domain Is a Highly Efficient Catalytic Machine
To obtain the maximal rates of the catalyzed nucleotide exchange reaction and a rough estimate of the catalytic efficiency of the complexes between RhoA-mantGDP and the DH-PH domains of p115, p190, PRG, or LARG, we performed single turnover stopped-flow measurements under the same conditions as described above. As shown in Fig. 3A, the decrease in fluorescence, which corresponds to the catalyzed mantGDP dissociation from RhoA, occurred incrementally faster with the addition of increasing amounts of the DH-PH domain of PRG (1–50 μm). Corresponding kobs values were plotted against the varied DH-PH concentrations (Fig. 3B, closed circles). The kinetic parameters of the PRG-accelerated nucleotide dissociation from RhoA were estimated from the hyperbolic kinetics with a Km of 425.7 μm and a kmax of 31.3 s−1 as described elsewhere (38). As in the case of PRG, a complete saturation was also not achieved for the reaction catalyzed by the p190 DH-PH tandem (Fig. 3C, closed circles). Nevertheless, p190 appears to have a much lower kmax (0.37 s−1) and a higher Km (143.5 μm) as compared with PRG DH-PH.
FIGURE 3.

Kinetics of catalyzed nucleotide dissociation reaction of RhoA by RhoGEFs PRG, p190, p115, and LARG using fluorescent nucleotides. A, kinetics of mantGDP dissociation from RhoA (0.1 μm) were measured in the presence of 20 μm non-fluorescent GDP and increasing concentrations of the DH-PH domain of PRG (1, 2, 5, 10, 20, and 50 μm) under the same condition as in Fig. 2. Observed rate constants (kobs) of the respective data were obtained by single exponential fitting. The dependence of the kobs values for the mantGDP dissociation on the concentrations of the DH-PH (closed circles) and the DH (open circles) domains of PRG (B), p190 (C), p115 (D), and LARG (E) was fitted to a hyperbolic curve to obtain the kinetic parameters of the GEF-catalyzed nucleotide dissociation from RhoA.
For LARG DH-PH, which also belongs to the RGS-containing Dbl protein family (Fig. 1), we obtained the highest catalytic activity of 75 s−1 at a protein concentration of 500 μm, which also was still far below saturation (Fig. 3E, closed circles). Similarly, the data obtained for p115 indicate that Km and kmax must be far beyond 100 μm and 0.15 s−1, respectively (Fig. 3D, closed circles). Hence, as the single concentration measurements already indicated, LARG and PRG are 2 orders of magnitude more efficient catalysts of the nucleotide exchange reaction as compared with p115 and p190. The overall acceleration of approximately 0.88 × 104-fold by p115, 2.17 × 104-fold by p190, most remarkably 1.84 × 106-fold by PRG, and at least 4.70 × 106-fold by LARG was calculated from the ratio of the respective kmax or maximal kobs values and the intrinsic dissociation rate of mantGDP (1.7 × 10−5 s−1; Fig. 2A). This clearly demonstrates that the GEFs of the Dbl family proteins are able to catalyze the nucleotide exchange of RhoGTPases as efficiently as was reported previously for the GEFs of Ran, Ras, and Rab (45–47).
PH-assisted Exchange Reaction of p115 and PRG but Not of p190 and LARG
The majority of Dbl family proteins comprise a characteristic tandem DH-PH organization, suggesting that the PH domain may provide an essential, conserved function in the regulation of DH domain activity. Therefore, we scrutinized the influence of the PH domains of the four RhoGEFs on the DH-catalyzed nucleotide exchange. Similarly to the tandem DH-PH, hyperbolic dependences of the mantGDP dissociation rate on DH concentration were observed for PRG and p190 (Fig. 3, B and C). They were saturated at a kmax value of 6.9 (PRG) and 0.28 s−1 (p190) and an apparent Km value of 516.7 (PRG) and 151.4 μm (p190), respectively (Fig. 3, B and C, open circles). It is important to note that a shorter segment of PRG DHs (residues 729–939), which has been reported to be inactive (16), could not be analyzed because it turned out to be insoluble (data not shown). A possible reason may be the truncated α13 at the C terminus (supplemental Fig. S2A). The PH domain of p115 also appeared to influence considerably the activity of its DH domain as the rate of nucleotide dissociation was ∼4-fold lower for each particular concentration of the corresponding protein constructs (Fig. 3D, open circles). However, because the kinetic parameters for the nucleotide exchange reaction of p115 DH on RhoA-mantGDP could not be determined, it remains unclear whether this effect results either from reduced activity or from reduced affinity. Also only minor differences were observed for the nucleotide exchange between the DH and DH-PH for LARG, indicating that the tandem PH domain of this GEF influences the acceleration of the nucleotide exchange reaction of RhoA only marginally (Fig. 3E, open circles).
Fluorescent RhoA Allows Monitoring RhoA-GDP Association of RhoGEFs
Although the use of mantGDP proved to be very useful for the elucidation of GEF-catalyzed nucleotide dissociation from RhoGTPases, it does not enable monitoring of events upon RhoGEF association. Therefore, we set out to extend our technical capacity by developing a method that enables us to measure the binding kinetics of RhoA-GDP with GEFs in real time. An alternative, fluorescence-based approach is the introduction of reporter groups into the interacting partner, RhoA or the DH domain of the RhoGEFs. Labeling of small GTPases with fluorescent reporter groups has so far only been used to study the interaction of Cdc42 with an effector (48) and the mechanism of NTF2-mediated Ran transport into the nucleus (49), with C-terminally dansyl-labeled Rab to study the interaction with the prenyltransferases (50), and with H-Ras and Rap1 to measure GTPase-activating protein binding (40).
Specific attachment of fluorophores on protein surfaces can be achieved via the modification of thiol groups of cysteines. C-terminally truncated RhoA (residues 1–181) contains five cysteines, but none of them is accessible from the solvent according to the structures of RhoA-GDP (51) and RhoA-GTPγS (52). This was verified by performing an Ellman reaction with GDP-bound and GppNHp-bound RhoA proteins, including RhoA(C20S) as a control, using Ellman's reagent (5,5′-dithiobis(nitrobenzoic acid); Sigma) (40, 53). We then inspected the structures of RhoA in complex with the DH-PH domains of PRG (16, 54, 55) and LARG (17) to identify the residues on the RhoA surface that are close to or at the edge of the binding interface but do not participate significantly in the interaction with DH-PH (Fig. 4A). From nine freely accessible residues, we chose the conserved Val33 (supplemental Fig. S2A) that was replaced by cysteine and labeled with the fluorescence reporter group AEDANS (see “Experimental Procedures”). AEDANS-labeled fluorescent RhoA(V33C), which is called, for simplicity, fRhoA, notably showed an incremental increase in fluorescence in the presence of increasing amounts of p115 DH-PH (Fig. 4B). This was not observed when we applied to fRhoA a Rac1-specific DH-PH of Tiam1 instead of p115 (data not shown). A rate constant for the association (kon) of 1.186 × 105 s−1 m−1 was calculated by plotting kobs values of the corresponding binding curves against the p115 DH-PH concentrations (Fig. 4C and supplemental Fig. S3, filled circles). The dissociation rate constant (koff) was determined by displacing fRhoA-GDP from its complex with p115 DH-PH in the presence of unlabeled, nucleotide-free RhoA, a reaction that led to rapid decrease in fluorescence (Fig. 4D and supplemental Fig. S3, black line). Nucleotide-free RhoA has a much higher affinity for the GEFs compared with the nucleotide-bound forms of the GTPases (56).5 The obtained dissociation rate constant (koff) of 1.7 s−1 divided by the kon value enabled us to calculate a dissociation constant (Kd) of 14.5 μm for the fRhoA/GDP interaction with the p115 DH-PH proteins.
FIGURE 4.

Real time monitoring of RhoGEF interactions with GDP-bound fRhoA. A, RhoA labeling strategy with the fluorescence reporter group AEDANS (inset). The van der Waals surface of nucleotide-free RhoA from the LARG DH-PH complex (17) (Protein Data Bank code 1X86) shows the solvent-accessible surrounding residues (green) around the interaction surface of LARG (orange). Valine 33 (V33) of RhoA substituted by cysteine and labeled with AEDANS (fRhoA) is shown in red. B, fRhoA allows monitoring of the RhoGEF association in real time. Rapid mixing of increasing p115 DH-PH concentrations (0.5–5 μm) with fRhoA-GDP (0.2 μm) resulted in an incremental increase in fluorescence corresponding to the association reaction. C, the association rate constants (kon) of fRhoA-GDP binding to the DH and DH-PH proteins of LARG and p115, respectively, clearly revealed differences in the binding properties of the two RhoGEFs. D, the dissociation rate constant (koff) of the DH and DH-PH proteins of LARG and p115, respectively, displaced from the fRhoA-GDP complex in the presence of excess amounts of unlabeled, nucleotide-free RhoA revealed an impact of p115 PH domain on the GEF dissociation kinetics. The kinetic data are shown in supplemental Fig. S3. The dissociation constant (Kd) was calculated from the kinetic parameters of dissociation and association reactions by the equation Kd = koff/kon. For convenience, the exact kon and koff values are given as numbers above the bars in C and D, respectively.
LARG Association with RhoA-GDP Is Strikingly Faster than p115
With fRhoA, we now have an attractive technique that enables us to better understand both the differential RhoA binding characteristics of the RhoGEFs and the role of the PH domains in the exchange reactions. As shown on Figs. 2 and 3, the largest differences in exchange efficiency and involvement of PH domain were observed for LARG and p115. Therefore, we determined the individual rate constants for the interaction of fRhoA-GDP with the DH and DH-PH domains of these two GEFs. Fig. 4C shows that LARG associated with fRhoA-GDP 15-fold faster than p115 independently of the presence or absence of the PH domain. Interestingly, this was not the case for the dissociation reaction (Fig. 4D). The koff values are similar for LARG DH and DH-PH but vary about 2-fold between p115 DH and DH-PH (Fig. 4D). This is consistent with our data on the involvement of the PH domain of p115 but not of LARG in nucleotide exchange catalysis (Fig. 3, D and E). Moreover, these data strongly suggest that the efficiency of LARG in catalyzing the mantGDP dissociation is in fact attributed to its faster association with fRhoA-GDP.
Short N-terminal DH Segment Is Critical for Binding and Catalysis
The results described above demonstrate clearly different binding capacities and catalytic efficiencies of the RhoGEFs investigated but did not reveal which regions in the DH domain are responsible for their association with the GDP-bound RhoA. To shed light on the molecular basis of the observed differences, we inspected individual amino acids of the DH domains of LARG, PRG, p190, and p115. We sorted out identical residues and selected variable residues between p115 versus LARG and PRG using a multiple sequence alignment of the DH domains (supplemental Fig. S2A). Based on the crystal structures of RhoA in complex with DH-PH of PRG (16) and of LARG (17), we further selected solvent-exposed residues that are close to or part of the interacting interface and identified eight potential residues (supplemental Fig. S2A, red underlined). Structural analysis of these amino acids interestingly showed that among these eight residues six are close to the switch I region and four of these, namely Asn768, Asp770, Arg775, and Gly780, are clustered at the very N-terminal segment (Fig. 5A).
FIGURE 5.

Critical role of N-terminal segment of DH domain in association and nucleotide exchange reactions. A, possible new signatures for the DH function. The crystal structure (17) (Protein Data Bank code 1X86) of the nucleotide-free RhoA (violet) in the complex with LARG DH-PH (turquoise) highlights eight residues (orange) in the DH domain that may be critical for the efficiency of LARG in both associating with GDP-bound RhoA and catalyzing nucleotide dissociation. Four of the eight residues are located in a short peptide called the N-terminal (N-term.) segment (green). Switch (Sw) regions I and II of RhoA and shown in blue and red, respectively. B, the kobs values highlight the association efficiency of the DH-PH variants of LARG and p115 (5 μm, respectively) with 0.2 μm fRhoA-GDP. C, the kobs values show the exchange reaction of mantGDP from RhoA (0.1 μm) catalyzed by the DH-PH variants of LARG and p115 (10 μm, respectively). For convenience, the exact kobs values are given as numbers above the bars in B and C.
Considering the fact that the crystal structures of RhoA-LARG and RhoA-PRG complexes are nucleotide-free complexes, it is rather tempting to speculate that these eight residues may play a role in the DH association with GDP-bound RhoA. Thus, we first generated a deletion mutant of the DH-PH of LARG (DH-PHΔN) lacking the N-terminal segment with its four putative association-determining residues Asn768, Asp770, Arg775, and Gly780 (Fig. 5A). We measured the properties of DH-PHΔN regarding its association with fRhoA-GDP and its activity in catalyzing the mantGDP dissociation from RhoA in comparison with LARG DH-PH. Fig. 5, B and C, show that a deletion of 16 amino acids at the N terminus of LARG DH-PH (DH-PHΔN; Fig. 1 and supplemental Fig. S2A) clearly and substantially affected its association efficiency and consequently its catalytic activity. Having partially proven our hypothesis, we mutated two further potential residues in DH-PHΔN, namely Asn946 and Lys949 to Ser946 and Gln949, which are the equivalent residues in p115 (DH-PHΔN2m; Figs. 1 and 5A and supplemental Fig. S2A). This protein revealed an 8-fold decrease in association with fRhoA-GDP and 25-fold decrease in catalyzing the mGDP dissociation from RhoA (Fig. 5, B and C). These data strongly suggest that the association of GTPase and GEF is strongly contributing to the catalytic efficiency of the exchange reaction.
Most recently, an acidic stretch upstream of the N-terminal segment that inhibits the catalytic activity of the PRG DH domain has been identified (37). The corresponding glutamic and aspartic acids are conserved in p115 (supplemental Fig. S2A). To determine the impact of both the acidic region and the N-terminal segment on p115 activity under the same experimental conditions, we set out to adjust its DH-PH domain assembly to the length of LARG DH-PH by shortening it at both termini (supplemental Fig. S2A, underlined sequence). DH-PHc and DH-PHcn revealed marginal changes in their ability to associate with fRhoA-GDP, but their nucleotide exchange activity was unexpectedly reduced (Fig. 5, B and C). To what extent these regions contribute structurally to the GEF activity remains unclear as all structures of PRG and LARG complexes with RhoA are shorter and do not contain these regions (16, 17, 55).
To address the question of what is the impact of the four variable residues within the N-terminal segment on the p115 activity, we analyzed the biochemical properties of p115 DH-PHcnΔN, the fragment equivalent to LARG DH-PHΔN. As shown in Fig. 5, B and C, neither the rates for association with fRhoA-GDP nor the rates for the catalyzed mantGDP dissociation from RhoA were grossly affected. This is in agreement with our consideration that the N-terminal segment may be an integral element for the catalytic efficiency of LARG and PRG versus p115 and p190.
N-terminal RGS-Linker Negatively Controls DH Activity
It appears to be a rule that the Rho family GEFs underlay an autoinhibitory mechanism (37, 57–61). Apart from the catalytic core, which dictates the nucleotide exchange in terms of the DH-PH of RhoGEFs, there are additional domains in the same polypeptides that are essential autoinhibitory elements (37, 57, 58, 60). A G-protein-mediated regulatory principle has been implicated by Sternweis et al. (24) for RhoA activation by RGS-containing RhoGEFs. In a recent report, Zheng et al. (37) have shown that the RGS domain and a unique sequence motif upstream of the DH domain of PRG (supplemental Fig. S2A) act cooperatively to bind the DH domain and to inhibit its catalytic activity.
To examine a direct modulation of the DH exchange activity by the RGS domain, we measured the catalytic activity of purified p115 RGS-Linker-DH protein under the same condition as described above. As shown in Fig. 6, the GEF activity of the RGS-Linker-DH was 28-fold reduced compared with the isolated DH domain (Fig. 6). Because such an inhibition of the GEF activity strongly suggests an autoinhibitory effect of the DH domain by the RGS domain, we further analyzed the DH-catalyzed nucleotide dissociation in the presence of isolated RGS. The observed kinetic data (Fig. 6) revealed no interference of the RGS domain with the DH activity at all. These findings suggested that p115-mediated regulation of Rho activation is not mainly controlled directly by the RGS interaction with the DH domain but also by the linker region between RGS and DH domain. To test this hypothesis, we measured the DH-catalyzed mantGDP dissociation from RhoA in the presence of the linker and a mixture of the linker and the RGS domain, respectively. However, we did not detect any inhibition of the DH activity by these isolated domains even in the presence of a 100-fold excess of the RGS-Linker over the DH domain (Fig. 6). The same result was obtained when the p115 DH-PH tandem was used instead of the DH domain (data not shown). Taken together, our data support the previous reports that p115 underlies an autoinhibitory mechanism (27) that seems to be partially different compared with that of PRG, which also utilizes a cluster of acidic residues immediately upstream of the DH domain (37).
FIGURE 6.
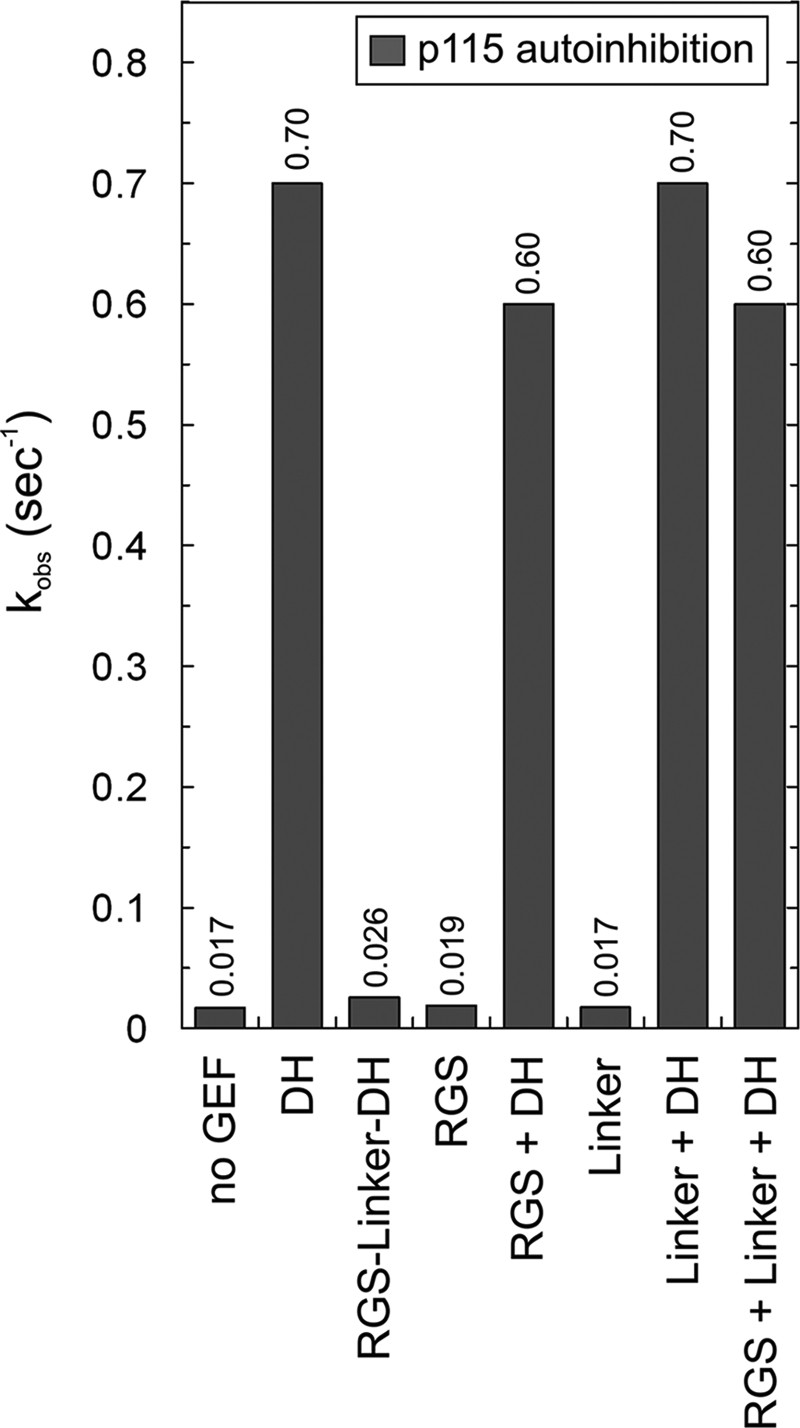
RGS-Linker-mediated autoinhibition of p115 DH Activity. The effects of various p115 domains on the intrinsic and the DH-catalyzed mantGDP dissociation from RhoA were measured under the same conditions as in Fig. 2. The following protein concentrations were used: 0.1 μm RhoA-mantGDP, 1 μm DH, 1 μm RGS-Linker-DH, 10 μm RGS, and 10 μm Linker. The observed rate constants (kobs) of both intrinsic and catalyzed reactions were obtained by single exponential fitting of the data. For convenience, the exact kobs values are given as numbers above the bars.
DISCUSSION
The cellular activity of small GTPases, such as RhoA, is determined by the nature of bound nucleotide and is strictly regulated. Activated GEFs, for example, accelerate the otherwise very slow exchange of GDP to GTP by several orders of magnitude. In this study, we used fluorescence spectroscopic methods to determine quantitatively (i) the specificity of four GEFs, p115, p190, PRG, and LARG on six RhoGTPases; (ii) their catalytic constants (kcat and Km) toward RhoA; (iii) the association of RhoGEF with GDP-bound fRhoA; and (iv) the influence of other domains, such as the PH and RGS domains, and an N-terminal segment on the DH capability in both binding fRhoA-GDP and catalyzing mantGDP dissociation from RhoA.
Rho Isoform Specificity of p115, p190, PRG, and LARG
We analyzed the activity of GEFs using mantGDP-bound RhoA isoforms and showed that isolated DH-PH domains represent the catalytic units of these GEFs. This is, in the first instance, consistent with previous results on p115 (25, 26, 28), p190 (39), PRG (16, 29, 62), and LARG (17, 30, 63) obtained by different kinds of assays. In addition, we clearly demonstrated that these GEFs do not exhibit any activity toward Rac1, Cdc42, and TC10 at all, suggesting their unique substrate specificity for the three isoforms RhoA, RhoB, and RhoC. GEFs of the Dbl family are mostly specific for one member or a subgroup of the Rho family GTPases (64). For example, hPEM-2/Collybistin, ITSN1, and Asef are specific for Cdc42 (44, 65, 66), whereas Tiam1 specifically activates Rac isoforms (41, 67, 68). On the other hand, there are GEFs with dual specificity, including Dbs as a GEF for RhoA and Cdc42 (69), Vav3 for RhoA and RhoG (70), and Trio for Rac1 and RhoG (71).
The high sequence conservation within both individual RhoGTPase members and various DH-PH domains (supplemental Fig. S2) raises the question of how the specificity of the RhoGEFs for the Rho isoforms is achieved. To address this important issue, we identified the contacting residues of the RhoA-PRG and RhoA-LARG complexes using the respective crystal structures (16, 17) and aligned them to various DH-PH tandems and to the G domains analyzed in this study (Fig. 7). Considering the interactions from the Rho side, five of 18 DH-PH-contacting residues (Arg5, Val33, Asp45, Glu54, and Asp76) are identical in the RhoA, RhoB, and RhoC isoforms but variable in Cdc42, Rac1, and TC10 (Fig. 7, residues with cyan background).
FIGURE 7.

Structure-based interaction sequence matrix illustrating specificity determining residues for RhoA interaction with its GEFs. Based on the crystal structures of RhoA (G domain) in the complex with DH-PH of PRG (16) (Protein Data Bank code 1XCG) and of LARG (17) (Protein Data Bank code 1X86) the interacting residues (colored background; <4 Å in distance) were determined and aligned onto the DH-PH tandem and the G domain of RhoGTPases. Residues with a light blue background are conserved in Rho-specific GEFs and critical in determining the specificity of the RhoA/DH-PH interaction. Variable residues with a black background may be critical in determining the catalytic efficiency of Rho-specific GEFs.
Strikingly, these specificity-determining residues, except for Val33, are not part of the switch regions (supplemental Fig. S2B) (4). Substitution of Val33 in RhoA (corresponding to Val33 in RhoB and RhoC, Glu31 in Rac1 and Cdc42, and Glu45 in TC10) by cysteine for fluorescence labeling did not show any significant influence on the GEF-catalyzed mantGDP dissociation either by LARG or p115 (data not shown). In addition, Val33 replacement by glutamate did not change the catalytic properties of LARG DH-PH (17). Val43 in RhoA and RhoB (Ile43 in RhoC) must also be included in this group of residues, which has been described previously to be critical for Rho recognition by GEFs (54). Its replacement by a Serine (equivalent to Ser41 in Rac1) resulted in a 25-fold reduction of the PRG DH-PH exchange activity, the most critical impairment among all tested mutants of RhoA (54).
Considering the interactions from the DH-PH side, three of 34 RhoA-contacting residues (supplemental Fig. S2A; N-terminal of the conserved region 3) are identical in Rho-specific PRG (Lys884, Arg868, and Asp873), LARG, p115, and p190 but variable in GEFs specific for other members of the Rho family, e.g. the Cdc42-specific ITSN1 and the Rac1-specific Tiam1 (Fig. 7, residues with cyan background). These specificity-determining residues from both the Rho-specific GEFs and the Rho isoforms are strikingly linked together via ionic and H-bonds (Fig. 7, red and orange fields). The corresponding contacts are Arg5 with Arg868 and Asp871, Asp45 and Glu54 with Arg868, and Asp76 with Lys844. Individual substitution of these residues of RhoA to Rac1 (R5A, D45N, E54N, and D76Q) or PRG to ITSN1 (R868G and D873S; Lys844 was not analyzed) has been shown to result in a drastic reduction of the PRG-catalyzed nucleotide dissociation up to 25-fold (54). The same study has shown that substitution of Rac1 and Cdc42 residues at four positions equivalent to RhoA residues (A3R, S41V, N45D, and N52E and T3R, A41V, T45D, and T52E, respectively) generates proteins whose nucleotide exchange can be significantly accelerated by PRG when compared with the wild type proteins.
Attributes of Catalytic Efficiency of RhoGEFs
A striking finding of this study is that PRG and LARG exhibited a GEF activity that was 2 orders of magnitude higher as compared with p115 and p190. This is particularly interesting because p115 belongs to the same subfamily of RGS-containing Dbl proteins as PRG and LARG (22, 72). An efficient catalytic activity of a GEF is dependent on at least two successive reactions: (i) association with RhoA-GDP and (ii) exchange of the bound GDP for GTP proceeding via a high affinity nucleotide-free GEF-RhoA reaction intermediate. To explain the catalytic efficiency of PRG and LARG versus p115, we first focused on the available structural data. From our structure-based interaction sequence matrix in which we inspected crystal structures of PRG (16) and LARG (17) in complex with RhoA, we selected nine variable residues contacting the nucleotide-free form of RhoA (Fig. 7, residues with black background). The three residues from the PH domain can be excluded because of the fact that this domain does not contribute to catalytic activity of LARG (see below). Within the other six residues, Ile876 substitution by proline (equivalent residue in LARG and p115) in PRG DH-PH has been shown in a comprehensive mutational study to generate a more efficient exchange factor (54), consistent with our observation that LARG exhibited a 4-fold higher activity than PRG. Two asparagines that are conserved in PRG (Asn715 and Asn928) and LARG (Asn767 and Asn928) appear to be critical for both DH-PH associations with RhoA and nucleotide exchange on RhoA (see below).
An obvious alternative explanation for the much lower efficiency of p115 GEF as compared with PRG and LARG is based on differences of the GEF association with the GDP-bound RhoA, which we found by developing a new method. Six of eight selected residues in LARG (Asn768, Asp770, Arg775, Gly780, Asn946, and Lys949) seem to play an important role in the association of LARG DH-PH with RhoA-GDP and for catalytic activity of LARG. Most of these residues are identical in PRG and in LARG, including Asn768/Asn946 and Asn715/Asn928 (Fig. 7, black background), and thus play a similar role in both the association and in the exchange reaction. The N-terminal segment of LARG DH-PH contains two short α-helices encompassing four of these residues (Asn768, Asp770, Arg775, and Gly780) from which only Asn768 contacts nucleotide-free RhoA (16). This suggests that the other three residues most likely contact RhoA in its nucleotide-bound form. We found that GEFs can equally recognize GDP- and GTP-bound RhoA, which also confirms the reversible character of the nucleotide exchange reaction. The molecular basis for the recognition of RhoA-GDP by GEFs seems to be mainly dependent on the β2-β3 regions (54, 66). There is only one crystal structure of a ternary complex (GTPase-GDP-GEF) known, which is the plant GTPase ROP4-GDP in complex with its GEF plant-specific ROP nucleotide exchanger (PRONE), that presents a common mechanism of catalyzed nucleotide exchange applicable to small GTPases in general (74). Rop4-contacting regions of PRONE are the P-loop, switch I, the β1 strand, part of switch II, and the end of the insert helix (supplemental Fig. S2B). This issue must be resolved structurally for the RhoA-related proteins.
Differential Roles of Tandem PH Domain
PH domains are best known for their ability to bind phosphoinositides with high affinity and specificity, although it is now clear that less than 10% of all PH domains share this property (75). Work with the Dbl family exchange factors consistently raises the question regarding the functional role of the tandem PH domain. Such an arrangement has been proposed to imply a crucial and unique functional interrelationship (5, 7). It has been shown that the PH domains of Trio, Dbs, and Dbl have a cooperative effect on the catalysis of the exchange reaction by the DH domain as its absence leads to a strong decrease in stimulation of the nucleotide dissociation (14, 76, 77). Our kinetic data of the exchange reaction imply that the PH domains contribute to the nucleotide exchange reaction mediated by the DH domain to different extents depending on the particular GEF. Compared with the activity of the isolated DH domain, the DH-PH tandem of PRG and p115 exhibited up to 5-fold enhanced exchange activities, respectively. This finding is supported by several previous studies. Wells et al. (28) reported that removal of the PH domain dramatically reduced the in vitro activity of p115 RhoGEF. The crucial role of the PH domain of PRG has been demonstrated previously for the catalysis of RhoA nucleotide exchange (16). Mutation of the PH binding residue of RhoA was shown to affect strongly the catalytic function of PRG DH-PH protein (78). In addition to the interaction with nucleotide-free GTPase, PRG PH domain can also interact with GTP-bound RhoA, regulating cellular PRG activity (60). In contrast to PRG and p115, the influence of the PH domain on the activity of the DH domain of p190 and LARG was rather insignificant and very similar to the Cdc42-specific Asef (44). These data indicate that the DH domains of p190 and LARG represent the entire catalytic machinery to accomplish Rho activation and that their PH domains do not contribute to Rho activation.
One possible role of the PH domain on the activity of the DH domain might be its direct interaction with the GTPase (16). For example, the x-ray structure of Dbl in complex with Cdc42 (14, 15) and structures of Dbs (13), PRG (16, 54, 55, 78), and LARG (17) in complex with the nucleotide-free RhoA revealed that the PH domains of these GEFs directly contact switch II and the α3 helix of the respective GTPase. On the other hand, no direct interaction between the PH domain and GTPase was observed in complex structures of Rac1-Tiam1 (20), Rac1-Trio (76), Cdc42-ITSN1 (14), and Cdc42-Collybistin (21) and in the ternary complex Gαq-p63RhoGEF-RhoA (79). Interestingly, despite no interaction of the PH domain of Trio in the complex structure with Rac1, its absence caused a 4-fold decrease in exchange activity (76). A nearly opposite scenario is observed for LARG that contacts the GTPase with its PH domain (17) in the same manner as shown for PRG (16). Although PRG PH clearly contributes to kinetics of nucleotide exchange, LARG PH is dispensable for the DH activity in vitro. Interestingly, a conserved hydrophobic patch of the LARG PH domain has been reported recently to be critical for RhoA activation in cells (80). It has been suggested that the LARG PH domain is involved in regulatory interactions with other proteins near the membrane surface. It is assumed that in cells GEFs are directionally translocated to the plasma membrane in response to extracellular signals (12) where they are localized to posttranslationally modified small GTPases. In an in vitro liposome reconstitution, Robbe et al. (81) have shown that Tiam1 DH-PH, which specifically accelerates nucleotide exchange of the Rac isoforms (41, 42), activates prenylated Rac1 much more efficiently in the presence of liposomes. A model for PH domain-assisted nucleotide exchange has been proposed for Dbs and also for other GEFs such as Dbl and Trio. Herein the PH domain serves multiple roles in signaling events anchoring GEFs to the membrane (via phosphoinositides), directing them toward their interacting GTPases, which are already attached to the membrane (14, 22, 77, 82–85).
p115 Autoinhibition
The RGS-containing RhoGEFs, including p115, PRG, and LARG, represent a distinct family of guanine nucleotide exchange factors for RhoA that are regulated by the Gα12/13 proteins. Experimental evidence indicates that the complex architecture of these RhoGEFs provides the structural basis for regulatory mechanisms mediated by protein-protein interactions (26, 86). Association of the RGS domain of p115 with Gα12/13 proteins was shown to partially activate its GEF activity toward RhoA, suggesting that the N terminus of p115 may contribute to autoinhibition of the DH-PH activity (26). Accordingly, we could demonstrate that a large protein fragment consisting of RGS-Linker-DH (residues 40–645) indeed exhibited significantly reduced GEF activity. This suggests that regions upstream of the DH domain may interact in intramolecular fashion with the DH domain and mask it from binding to RhoA-GDP. Such an apparent autoinhibition of p115 DH activity could not be verified when its N-terminal regions, including RGS, Linker, and RGS-Linker, were separately mixed with the DH domain. Even at very high concentrations of the respective proteins (100-fold above the DH domain), we could not detect any trans inhibition of the DH activity by the N-terminal regions. The most recent structural analysis of p115 has shown that an N-terminal extension of the DH domain appears to play a critical role in p115 autoinhibition (60). Similarly, structural and biochemical analysis of various PRG proteins reported by Zheng et al. (37) has provided new insight into the molecular nature of such an intramolecular interaction. PRG utilizes an electrostatic patch immediately upstream of the DH domain and contributes in part to an autoinhibitory mechanism that appears to require additional regions of the full-length protein, including the RGS domain.
In summary, our data strongly support the conclusion that the DH domain of the RhoGEF itself determines the specificity for binding RhoGTPases and represents very efficient catalytic machinery for the nucleotide exchange in a cell-free and membrane-free system. In cells, however, a set of additional domains and interactions are required for the shuttling, localization, and activation of the GEFs (11, 81, 73). Complex formation of GEFs with receptors (e.g. semaphorin receptors/plexins) and G-proteins (e.g. Gα12/13) at the membrane are required for the functional activation and the regulation of cellular processes, including adhesion, contraction, and motility.
Supplementary Material
Acknowledgments
We gratefully thank G. Bollag and P. C. Sternweis for providing the cDNA of human p115, J. J. Tesmer for the LARG DH and DH-PH constructs, W. Moolenaar for providing us the cDNA of murine p190, and K. H. Jakobs for the cDNA of human PRG.
This work was supported in part by Deutsche Forschungsgemeinschaft Grants AH 92/3-1 and AH 92/5-1, by the E-Rare project NSEuroNet, and by Forschungskommission der Medizinischen Fakultät der Heinrich-Heine-Universität Düsseldorf Grant 9772337-09.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
Z. Guo and M. R. Ahmadian, unpublished data.
- GTPase
- GDP/GTP-binding protein
- GEF
- guanine nucleotide exchange factor
- PRG
- PDZ-RhoGEF
- LARG
- leukemia-associated RhoGEF
- RGS
- regulator of G-protein signaling
- DH
- Dbl homology
- PH
- pleckstrin homology
- ITSN1
- Intersectin1
- 1,5-I-AEDANS (AEDANS)
- N-(iodoacetaminoethyl)-1-naphthylamine-5-sulfonic acid
- GppNHp
- guanosine 5′-(β,γ-imido)triphosphate
- mant
- methylanthraniloyl
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- GDPγS
- guanosine 5′-3-O-(thio)triphosphate
- fRhoA
- fluorescent RhoA
- PDZ
- postsynaptic density-95, discs large, and zona occludens-1.
REFERENCES
- 1. Etienne-Manneville S., Hall A. (2002) Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Raftopoulou M., Hall A. (2004) Dev. Biol. 265, 23–32 [DOI] [PubMed] [Google Scholar]
- 3. Vetter I. R., Wittinghofer A. (2001) Science 294, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 4. Dvorsky R., Ahmadian M. R. (2004) EMBO Rep. 5, 1130–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whitehead I. P., Campbell S., Rossman K. L., Der C. J. (1997) Biochim. Biophys. Acta 1332, F1–F23 [DOI] [PubMed] [Google Scholar]
- 6. Stam J. C., Collard J. G. (1999) Prog. Mol. Subcell. Biol. 22, 51–83 [DOI] [PubMed] [Google Scholar]
- 7. Zheng Y. (2001) Trends Biochem. Sci. 26, 724–732 [DOI] [PubMed] [Google Scholar]
- 8. Schmidt A., Hall A. (2002) Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 9. Hoffman G. R., Cerione R. A. (2002) FEBS Lett. 513, 85–91 [DOI] [PubMed] [Google Scholar]
- 10. Erickson J. W., Cerione R. A. (2004) Biochemistry 43, 837–842 [DOI] [PubMed] [Google Scholar]
- 11. Rossman K. L., Der C. J., Sondek J. (2005) Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 12. Schiller M. R. (2006) Cell. Signal. 18, 1834–1843 [DOI] [PubMed] [Google Scholar]
- 13. Snyder J. T., Worthylake D. K., Rossman K. L., Betts L., Pruitt W. M., Siderovski D. P., Der C. J., Sondek J. (2002) Nat. Struct. Biol. 9, 468–475 [DOI] [PubMed] [Google Scholar]
- 14. Rossman K. L., Worthylake D. K., Snyder J. T., Siderovski D. P., Campbell S. L., Sondek J. (2002) EMBO J. 21, 1315–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rossman K. L., Cheng L., Mahon G. M., Rojas R. J., Snyder J. T., Whitehead I. P., Sondek J. (2003) J. Biol. Chem. 278, 18393–18400 [DOI] [PubMed] [Google Scholar]
- 16. Derewenda U., Oleksy A., Stevenson A. S., Korczynska J., Dauter Z., Somlyo A. P., Otlewski J., Somlyo A. V., Derewenda Z. S. (2004) Structure 12, 1955–1965 [DOI] [PubMed] [Google Scholar]
- 17. Kristelly R., Gao G., Tesmer J. J. G. (2004) J. Biol. Chem. 279, 47352–47362 [DOI] [PubMed] [Google Scholar]
- 18. Nimnual A. S., Yatsula B. A., Bar-Sagi D. (1998) Science 279, 560–563 [DOI] [PubMed] [Google Scholar]
- 19. Bellanger J. M., Estrach S., Schmidt S., Briançon-Marjollet A., Zugasti O., Fromont S., Debant A. (2003) Biol. Cell 95, 625–634 [DOI] [PubMed] [Google Scholar]
- 20. Worthylake D. K., Rossman K. L., Sondek J. (2000) Nature 408, 682–688 [DOI] [PubMed] [Google Scholar]
- 21. Xiang S., Kim E. Y., Connelly J. J., Nassar N., Kirsch J., Winking J., Schwarz G., Schindelin H. (2006) J. Mol. Biol. 359, 35–46 [DOI] [PubMed] [Google Scholar]
- 22. Aittaleb M., Boguth C. A., Tesmer J. J. G. (2010) Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998) Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 24. Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y. F., Singer W. D. (2007) Adv. Protein Chem. 74, 189–228 [DOI] [PubMed] [Google Scholar]
- 25. Hart M. J., Sharma S., elMasry N., Qiu R. G., McCabe P., Polakis P., Bollag G. (1996) J. Biol. Chem. 271, 25452–25458 [DOI] [PubMed] [Google Scholar]
- 26. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., Bollag G. (1998) Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 27. Wells C. D., Liu M. Y., Jackson M., Gutowski S., Sternweis P. M., Rothstein J. D., Kozasa T., Sternweis P. C. (2002) J. Biol. Chem. 277, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 28. Wells C. D., Gutowski S., Bollag G., Sternweis P. C. (2001) J. Biol. Chem. 276, 28897–28905 [DOI] [PubMed] [Google Scholar]
- 29. Fukuhara S., Murga C., Zohar M., Igishi T., Gutkind J. S. (1999) J. Biol. Chem. 274, 5868–5879 [DOI] [PubMed] [Google Scholar]
- 30. Fukuhara S., Chikumi H., Gutkind J. S. (2000) FEBS Lett. 485, 183–188 [DOI] [PubMed] [Google Scholar]
- 31. Jackson M., Song W., Liu M. Y., Jin L., Dykes-Hoberg M., Lin C. I., Bowers W. J., Federoff H. J., Sternweis P. C., Rothstein J. D. (2001) Nature 410, 89–93 [DOI] [PubMed] [Google Scholar]
- 32. Booden M. A., Siderovski D. P., Der C. J. (2002) Mol. Cell. Biol. 22, 4053–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vázquez-Prado J., Basile J., Gutkind J. S. (2004) Methods Enzymol. 390, 259–285 [DOI] [PubMed] [Google Scholar]
- 34. Vogt S., Grosse R., Schultz G., Offermanns S. (2003) J. Biol. Chem. 278, 28743–28749 [DOI] [PubMed] [Google Scholar]
- 35. Swiercz J. M., Kuner R., Dettlaff D. A., Behrens J., Offermanns S. (2002) Naunyn Schmiedebergs Arch. Pharmacol. 365, R53 [Google Scholar]
- 36. Swiercz J. M., Kuner R., Behrens J., Offermanns S. (2002) Neuron 35, 51–63 [DOI] [PubMed] [Google Scholar]
- 37. Zheng M., Cierpicki T., Momotani K., Artamonov M. V., Derewenda U., Bushweller J. H., Somlyo A. V., Derewenda Z. S. (2009) BMC Struct. Biol. 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hemsath L., Ahmadian M. R. (2005) Methods 37, 173–182 [DOI] [PubMed] [Google Scholar]
- 39. van Horck F. P., Ahmadian M. R., Haeusler L. C., Moolenaar W. H., Kranenburg O. (2001) J. Biol. Chem. 276, 4948–4956 [DOI] [PubMed] [Google Scholar]
- 40. Kraemer A., Brinkmann T., Plettner I., Goody R., Wittinghofer A. (2002) J. Mol. Biol. 324, 763–774 [DOI] [PubMed] [Google Scholar]
- 41. Haeusler L. C., Blumenstein L., Stege P., Dvorsky R., Ahmadian M. R. (2003) FEBS Lett. 555, 556–560 [DOI] [PubMed] [Google Scholar]
- 42. Haeusler L. C., Hemsath L., Fiegen D., Blumenstein L., Herbrand U., Stege P., Dvorsky R., Ahmadian M. R. (2006) Methods Enzymol. 406, 1–11 [DOI] [PubMed] [Google Scholar]
- 43. Ahmadian M. R., Wittinghofer A., Herrmann C. (2002) Methods Mol. Biol. 189, 45–63 [DOI] [PubMed] [Google Scholar]
- 44. Gotthardt K., Ahmadian M. R. (2007) Biol. Chem. 388, 67–71 [DOI] [PubMed] [Google Scholar]
- 45. Klebe C., Prinz H., Wittinghofer A., Goody R. S. (1995) Biochemistry 34, 12543–12552 [DOI] [PubMed] [Google Scholar]
- 46. Lenzen C., Cool R. H., Prinz H., Kuhlmann J., Wittinghofer A. (1998) Biochemistry 37, 7420–7430 [DOI] [PubMed] [Google Scholar]
- 47. Itzen A., Rak A., Goody R. S. (2007) J. Mol. Biol. 365, 1359–1367 [DOI] [PubMed] [Google Scholar]
- 48. Nomanbhoy T., Cerione R. A. (1999) Biochemistry 38, 15878–15884 [DOI] [PubMed] [Google Scholar]
- 49. Ribbeck K., Lipowsky G., Kent H. M., Stewart M., Görlich D. (1998) EMBO J. 17, 6587–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alexandrov K., Scheidig A. J., Goody R. S. (2001) Methods Enzymol. 329, 14–31 [DOI] [PubMed] [Google Scholar]
- 51. Wei Y., Zhang Y., Derewenda U., Liu X., Minor W., Nakamoto R. K., Somlyo A. V., Somlyo A. P., Derewenda Z. S. (1997) Nat. Struct. Biol. 4, 699–703 [DOI] [PubMed] [Google Scholar]
- 52. Ihara K., Muraguchi S., Kato M., Shimizu T., Shirakawa M., Kuroda S., Kaibuchi K., Hakoshima T. (1998) J. Biol. Chem. 273, 9656–9666 [DOI] [PubMed] [Google Scholar]
- 53. Riddles P. W., Blakeley R. L., Zerner B. (1983) Methods Enzymol. 91, 49–60 [DOI] [PubMed] [Google Scholar]
- 54. Oleksy A., Opaliński Ł., Derewenda U., Derewenda Z. S., Otlewski J. (2006) J. Biol. Chem. 281, 32891–32897 [DOI] [PubMed] [Google Scholar]
- 55. Cierpicki T., Bielnicki J., Zheng M., Gruszczyk J., Kasterka M., Petoukhov M., Zhang A., Fernandez E. J., Svergun D. I., Derewenda U., Bushweller J. H., Derewenda Z. S. (2009) Protein Sci. 18, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cool R. H., Schmidt G., Lenzen C. U., Prinz H., Vogt D., Wittinghofer A. (1999) Mol. Cell. Biol. 19, 6297–6305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitin N., Betts L., Yohe M. E., Der C. J., Sondek J., Rossman K. L. (2007) Nat. Struct. Mol. Biol. 14, 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yohe M. E., Rossman K. L., Gardner O. S., Karnoub A. E., Snyder J. T., Gershburg S., Graves L. M., Der C. J., Sondek J. (2007) J. Biol. Chem. 282, 13813–13823 [DOI] [PubMed] [Google Scholar]
- 59. Li P., Martins I. R., Amarasinghe G. K., Rosen M. K. (2008) Nat. Struct. Mol. Biol. 15, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Z., Guo L., Sprang S. R., Sternweis P. C. (2011) Modulation of a GEF Switch: Autoinhibition of the Intrinsic Guanine Nucleotide Exchange Activity of p115-RhoGEF. Protein Science 20, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu B., Martins I. R., Li P., Amarasinghe G. K., Umetani J., Fernandez-Zapico M. E., Billadeau D. D., Machius M., Tomchick D. R., Rosen M. K. (2010) Cell 140, 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rümenapp U., Blomquist A., Schwörer G., Schablowski H., Psoma A., Jakobs K. H. (1999) FEBS Lett. 459, 313–318 [DOI] [PubMed] [Google Scholar]
- 63. Reuther G. W., Lambert Q. T., Booden M. A., Wennerberg K., Becknell B., Marcucci G., Sondek J., Caligiuri M. A., Der C. J. (2001) J. Biol. Chem. 276, 27145–27151 [DOI] [PubMed] [Google Scholar]
- 64. Karnoub A. E., Symons M., Campbell S. L., Der C. J. (2004) Breast Cancer Res. Treat. 84, 61–71 [DOI] [PubMed] [Google Scholar]
- 65. Reid T., Bathoorn A., Ahmadian M. R., Collard J. G. (1999) J. Biol. Chem. 274, 33587–33593 [DOI] [PubMed] [Google Scholar]
- 66. Karnoub A. E., Worthylake D. K., Rossman K. L., Pruitt W. M., Campbell S. L., Sondek J., Der C. J. (2001) Nat. Struct. Biol. 8, 1037–1041 [DOI] [PubMed] [Google Scholar]
- 67. Michiels F., Habets G. G., Stam J. C., van der Kammen R. A., Collard J. G. (1995) Nature 375, 338–340 [DOI] [PubMed] [Google Scholar]
- 68. Fiegen D., Haeusler L. C., Blumenstein L., Herbrand U., Dvorsky R., Vetter I. R., Ahmadian M. R. (2004) J. Biol. Chem. 279, 4743–4749 [DOI] [PubMed] [Google Scholar]
- 69. Whitehead I. P., Lambert Q. T., Glaven J. A., Abe K., Rossman K. L., Mahon G. M., Trzaskos J. M., Kay R., Campbell S. L., Der C. J. (1999) Mol. Cell. Biol. 19, 7759–7770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Movilla N., Bustelo X. R. (1999) Mol. Cell. Biol. 19, 7870–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bellanger J. M., Astier C., Sardet C., Ohta Y., Stossel T. P., Debant A. (2000) Nat. Cell Biol. 2, 888–892 [DOI] [PubMed] [Google Scholar]
- 72. Fukuhara S., Chikumi H., Gutkind J. S. (2001) Oncogene 20, 1661–1668 [DOI] [PubMed] [Google Scholar]
- 73. García-Mata R., Burridge K. (2007) Trends Cell Biol. 17, 36–43 [DOI] [PubMed] [Google Scholar]
- 74. Thomas C., Fricke I., Scrima A., Berken A., Wittinghofer A. (2007) Mol. Cell 25, 141–149 [DOI] [PubMed] [Google Scholar]
- 75. Lemmon M. A. (2007) Biochem. Soc. Symp. 74, 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chhatriwala M. K., Betts L., Worthylake D. K., Sondek J. (2007) J. Mol. Biol. 368, 1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu X., Wang H., Eberstadt M., Schnuchel A., Olejniczak E. T., Meadows R. P., Schkeryantz J. M., Janowick D. A., Harlan J. E., Harris E. A., Staunton D. E., Fesik S. W. (1998) Cell 95, 269–277 [DOI] [PubMed] [Google Scholar]
- 78. Gasmi-Seabrook G. M., Marshall C. B., Cheung M., Kim B., Wang F., Jang Y. J., Mak T. W., Stambolic V., Ikura M. (2010) J. Biol. Chem. 285, 5137–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lutz S., Shankaranarayanan A., Coco C., Ridilla M., Nance M. R., Vettel C., Baltus D., Evelyn C. R., Neubig R. R., Wieland T., Tesmer J. J. (2007) Science 318, 1923–1927 [DOI] [PubMed] [Google Scholar]
- 80. Aittaleb M., Gao G., Evelyn C. R., Neubig R. R., Tesmer J. J. G. (2009) Cell. Signal. 21, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Robbe K., Otto-Bruc A., Chardin P., Antonny B. (2003) J. Biol. Chem. 278, 4756–4762 [DOI] [PubMed] [Google Scholar]
- 82. Skowronek K. R., Guo F., Zheng Y., Nassar N. (2004) J. Biol. Chem. 279, 37895–37907 [DOI] [PubMed] [Google Scholar]
- 83. Hart M. J., Eva A., Zangrilli D., Aaronson S. A., Evans T., Cerione R. A., Zheng Y. (1994) J. Biol. Chem. 269, 62–65 [PubMed] [Google Scholar]
- 84. Baumeister M. A., Rossman K. L., Sondek J., Lemmon M. A. (2006) Biochem. J. 400, 563–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Snyder J. T., Rossman K. L., Baumeister M. A., Pruitt W. M., Siderovski D. P., Der C. J., Lemmon M. A., Sondek J. (2001) J. Biol. Chem. 276, 45868–45875 [DOI] [PubMed] [Google Scholar]
- 86. Chikumi H., Barac A., Behbahani B., Gao Y., Teramoto H., Zheng Y., Gutkind J. S. (2004) Oncogene 23, 233–240 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.