

Abstract
Neuroglobin is a highly conserved hemoprotein of uncertain physiological function that evolved from a common ancestor to hemoglobin and myoglobin. It possesses a six-coordinate heme geometry with proximal and distal histidines directly bound to the heme iron, although coordination of the sixth ligand is reversible. We show that deoxygenated human neuroglobin reacts with nitrite to form nitric oxide (NO). This reaction is regulated by redox-sensitive surface thiols, cysteine 55 and 46, which regulate the fraction of the five-coordinated heme, nitrite binding, and NO formation. Replacement of the distal histidine by leucine or glutamine leads to a stable five-coordinated geometry; these neuroglobin mutants reduce nitrite to NO ∼2000 times faster than the wild type, whereas mutation of either Cys-55 or Cys-46 to alanine stabilizes the six-coordinate structure and slows the reaction. Using lentivirus expression systems, we show that the nitrite reductase activity of neuroglobin inhibits cellular respiration via NO binding to cytochrome c oxidase and confirm that the six-to-five-coordinate status of neuroglobin regulates intracellular hypoxic NO-signaling pathways. These studies suggest that neuroglobin may function as a physiological oxidative stress sensor and a post-translationally redox-regulated nitrite reductase that generates NO under six-to-five-coordinate heme pocket control. We hypothesize that the six-coordinate heme globin superfamily may subserve a function as primordial hypoxic and redox-regulated NO-signaling proteins.
Keywords: Enzyme Kinetics, Hemoglobin, Myoglobin, Neurobiology, Nitric Oxide, Nitric-oxide Synthase, Neuroglobin, Nitrite, Six-coordinate Heme
Introduction
A phylogenic analysis of the heme-globin family indicates that the well characterized proteins hemoglobin and myoglobin were antedated by neuroglobin, which already existed 800 million years ago (1, 2). Neuroglobin (Ngb)4 sequences remained highly conserved throughout mammalian evolution, suggesting a strongly selected vital functionality (3). This heme-containing, monomeric 16.9-kDa protein shares 21 and 25% sequence similarity with myoglobin and hemoglobin. However, unlike myoglobin and hemoglobin, it possesses a bis-histidine six-coordinate heme geometry, such that the proximal and distal histidines in the heme pocket are directly bonded to the heme iron (both Fe2+ or Fe3+ oxidation states) (4). Indeed, at equilibrium the concentration of the five-coordinate neuroglobin is very low, reported from 0.1 to 5% (5). Binding of oxygen or other gas ligands, such as nitric oxide (NO) or carbon monoxide, to the heme iron occurs upon displacement of the 6th coordination bond with the distal histidine 64 residue (6, 7). Despite this structural difference with myoglobin, neuroglobin displays comparable α-helix globin folding and high oxygen affinity (P50 about 1–2 mm Hg at 20 °C) (8, 9). However, the low tissue concentration of neuroglobin and the rapid auto-oxidation of the oxygen-bound species suggest neuroglobin has not evolved to store and supply oxygen, leading to a number of different hypotheses about its molecular functionality (2, 10).
In vitro and in vivo expression of neuroglobin produces cytoprotective effects, limiting neuronal cell death during glucose deprivation and hypoxia and limiting the volume of brain infarction in stroke models (11–14). Understanding the functionality of neuroglobin could provide a paradigm shift in both biology and therapeutics because several highly conserved heme-globins, ubiquitous in plants and animals, exist in equilibrium between dominant six-coordinate heme geometry and a less frequent five-coordinate state. Examples include cytoglobin, cytochrome c, Drosophila melanogaster hemoglobin, and the nonsymbiotic plant hemoglobins (15–17).
Over the past 5 years, our groups have examined the ability of deoxygenated hemoglobin and myoglobin to react with and reduce nitrite to NO (18, 19). We have proposed that this reaction subserves a function similar to the bacterial nitrite reductases, in which a coupled electron and proton transfer to nitrite generates NO.
In the heart, myoglobin can reduce nitrite to NO to regulate hypoxic mitochondrial respiration and enhance the cellular resilience to prolonged ischemia, analogous to the cytoprotective effects of neuroglobin (19). Studies using the myoglobin knock-out mouse have now confirmed that myoglobin is necessary for the following: 1) nitrite-dependent NO and cGMP generation in the heart; 2) nitrite-dependent cytoprotection after ischemia/reperfusion, and 3) nitrite-dependent control of hypoxic cellular respiration (20). It is therefore apparent that both myoglobin and neuroglobin may have roles in limiting cell death after ischemia-reperfusion injury. Of relevance to neuroglobin, we have recently discovered that the mitochondrial protein cytochrome c can reduce nitrite to NO more rapidly than either hemoglobin or myoglobin, but only when it assumes the five-coordinate conformation (21). This conformation only occurs during the interaction with anionic phospholipids or upon oxidation or nitration of protein residues, suggesting a post-translational tertiary structure regulation of nitrite reduction and NO generation.
Interestingly, human neuroglobin contains two surface cysteines (Cys-46 and Cys-55) that form a disulfide bridge upon oxidation (22). Disulfide bond formation is accompanied by a decrease in the distal histidine binding affinity to heme iron (KHis has been shown to decrease from ∼3000 to 280, and values are calculated as kon/koff and are dimensionless) (23). This in turn increases the subpopulation of five-coordinate neuroglobin and increases the affinity for endogenous ligands such as oxygen (P50 shift from about 9 to 1 mm Hg) (22). Nicolis et al. (24) reported that the oxidized disulfide-bridged neuroglobin also exhibits a higher affinity for nitrite than the thiol-reduced form.
We therefore hypothesized that neuroglobin, and more generally the six-coordinate heme globins, may function as post-translationally redox-regulated nitrite reductases that generate NO under control of the six-to-five-coordinate heme iron transition. Such functionality may underlie hypoxic neuroprotective signaling and the control of hypoxic cellular respiration.
EXPERIMENTAL PROCEDURES
Reagents and Standards Sample Preparation
All reagents were purchased from Sigma unless otherwise specified. UV-visible spectra and kinetic data were recorded on an HP8453 UV-visible spectrophotometer (Agilent Technologies, Palo Alto, CA). Superdex S200 gel filtration columns were purchased from GE Healthcare. Solutions of sodium dithionite and nitrite were prepared with argon-degassed 0.1 m phosphate buffer, pH 7.4, and kept at 25 °C under inert gas. Purchased horse heart myoglobin was further purified by passing through a Sephadex G-25 gel filtration column and eluting with 0.1 m phosphate buffer, pH 7.4. Neuroglobin was oxidized with excess potassium ferricyanide or reduced by incubation with 500 mm sodium dithionite; excess reagents were removed by passing the mixture through two sequential Sephadex G-25 desalting columns. Met-Ngb concentrations were estimated by measuring the absorbance of the heme Soret band using ϵ414 = 129 mm−1 cm−1. Standard reference species of recombinant Ngb for spectral deconvolution were prepared following procedures previously described for hemoglobin and myoglobin (18, 19, 25). Reference spectra were recorded for deoxy-Ngb, iron-nitrosyl-Ngb, met-Ngb, and oxy-Ngb. When necessary, anaerobically reduced Ngb samples were prepared in glovebox under a 2–4% H2 atmosphere of catalyst-deoxygenated nitrogen, collected directly in cuvettes, and sealed with rubber septa inside the glovebox. To reduce the intramolecular Ngb disulfide bond, Ngb solutions were dialyzed in PBS containing 10 mm DTT dissolved in degassed 100 mm phosphate buffer and 0.5 mm EDTA as described previously (24).
Cloning, Expression, and Purification of Recombinant Ngb
Molecular biology was performed using standard techniques. For the expression of the 151-amino acid polypeptides of human Ngb, the cDNA SC122910 was cloned in BL21(DE3)pLysS(pET28a). Cells were grown in LB broth containing 30 μg/ml kanamycin and 25 μg/ml chloramphenicol; expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and carried out for 4 h at 37 °C, including δ-aminolevulinic acid (0.4 mm) in the media. Purification was carried out as described with minor modifications (26). To increase purification yield, human Ngb cDNA was fused with a His6 tag in the N termini and cloned into Escherichia coli BL21(DE3)pET28a for protein overexpression, and His-tagged human Ngb was purified using nickel-nitrilotriacetic acid-agarose (Qiagen, Valencia, CA) affinity column according to the manufacturer's instructions. The eluted protein was dialyzed against PBS at 4 °C, concentrated with 10-kDa cutoff filter, and stored in aliquots at −80 °C. The additional amino acids at the N terminus of His-tagged Ngb were removed using a thrombin cleavage capture kit (Novagen, Gibbstown, NJ). The purity of each recombinant Ngb batch prepared was assessed by SDS-PAGE and UV-visible spectroscopy. The number of accessible thiol groups per heme was measured by the 4,4′-dithiodipyridine assay (27).
Mutagenesis of Recombinant Ngb
Site-directed mutagenesis was performed using QuikChange II kit (Stratagene, Palo Alto, CA). The oligonucleotides for mutation C46A, C55A, H64L, and H64Q are reported in supplemental Table 1. The template used for C46A and C55A was pCMV-1A and for H64L and H64Q was pET28a. Clones were sequenced to confirm the desired mutations. Expression and purification of mutant Ngb were carried out using the same procedures as for wild type Ngb.
Anaerobic Reactions of Globins with Excess Nitrite
Reaction kinetics of known amounts of Mb or Ngb with nitrite were monitored by absorption spectroscopy for the indicated time in a cuvette in the presence or in the absence of 2–4 mm sodium dithionite. All reactions were run at 25 or 37 °C in 0.1 m phosphate buffer at controlled pH. Deoxygenated nitrite was added, using an airtight syringe, to a sealed anaerobic cuvette to initiate the reaction. Oxygen contamination was prevented by application of positive argon pressure without a channel for gas escape. Concentrations of single species during reactions were determined by least squares deconvolution of the visible absorption spectrum into standard reference spectra using Microsoft Excel analysis. Oxy-Ngb was included to confirm successful deoxygenation before the reaction. To vary pH values, deoxy-Ngb and nitrite were prepared in phosphate buffer adjusted to the target pH values. Fast kinetic studies were performed using an Applied Photophysics SX-20 stopped-flow instrument equipped with rapid-scanning diode array detection (Applied Photophysics Ltd., Leatherhead, Surrey, UK). Experiments were carried out at 25 °C by rapidly mixing a solution of reduced deoxy-Ngb containing 2 mm dithionite with a known solution of nitrite at controlled pH. To determine bimolecular rate constants, all reactions were analyzed with Pro-K software (Applied Photophysics Ltd., Leatherhead, Surrey, UK) using singular value decomposition followed by fitting of the reduced data matrix to a pseudo first-order kinetic model.
Model of the Wild Type Human Ngb Structure
Crystallization of the wild type human Ngb is hindered by aggregation and precipitation problems. Mutation of the three cysteine residues yielded a protein suitable for crystallization studies (28). The reported structure (Protein Data Bank code 1OJ6) thus includes the mutations C46G, C55S, and C120S. To assess the possible structure of the wild type enzyme, a homology model was built using the Swiss-Model server (29) with the sequence of the wild type Ngb and the available human structure as template. The coordinates of the heme molecule were copied from the 1OJ6 structure.
Determination of the Midpoint Redox Potential of the Thiol/Disulfide Couple in Ngb
Wild type and C55A mutant Ngb (50–60 μm) were incubated at 37 °C in an anaerobic glovebox with solutions containing various ratios of reduced (GSH) and oxidized (GSSG) glutathione, with the total GSH and GSSG concentration fixed at 20 mm in 0.1 m phosphate buffer, pH 7.0 (30). The GSH/GSSG ratio was varied to establish a gradient of redox potentials between −130 and −250 mV, calculated by the Nernst equation according to a midpoint reduction potential of −240 mV. After 1 or 2 h of incubation, glutathione was removed anaerobically by passage through a G-25 column, and Ngb was reacted immediately with 10 mm nitrite in 0.1 m phosphate buffer, pH 7.0, as described above. The observed rate constant determined at each glutathione ratio was fitted using the Nernst equation, and the midpoint reduction potential of the thiol/disulfide couple of Ngb was calculated.
Determination of Nitrite Binding Constants
The binding constant of nitrite to met-Ngb was determined by incubation of 10 μm wild type or mutant Ngb with increasing concentrations of nitrite in 200 mm phosphate buffer, pH 7.4, in a cuvette at 25 °C, and the UV-visible spectra were recorded after each increase in nitrite concentration. The dissociation constant Kd for each protein was determined by interpolation of the absorbance difference data following procedures in Nicolis et al. (24). The reaction rate of nitrite and met-Ngb was then determined under the same conditions at 100 mm nitrite.
NMR Spectroscopy
1H NMR spectra in 1H2O were collected at 29 °C on a Bruker DRX-600 NMR spectrometer (Bruker, Billerica, MA) operating at 599.79 MHz with a 5-mm triple resonance probe using a water presaturation pulse sequence with 1-s irradiation time. Samples of wild type and mutant 250–300 μm met-Ngb were prepared in 0.1 m phosphate buffer, pH 7.4. Typically, 1024 transients were averaged, using 90° pulses, spectral width of 80 ppm, and 16 K time domain points. Spectra are referenced indirectly through the resonance of the water, which occurs at 4.76 ppm downfield from the methyl resonance of 2,2-dimethyl-2-silapentane-5-sulfonate.
Electron Paramagnetic Resonance Spectroscopy
Iron nitrosyl species were measured by EPR spectroscopy using a Bruker EMX 10/12 spectrometer (Bruker, Billerica, MA) operating at 9.4 GHz, 5-G modulation, 10.1-milliwatt power, 327.68-ms time constant, and 163.84-s scan over 600 G at 110 K as described previously (21, 31). The concentrations of Mb and Ngb species were determined by performing the double integral calculation and comparing with standard samples.
Direct Measurement of NO Release by Chemiluminescence
Deoxy-Ngb (final concentration 20 μm) was injected in a reaction vessel containing 100 mm phosphate buffer, pH 7.4 (3 ml), pre-equilibrated, and purged with helium and connected in line to a nitric oxide analyzer (NOA 280i) (Sievers, GE Analytical Instruments, Boulder, CO). Once a stable base line was established, Ngb was reacted with a known amount of nitrite as described previously (18). Traces were smoothed by a running average spanning 2 s.
Respiration of Isolated Mitochondria in the Presence of Ngb
Mitochondria were isolated from the livers of male Sprague-Dawley rats and incubated with wild type or mutant Ngb proteins in a sealed, stirred chamber at 37 °C. State 3 respiration was stimulated with succinate (15 mm) and ADP (1 mm), and oxygen consumption was measured with a Clark-type oxygen electrode. All experiments with respiring mitochondria were performed according to previously reported procedures (19). Similar experiments were performed with SH-SY5Y cells suspended in the respirometer and treated with the uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (5 μm) to measure hypoxic inhibition of cellular respiration.
Immunoblotting of Ngb Expression in SH-SY5Y Neuronal Cells
Equal amounts of denatured total proteins (25 μg) from the SH-SY5Y neuronal cells expressing GFP vector, wild type and H64L mutant Ngb, were subjected to 4–15% SDS-polyacrylamide gradient gels and immunoblotted with anti-GFP monoclonal antibody (Santa Cruz Biotechnologies, Inc., Santa Cruz, CA) and then scanned using a Odyssey imaging system (LI-COR Biosciences, Lincoln, NE).
Statistical Analysis
Each experiment was performed at least in triplicate, and values are representative of two or more independent determinations using different batches of protein purified separately. Data were analyzed using Origin 8.0 (OriginLab Corp., Northampton, MA) and expressed as mean ± S.D. Analysis for statistically significant differences among mean values was done, when applicable (Figs. 2B, 5B, and 6B), using the one-way analysis of variance.
FIGURE 2.

Redox state of cysteines 46 and 55 modulates nitrite reductase reactivity. A, model of the wild type human neuroglobin structure with the indicated reduced cysteines Cys-46, Cys-55, and Cys-120. B, determination of the number of reduced cysteines by the 4,4′-dithiodipyridine assay (see “Experimental Procedures.”). C, comparison of the decrease of deoxy-Ngb and the formation of iron-nitrosyl Ngb over time for wild type Ngb with oxidized (SS) and reduced (SH) thiol and C46A and C55A mutant Ngb. D, observed nitrite reductase rate constants versus determined redox potentials. Data were fit using the Nernst equation. The midpoint redox potential of the thiol/disulfide couple in wild type Ngb is −194 ± 3 mV. SHE, standard hydrogen electrode. E, comparison of the NMR spectrum of wild type and C55A mutant met-Ngb. F, nitrite binding affinity constant determination by difference spectral titration for wild type, DTT-reduced, and C55A mutant Ngb by differential spectra after indicated nitrite addition.
FIGURE 5.

Nitrite reduction by deoxyneuroglobin generates NO gas. A, representative chemiluminescence traces of NO detection in gas phase released during the anaerobic reaction of nitrite with buffer only (blue) or 20 μm deoxy-Ngb wild type (black), H64L (red) or C55A (green). B, quantification of the rate of NO detected per min. C, nitric oxide signal measured during incubation of 30 μm H64L deoxy-Ngb and increasing concentrations of nitrite.
FIGURE 6.

Deoxyneuroglobin nitrite reduction mediates intracellular NO signaling. A, traces of oxygen consumption by isolated mitochondria showing nitrite-dependent inhibition of respiration; the early rise in oxygen tension indicates NO-dependent inhibition of cellular respiration, which is maximal for cyanide. B, comparison of percentage of extent of inhibition (cyanide defined as 100% inhibition) as measured in A for isolated mitochondria. C, quantification of expression of GFP only, wild type Ngb, and H64L mutant Ngb in lentivirus-transfected and cloned SH-SY5Y cells by Western blot of 4–15% SDS-polyacrylamide gradient gel. D, mean extent of hypoxic inhibition of cellular respiration by incubation of SH-SY5Y cells expressing GFP, wild type Ngb, or H64L Ngb with 20 μm nitrite (*, p < 0.01; **, p < 0.001, compared with control).
RESULTS
Nitrite Is Reduced to NO via Reaction with Deoxygenated Human Neuroglobin
To examine the reaction of nitrite with neuroglobin, we expressed and purified recombinant human neuroglobin. Spectrophotometric analysis of our proteins confirmed the six-coordinate heme structure in both the ferrous and ferric states of Ngb, with visible α- and β-peaks around the 550 nm wavelength (supplemental Fig. 1A). We prepared ferrous deoxy-Ngb in an anaerobic glovebox as detailed under “Experimental Procedures” and recorded the visible spectra of the reaction between 10 μm deoxy-Ngb and 10 mm nitrite at 25 °C at constant intervals in a sealed air-tight cuvette under external argon pressure (Fig. 1A). The time-dependent changes of deoxy-Ngb, ferric met-Ngb, and iron-nitrosyl-Ngb (Fe2+-NO) species (Fig. 1B) were calculated by least squares deconvolution of the reaction spectra using standard reference spectra (supplemental Fig. 1A). In an anaerobic environment, nitrite is reduced to NO according to Reaction 1, and the NO generated has very high affinity for the ferrous Ngb heme (Fe2+) (WT Ngb: kon = 1.5 × 108 m−1 s−1 and koff = 2 × 10−4 s−1; H64L Ngb: kon = 2.7 × 108 m−1s−1 and koff = 2 × 10−4 s−1; H64Q Ngb: kon = 1.9 × 108 m−1 s−1 and koff = 3 × 10−4 s−1(32)) thus yielding ferric Ngb heme (Fe3+) and iron-nitrosyl-heme (Fe2+-NO) as a final reaction product (Reaction 2).
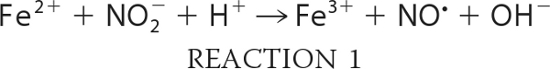 |
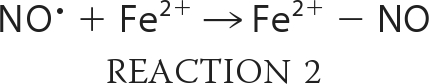 |
FIGURE 1.

Anaerobic reaction of deoxyneuroglobin with nitrite in the absence and presence of dithionite. A, selected visible spectra of the reaction between 10 μm deoxy-Ngb and 10 mm nitrite at 1-min intervals. B, time-dependent changes of deoxy-Ngb (blue), iron-nitrosyl-Ngb (green), and total met-Ngb (red) concentration during the reaction. C and D, as in A and B, respectively, for the reaction in the presence of 3 mm dithionite. E, plot of observed rate constants (kobs) versus nitrite concentration; the second-order bimolecular rate constant obtained from the linear fit of the data is 0.12 ± 0.02 m−1 s−1. F, effect of pH on the nitrite reductase reaction rates. Inset, bimolecular rate constant is linear with the proton concentration, and it extends through the zero point (line shows linear regression analysis of the data). All measurements were made in 100 mm phosphate buffer and at 25 °C as described under “Experimental Procedures.”
We observed a reaction stoichiometry consistent with the reaction of nitrite with hemoglobin or myoglobin, with two deoxy-Ngb molecules forming one iron-nitrosyl-Ngb and one ferric Ngb (Fig. 1B). Analysis of the bimolecular rate constant indicated that the reaction of nitrite with Ngb proceeds overall at 0.12 ± 0.02 m−1 s−1 at 25 °C, pH 7.4 (0.26 ± 0.02 m−1s−1 at 37 °C). A recent study (33) reported that the reaction of deoxy mouse neuroglobin with nitrite in the range 7–230 μm generated ferric met-Ngb in excess of ferrous nitrosyl-Ngb at apparent second-order rate constant of 5.1 ± 0.4 m−1 s−1; however, our experimental conditions with human neuroglobin differ considerably.
Both Gladwin and co-workers (34) and Salhany (35) have shown that the reaction of nitrite with hemoglobin in the presence of dithionite proceeds via Reactions 1 and 2, but the ferric heme that is formed is reduced back to the ferrous form to continue the reaction. Thus, iron-nitrosyl-heme forms at the same rate as deoxyheme is consumed, and the overall stoichiometry is one deoxy-Ngb forming one iron-nitrosyl-Ngb. Performing the reaction in the presence of dithionite limits the auto-oxidation of the ferrous heme prior to the reaction with nitrite and allows for facile assessments of anaerobic reaction mechanisms and kinetics. By complementary studies using myoglobin, we verified that the rate-limiting step of the reaction in the presence of dithionite is the heme iron catalyzed conversion of nitrite to NO (supplemental Fig. 2, A and B). We then performed the reaction of anaerobic nitrite and deoxy-Ngb (10 mm and 10 μm, respectively) as described above in the presence of 3 mm excess dithionite at pH 7.4 in 100 mm phosphate buffer (Fig. 1, C and D). The stoichiometry was consistent with one deoxy-Ngb forming one iron-nitrosyl-Ngb, and the calculated bimolecular rate constant was 0.11 ± 0.01 m−1 s−1, in accordance with the value obtained in the absence of dithionite. We further investigated the reactivity of deoxy-Ngb with nitrite in the concentration range 0.25–20 mm (Fig. 1E). The second-order bimolecular rate constant derived from the linear fit of the observed rate constants versus nitrite concentration is 0.12 ± 0.02 m−1 s−1 in agreement with the calculated instantaneous reaction rate.
Proton Dependence of the Nitrite Reductase Reaction with Neuroglobin
We next explored whether deoxy-Ngb-dependent nitrite reduction requires a proton (Reaction 1). We determined the pH dependence of the bimolecular rate constant of the nitrite reductase reaction near the physiological range, pH 6.5–8.0 (Fig. 1F). We found that increasing concentrations of protons accelerates the reaction rate by 10-fold for each pH unit decrease. The slope of the linear fit, which represents the order of rate dependence on [H+], is 0.96, close to the ideal of 1.0, and it extends through the zero point (Fig. 1F, inset) indicating the requirement for one proton in the reaction. We conclude that the reaction constitutes a concerted electron and proton transfer to nitrite to form NO analogous to bacterial nitrite reductase.
Surface Cysteines Cys-46 and Cys-55 Regulate the Heme Pocket Coordination and the Rate of Nitrite Reduction to NO
Unlike most other globins, human Ngb displays three conserved cysteines (notable exception being mouse Ngb) at positions 46, 55, and 120 located on the protein surface as shown in the wild type thiol-reduced human Ngb structure model (Fig. 2A). When oxidized, cysteines 46 and 55 form an intramolecular disulfide bond (36), which influences the position of the E-helix containing the distal histidine (22) and regulates the heme ligand binding equilibrium. Reduction of the disulfide bond allows additional structural freedom in the orientation of the E-helix (Fig. 2A), which leads to an increased proportion of molecules in the six-coordinate state and thus reduced oxygen and nitrite binding affinities (22, 24). We determined by the 4,4′-dithiodipyridine assay the number of accessible thiols per heme in our wild type Ngb, as purified, reduced by DTT, and in the Cys-55 to alanine mutant Ngb (Fig. 2B). The results are consistent with the quantitative formation of a disulfide bond during protein purification and the presence of the single reduced Cys-120 in the oxidized thiol form. To determine whether the rate of nitrite reduction is influenced by the redox state of cysteines 46 and 55, we first reduced the cysteines by incubation with 10 mm DTT and then measured the rate of nitrite reduction after anaerobic DTT removal. Fig. 2C shows that reduction of the disulfide bond slows down the rate by about 2-fold (0.062 ± 0.005 m−1 s−1 at 25 °C, pH 7.4). To directly test the hypothesis that disulfide bridge reduction affects the nitrite reactivity of neuroglobin, we generated recombinant mutants with cysteine 55 or 46 replaced by alanine (C55A and C46A), which slowed down the rate of nitrite reduction to similar rates observed with Ngb having fully reduced cysteines (Fig. 2C). For a direct comparison, we report the bimolecular reaction rates for WT and mutant Ngb proteins in Table 1 together with values for myoglobin and hemoglobin.
TABLE 1.
Summary of bimolecular reaction rates for the reaction of heme-containing deoxyglobins with nitrite (Hb, human hemoglobin; sw Mb, sperm whale myoglobin; Ngb, neuroglobin)
All reactions were studied in 100 mm sodium phosphate buffer, pH 7.4. Hemoglobin values were determined at 37 °C. Myoglobin and neuroglobin values were determined at 25 °C.
| Protein | k |
|---|---|
| m−1s−1 | |
| Hb (T state) | ≈0.12a |
| Hb (R state) | ≈6a |
| Horse Mb | 2.9 ± 0.2 |
| sw Mb WT | 5.6 ± 0.6 |
| sw Mb H64A | 1.8 ± 0.3 |
| sw Mb H64L | Very slowb |
| Ngb WT SS | 0.12 ± 0.02 |
| Ngb WT SH | 0.062 ± 0.005 |
| Ngb C55A | 0.060 ± 0.008 |
| Ngb C46A | 0.058 ± 0.006 |
| Ngb H64L | 259 ± 8 |
| Ngb H64Q | 267 ± 16 |
a Values are from Ref. 18.
b The reaction of Mb H64L is significantly (more than 10-fold) slower than wild type or H64A sperm whale Mb and apparently independent of [NO2−] in the concentration range studied (1–5 mm).
Physiological Redox Control of the Cys-46 to Cys-55 Disulfide Bond Regulates the Rate of Nitrite Reduction to NO
We next determined if the formation of a disulfide bond between Cys-46 and Cys-55 is redox-regulated within the physiological range of cellular redox state. We incubated wild type and C55A Ngb with increasing ratios of reduced/oxidized glutathione that established a gradient of ambient redox potentials (37). After 120 min of incubation, we removed glutathione anaerobically by passage through a G-25 column and measured the rates of nitrite reduction (Fig. 2D). We found that there was a sudden and substantial drop in the observed nitrite reductase rate constants (kobs) with decreasing redox potential only for the wild type protein. Fitting the data to the Nernst equation provided a midpoint reduction potential of the Cys-46/Cys-55 thiol/disulfide redox couple of −194 ± 3 mV. This value is within the range of cellular redox potentials (37).
To directly examine whether the cysteines redox state causes changes in heme pocket molecular and electronic structure, we compared the NMR spectrum of wild type and C55A mutant met-Ngb (Fig. 2E). Characteristic NMR signals for the heme methyls are visible in the spectral regions around 36 and 23 ppm and 20 to 12 ppm and were assigned by comparison with the published spectra (38, 39). The two spectra are largely similar, but a few marked differences in the positions of several heme methyl resonances (M8-B, M5-A, M1-A, and M5-B) as well of several hyperfine shifted resonances between 18 and 12 ppm (region marked with an asterisk in Fig. 2E) are evident. Also, some unassigned ring currents shifted resonances around −2 ppm are different. We conclude that the thiol mutation C55A affects the geometry of the heme pocket environment.
Nicolis et al. (24) reported that the oxidized disulfide-bridged (SS) met-Ngb exhibits a higher affinity for nitrite than the thiol-reduced (SH) form. We then determined the nitrite affinity for the wild type SS- and SH-Ngb and for C55A mutant met-Ngb by difference spectra titration (Fig. 2F). Because of the small absorbance changes, the calculated dissociation constants (Kd), reported in Table 2, show large standard deviations; nevertheless, there is an apparent influence of the redox state of the cysteines on the distal histidine and nitrite affinity to the heme iron. During these experiments, we noticed that met-Ngb very slowly reacts with nitrite to produce nitrosyl-Ngb (bimolecular rate constants reported in Table 2). The slow rates of reaction produce a detectable spectroscopic effect only at high nitrite concentrations, approaching 0.1 m, and result in an artificial decrease of maximal absorbance difference that has previously been assigned to a second low affinity binding constant (24). These experiments indicate that the redox state of cysteines 46 and 55 regulates both the five-to-six-coordinate equilibrium and the rate of nitrite conversion to NO. Intriguingly, an analogous effect is observed with hemoglobin, in which oxidation of cysteine 93 speeds up the rate of nitrite reduction to NO, and reduction slows the rate (40). This effect has been attributed to the effect of thiol oxidation on decreasing the heme redox potential.
TABLE 2.
Nitrite dissociation constants (Kd) and bimolecular rate constants (k) for reactions of met-Ngb with nitrite determined at 25 °C in 200 mm phosphate buffer, pH 7.4
| Protein | Kd (NO2−) | Rate of nitrite ferric heme reduction |
|---|---|---|
| mm | m−1s−1 | |
| Ngb WT SS | 6.2 ± 2.1 | 0.0005 ± 0.0005 |
| Ngb WT SH | 12.6 ± 3.3 | 0.0002 ± 0.0005 |
| Ngb C55A | 30.1 ± 4.5 | 0.0002 ± 0.0004 |
| Ngb H64L | 8.3 ± 1.8 | 0.032 ± 0.002 |
| Ngb H64Q | 12.5 ± 1.8 | 0.016 ± 0.009 |
Rate of Nitrite Reduction Is Maximal in the Five-coordinate State of Neuroglobin
To test the hypothesis that a change in the equilibrium between the five- and six-coordinate Ngb subpopulations mediates the control of the nitrite reduction rate, we generated recombinant Ngbs with His-64 replaced by Leu or Gln. The absorbance spectra analysis of oxygenated and deoxygenated ferrous H64L and H64Q Ngb and the ferric species (supplemental Fig. 1) confirmed that both mutants are “locked” in the five-coordinate conformation (41) and have very similar spectral characteristics to the classic five-coordinate heme protein myoglobin (supplemental Fig. 1). We examined the reaction of nitrite with deoxygenated H64L Ngb in the presence of excess dithionite similarly to experiments with wild type Ngb but using 100 μm nitrite (Fig. 3, A and B). To our surprise, the rate of deoxy-Ngb conversion to nitrosyl-Ngb was extremely fast, and the bimolecular rate constant was ∼2000-fold higher than for the wild type Ngb. We then used fast mixing stopped-flow spectroscopy to determine the rates of the reaction in the range 10–1000 μm nitrite (Fig. 3C). The observed rate constants increased linearly with increasing nitrite concentrations, and the bimolecular rate constant derived from the linear least square fit was 259 ± 8 m−1 s−1 at 25 °C, pH 7.4. Examination of the reaction at different pH values (Fig. 3D) indicates that the reaction requires a proton similar to the reaction with wild type Ngb. Remarkably, the rate increases above 2,500 m−1 s−1 at pH 6.5 and 25 °C. The H64Q mutant showed a similar behavior, with a rate constant of 267 ± 16 m−1 s−1 at 25 °C, pH 7.4 (Table 1, supplemental Fig. 3), and rate constants above 2,000 m−1 s−1 at pH 6.5. These are the fastest reactions of nitrite with a heme-globin ever reported and confirm our hypothesis that the six-to-five-coordinate heme pocket transition regulates the rate of nitrite reduction to NO.
FIGURE 3.

Kinetics of nitrite reaction with mutant H64L Ngb. A and B, spectrophotometric analysis of the anaerobic reaction of 10 μm H64L deoxy-Ngb with 100 μm nitrite, pH 7.4, at 25 °C and 3 mm dithionite. C, plot of kobs versus nitrite concentration (10 μm to 1 mm) for H64L Ngb-mediated reduction of nitrite and formation of Ngb Fe2+-NO at pH 7.4 and at 25 °C. The bimolecular rate constant derived from the linear fit of the data is 259 ± 8 m−1 s−1. D, effect of different pH values on the nitrite reductase rates. Inset, bimolecular reaction rate is linear with the proton concentration. E, nitrite binding affinity constant determination for H64L and H64Q. F, comparison of representative traces of Ngb wild type (with reduced and oxidized surface thiols) and mutants H64L and H64Q. The absorbance decreases of the Soret peak (425 nm) are plotted as the percentage of the total absorbance change for human Ngb H64L measured at 25 °C, pH 7.4.
We next determined the nitrite binding affinity for H64L and H64Q met-Ngb by fitting of the maximal changes in the Soret band as a function of nitrite concentration (Fig. 3E, normalized difference spectra titration compared with wild type SS-Ngb). The calculated Kd values for these mutants are comparable with the wild type Ngb (Table 2); however, the total maximal absorbance difference for H64L and H64Q mutants is more than 20-fold greater than wild type (supplemental Fig. 4). These results suggest that in wild type Ngb His-64 binding may outcompete nitrite binding even at high concentrations so only the ≈1% of five-coordinate molecules may bind nitrite, whereas for H64L and H64Q a fully bound situation is possible. The spectral changes for H64L and H64Q also indicate differences in the nitrite binding modes. For the H64L mutant, there is a large increase in the Soret peak and little change in the 500–600 nm region. In the case of H64Q there is a decrease in the Soret peak accompanied by a shift to longer wavelengths and also changes in the 500–600 nm region that resemble the nitrite binding to Hb or Mb (supplemental Fig. 4).
Finally, we compared the reaction of 1 mm nitrite with our wild type Ngb (with oxidized and reduced cysteines), mutant H64L, H64Q, and C55A Ngb (absorbance decrease of the Soret peak at 425 nm) in 0.1 m HEPES, pH 7.4. The relative percentage of the total absorbance change occurring in the first 60 min of the reaction is shown in Fig. 3F (with H64L or H64Q Ngb normalized to 100%, wild type SS-Ngb 38%, wild type SH-Ngb 20%, and C55A Ngb 18%). For both H64L and H64Q Ngb, the reaction of the five-coordinate mutant proteins reached the end point in the first minute of the reaction and are expanded in the inset of Fig. 3F.
Confirmation of Reaction Kinetics Using Electron Paramagnetic Resonance Spectrometry
EPR spectrometry allows for direct measurement of the paramagnetic NO-heme (iron-nitrosyl) species and provides confirmation of NO formation during the reaction of nitrite with Ngb. We evaluated the Fe2+-NO build up following reaction of 1 mm nitrite with wild type SS-Ngb, SH-Ngb, and mutants H64L and H64Q Ngb (40 ± 5 μm) and compared it with the rate of iron-nitrosyl-myoglobin formation (Fig. 4, A and B). EPR spectra analysis confirmed that the reduction of the cysteines (stabilizing the six-coordinate heme geometry) slowed the rate of iron-nitrosyl-Ngb formation, although replacement of the distal histidine with leucine (five-coordinate stabilization) dramatically increased the rate of NO formation. In particular, experiments using H64L and H64Q Ngb mutants and 1 mm nitrite were almost complete in 1 min, and to allow assessment of the reaction kinetics, lower concentrations of Ngb (10 μm) and nitrite (50 μm) were necessary (Fig. 4, C and D). The calculated rates of nitrosyl-Ngb formation are similar to data obtained by absorbance spectrometry.
FIGURE 4.

EPR spectroscopy. A and C, EPR spectra showing Fe2+-NO build up following addition of amount of nitrite. B and D, rate of formation of iron-nitrosyl-heme (Fe2+-NO) species measured by EPR. The concentrations were determined by performing the double integral calculation and comparing with standard samples.
Nitrite Reduction by Deoxyneuroglobin Generates NO
The reaction of nitrite with deoxy-Ngb generates NO and ferric Ngb. Although in our in vitro conditions deoxy-Ngb can recapture the NO, we next explored if free NO gas can escape at measurable rates. We mixed anaerobic Ngb (20 μm) and nitrite (1 mm) in a vessel purged with helium and carried in-line to a chemiluminescent NO analyzer. In these conditions, the anaerobic mixture generated NO in gas phase (Fig. 5A), and the rate of NO formation was again regulated by the cysteines 46–55 disulfide bond and by the heme pocket six-to-five-coordination equilibrium. Fig. 5B shows that the rate of NO detected was significantly decreased in reactions with six-coordinate C55A Ngb and increased in reactions with the five-coordinate H64L Ngb, consistent with the hypothesis of six-to-five-coordinate heme pocket control of nitrite reduction. Finally, we incubated mutant H64L Ngb (30 μm) with increasing amounts of nitrite, starting with physiologically relevant concentrations (10 and 25 μm) up to 1 mm, and we observed a rapid NO generation response roughly proportional to the nitrite concentration injected (Fig. 5C).
Comparison of Mutant Myoglobin and Neuroglobin Confirms Unique Fast Reactivity of Five-coordinate Neuroglobin with Nitrite
To elucidate the factors that confer increased nitrite reductase activity to the Ngb H64L mutant, we characterized the nitrite reductase activities of Mb mutants H64L and H64A and Ngb mutant H64Q (summarized in Table 1). Despite the structural similarities between the two globins, the difference in reactivity for comparable His-64 mutations took opposite directions. Replacement of Mb His-64 with Leu or Ala leads to decreased reactivity. This may be related to His-64 stabilizing the heme ligands through hydrogen bonding as highlighted by previous reports (42, 43). However, Ngb H64Q shows similar rates to the H64L mutant. This indicates that the removal of the 6th ligand and formation of a stable five-coordinate neuroglobin is the major determinant of the increased reactivity, and the residue polarity constitutes a lesser effect. Possible explanations for these effects are discussed below.
Nitrite Reduction by Deoxyneuroglobin Mediates NO Signaling
To test whether Ngb-generated NO inhibits mitochondrial respiration during hypoxia, isolated rat liver mitochondria were placed in a sealed, stirred respirometer, and substrates were added to stimulate respiration as described previously (19). Mitochondria were allowed to respire until the ambient oxygen tension dropped below detection levels. At this point the respirometer is opened to air oxygen, and cyanide is added to evaluate the time to complete inhibition of respiration, as determined by the increase in oxygen tensions measured with a Clark-type oxygen electrode (Fig. 6A). The extent of mitochondrial inhibition for all experiments (without cyanide) is then compared with the effect of cyanide. We detected no significant inhibition of respiration when nitrite (20 μm) or purified wild type Ngb (5 μm) alone were incubated with respiring mitochondria. However, when the same concentrations of nitrite and protein were allowed to react together, we observed 78 ± 6% inhibition of respiration. As expected, the extent of inhibition was increased significantly by the H64L mutant Ngb (96 ± 2% inhibition) and decreased by the C55A mutant Ngb (62 ± 4% inhibition) (Fig. 6B). To evaluate this in cells, the neuronal cell line SH-SY5Y was stably transfected using a lentivirus vector with GFP-tagged wild type and H64L mutant Ngb (Fig. 6C) and was used to perform similar experiments. One million intact SH-SY5Y cells were suspended in the respirometer, and maximal respiration rate was stimulated by addition of the uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone. Then nitrite was added to cells transfected with GFP only (negative control) and cells expressing wild type Ngb or the H64L mutant Ngb. In Fig. 6D, we compare the extent of respiration inhibition to the cyanide effect (complete inhibition); cells with GFP only exhibited no significant inhibition, but we observed about 15 and 40% inhibition, respectively, for wild type and H64L Ngb.
DISCUSSION
Altogether, our experiments reveal the following: 1) neuroglobin can function as a nitrite reductase, producing NO from nitrite; 2) the redox state of the surface thiols of Cys-46 and Cys-55 modulates the heme coordination, nitrite affinity, and NO generation and signaling; 3) replacement of the His-64 side chain locks the heme in a five-coordinate geometry with increased reactivity toward nitrite; and 4) under hypoxic conditions neuroglobin can inhibit mitochondrial respiration in the presence of nitrite.
We also show that the reaction of deoxy-neuroglobin with nitrite proceeds with stoichiometry formally similar to the reaction of myoglobin or hemoglobin (44). However, in neuroglobin, the process is further regulated by the six-to-five-coordinate equilibrium.
A mechanistic proposal is presented in Scheme 1 where the first step of the reaction is the dissociation of His-64 from the heme iron. The results obtained with Ngb H64L and H64Q mutants indicate that this process directly limits the nitrite reduction rate. The five-coordinate heme can now bind to external ligands such as nitrite. As evidenced in recent reports (45, 46) by experimental and density functional theoretical studies, nitrite may bind to the heme in either N-nitro- or O-nitrito-conformations (Scheme 1, second step). We cannot conclude from our results if either binding mode is preferred, and given the differences in the heme pocket of Ngb, as compared with Mb (supplemental Fig. 5), the observations made in Mb may not extrapolate (47). Addition of a proton to the nitrite yields the nitrous acid-bound species and those can be directly formed by nitrite at low pH. An alternative possibility is indeed the initial binding of HONO to the five-coordinate heme iron. In the N-bound route, loss of OH− and electron transfer yields the Fe3+-NO species with subsequent NO dissociation. In the O-bound route, NO dissociates from the bound nitrous acid, and Fe3+-OH− is formed. This met-hydroxide complex can be then protonated to the aquomet form. Independently of the route, the end products of the reaction are eventually the same, Fe3+ heme and NO. Interestingly, the dependence of this reaction on the concentration of proton is opposite to that of the reductive nitrosylation reaction, where the reaction rate increases with the concentration of OH− (48). These data show that the nitrite reductase reaction is the reverse of reductive nitrosylation. From Scheme 1, the relationship between both reactions is apparent, although it must be noted that the N- and O-bound routes indicate two different reductive nitrosylation reactions, one involving Fe3+-NO + OH− and another with Fe3+-OH− + NO. It is conceivable that depending on the protein (and the concentrations of NO and OH−), one route may be more important than the other.
SCHEME 1.

Mechanistic proposal for the reaction of neuroglobin with nitrite (N-bound versus O-bound routes). The first step of the reaction is the dissociation of His-64 from the heme iron (left). The five-coordinate heme can now bind nitrite. Two binding modes for nitrite are possible, N-binding (top right section) or O-binding (bottom right section). Subsequent addition of a proton yields the nitrous acid-bound species (those can be directly formed by nitrite at low pH). In the N-bound route, loss of OH− and electron transfer yields the Fe3+-NO species with subsequent NO dissociation. In the O-bound route, NO dissociates from the bound nitrous acid, and Fe3+-OH− is formed. This met-hydroxide complex can be then protonated to the aquomet form.
In myoglobin, the H64L mutation severely impairs ligand binding of cyanide and azide (42, 49) as hydrogen bonding appears to be necessary to stabilize the ligand. The situation for neuroglobin appears to be very different, and once the His-64-heme interaction is disrupted, the ligand affinity increases. Ngb H64L and H46Q show no significant differences in the nitrite reduction rates, and thus nitrite reduction appears to be unrelated to hydrogen bonding. We speculate that the differences in the nitrite reactivity for Ngb and Mb/Hb (and the different requirements for ligand hydrogen binding) can be related to differences in the preferred nitrite-binding mode. This, in turn, may be related to the electronic properties of the heme. These concepts deserve further research; resolving the crystal structure of the nitrite-bound ferric Ngb complex could provide definitive experimental insight into the nature of this rather complex nitrite-heme interaction.
Modeling of Nitrite Reduction by Five-coordinate Neuroglobin
Taking into account the proposed mechanism of reaction in Scheme 1, we can expand and rewrite our reaction model as shown in Reactions 3–7,
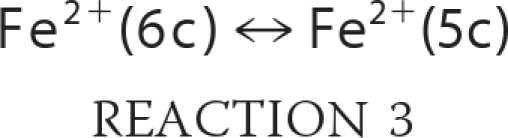 |
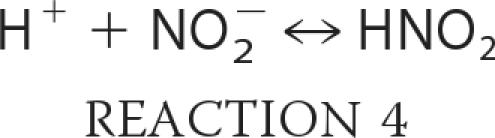 |
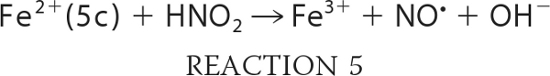 |
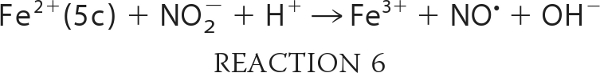 |
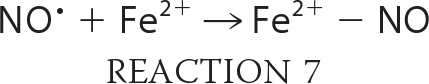 |
Then the rate of reduction of nitrite by Ngb can be written as shown in Equation 1,
where KHis is the ratio of hexacoordinate to pentacoordinate Ngb (Reaction 3); Ka is the equilibrium constant for nitrous acid and nitrite anion (about 10−3.15 m; Reaction 4); k0 is the rate of reaction of nitrous acid with Ngb (Reaction 5), and k′ is the rate that nitrite anion reacts with Ngb (Reaction 6). Equation 1 follows that used by Doyle for the reduction of nitrite by hemoglobin (50), combined with the idea that the heme must be at least transiently pentacoordinate for the reaction to occur. It describes the reaction to be bimolecular in nitrite and Ngb with the bimolecular rate constant k being equal to the term multiplying the concentration of these reactants;
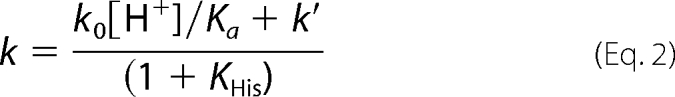 |
The rate of the reaction of Ngb with nitrite anion, like Hb, is small compared with that with nitrous acid except at very high pH. In principle, k0 could be different for the WT and H64L or other Ngb mutants. However, if the only or dominating effect of the mutation is the formation of a permanently pentacoordinate heme (KHis = 0), then one would expect k0 to be the same for both species. However, if the mutation affects more than the heme coordination state (such as hydrophobicity in the heme pocket) then we would expect it to change k0 as well.
To test the simple model and determine whether the H64L mutation does more than affect the coordination of the heme, we modeled our bimolecular rate constants as a function of pH. Fig. 7A shows the data and fit for the H64L mutant, where KHis is taken as zero. The least squares fit gave k0 as 6.5 × 106 m−1 s−1 and k′ as 49 m−1 s−1, confirming that k0 dominates. Next, we used these values to fit the WT data (Fig. 7B), with the only free parameter now being KHis, which we found to be 4200. The model fits the data very well, and the value obtained for KHis is very much in agreement with those determined previously (for example 3000 (8)). Additional calculations with the H64Q mutant (supplemental Fig. 3) yielded values of k0 as 4.6 × 106 m−1 s−1 and k′ as 50 m−1 s−1, in remarkable agreement given the values determined for H64L. In conclusion, our results support the idea that the rate differences between WT and H64L/H64Q Ngb can be primarily determined by the heme coordination (i.e. the six-coordinated to five-coordinated transition), and the nitrite reactivity of the WT enzyme could be vastly increased by changes in the KHis parameter.
FIGURE 7.

Fitting of bimolecular rate constants as a function of pH. Equation 2 was used to fit the data with Ka taken as 7 × 10−4 m−1. A, data for H64L were fit allowing k0 and k′ to vary. B, data for WT were fit allowing only KHis to vary using the values obtained from fitting of H64L data (A) for the other parameters.
Physiological Relevance of the Nitrite Reductase Activity of Neuroglobin
Neuroglobin has been shown to promote survival of neurons in hypoxic conditions, but its physiological function is still unclear. Some of the proposed functions include oxygen supply, reactive oxygen species/reactive nitrogen species detoxification, regulation of signaling pathways (10), and more recently, inhibition of cytochrome c-induced apoptosis (52). Ngb is expressed in metabolically active cells and organs (neurons, endocrine organs, retina, etc.) and has been hypothesized to interact with mitochondria and mediate cytoprotective responses to ischemic stress (53). NO binding to cytochrome c oxidase has been shown to reversibly inhibit electron transport at low oxygen tensions, in a process thought to contribute physiologically to hypoxic vasodilation and to the extension of oxygen diffusion gradients (54, 55). There is also increasing evidence of neuroprotective effects of NO and nitrite/nitrate (51, 56). We therefore hypothesized that the nitrite reductase activity of Ngb may regulate the hypoxic inhibition of cellular respiration by NO binding to cytochrome c oxidase. This pathway provides an alternative explanation of the protective action of neuroglobin.
NO can inhibit mitochondrial respiration and activate soluble guanylyl cyclase at picomolar concentrations. For this reason, it is possible that changes in the disulfide bond may produce enough NO to sustain physiologically relevant NO synthesis rates (Fig. 8).
FIGURE 8.

Neuroglobin act as a nitrite reductase under oxidative stress conditions. In normal conditions, cells keep a high concentration of reduced glutathione (GSH) and low oxidized glutathione (GSSG). In these circumstances, the disulfide bond of Ngb is not formed, and the protein has a low nitrite reductase activity (left). As oxidative stress conditions develop (right), reduced glutathione is consumed, and the number of neuroglobin molecules with formed disulfide bonds increases. The production of NO from nitrite increases, causing the inhibition of respiratory enzymes and limiting oxygen consumption and reactive oxygen species-producing reactions.
Consistent with this thesis, our data (Fig. 5) demonstrate an interaction between nitrite and deoxygenated neuroglobin that generates bioavailable NO. In Fig. 6A, using isolated mitochondria in the presence of nitrite, we observe more effective inhibition of respiration with wild type Ngb than the C55A mutant. However, the H64L and H64Q mutants indicate that fast rates of NO generation are within the reach of the enzyme. The extent of mitochondrial inhibition is dependent on the heme coordination structure of neuroglobin and intrinsic nitrite reductase activity. We speculate that the observed redox effects may be amplified by external effects such as changes in pH and protein-protein modifications, namely protein phosphorylation and/or protein-protein interactions, that further open up the enzyme under allosteric control.
In conclusion, the molecular examination of critical heme pocket and surface thiol amino acids, using site-directed mutagenesis, provides a novel understanding of neuroglobin functionality as an enzyme with a redox-regulated six-to-five-coordinate iron heme transition that directs nitrite in the heme pocket for controlled electron and proton transfer reactions to form NO. The results presented in this study support the provocative hypothesis that the cellular six-coordinate heme globins, neuroglobin, cytoglobin, D. melanogaster hemoglobin, and plant hemoglobins, may subserve a function as primordial allosterically redox-regulated NO-signaling proteins. The identification of other allosteric regulators of the six-to-five coordination of the neuroglobin heme pocket may reveal new intracellular mechanisms for controlling NO signaling via nitrite reduction.
Supplementary Material
Acknowledgments
We thank Dr. Li Yi for helpful advice and discussions on disulfide couple redox potential determination and Dr. Yinna Wang for excellent technical assistance. Sperm whale myoglobins were a gift from John S. Olson (Rice University).
This work was supported, in whole or in part, by National Institutes of Health Grants HL058091 (to D. B. K.-S.), GM084614 (to C. H.), and HL098032 (to M. T. G.). This work was also supported by Institute for Transfusion Medicine, Hemophilia Center of Western Pennsylvania (to M. T. G. and S. S.), and American Heart Association Grant 109SDG2150066 (to S. S.). M. Tiso, M. T. Gladwin, and D. B. Kim-Shapiro are listed as co-inventors on a patent application entitled “Neuroglobin as a Six-to-Five-coordinate Regulated Nitrite Reductase.”

The on-line version of this article (available at http://www.jbc.org) contains Table S1, Figs. S1–S5, Equations S1–S20, and the detailed derivation of Equation 1.
- Ngb
- neuroglobin
- Mb
- myoglobin.
REFERENCES
- 1. Hankeln T., Ebner B., Fuchs C., Gerlach F., Haberkamp M., Laufs T. L., Roesner A., Schmidt M., Weich B., Wystub S., Saaler-Reinhardt S., Reuss S., Bolognesi M., De Sanctis D., Marden M. C., Kiger L., Moens L., Dewilde S., Nevo E., Avivi A., Weber R. E., Fago A., Burmester T. (2005) J. Inorg. Biochem. 99, 110–119 [DOI] [PubMed] [Google Scholar]
- 2. Brunori M., Vallone B. (2007) Cell. Mol. Life Sci. 64, 1259–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burmester T., Haberkamp M., Mitz S., Roesner A., Schmidt M., Ebner B., Gerlach F., Fuchs C., Hankeln T. (2004) IUBMB Life 56, 703–707 [DOI] [PubMed] [Google Scholar]
- 4. Dewilde S., Kiger L., Burmester T., Hankeln T., Baudin-Creuza V., Aerts T., Marden M. C., Caubergs R., Moens L. (2001) J. Biol. Chem. 276, 38949–38955 [DOI] [PubMed] [Google Scholar]
- 5. Uzan J., Dewilde S., Burmester T., Hankeln T., Moens L., Hamdane D., Marden M. C., Kiger L. (2004) Biophys. J. 87, 1196–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Capece L., Marti M. A., Bidon-Chanal A., Nadra A., Luque F. J., Estrin D. A. (2009) Proteins 75, 885–894 [DOI] [PubMed] [Google Scholar]
- 7. Kriegl J. M., Bhattacharyya A. J., Nienhaus K., Deng P., Minkow O., Nienhaus G. U. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7992–7997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiger L., Uzan J., Dewilde S., Burmester T., Hankeln T., Moens L., Hamdane D., Baudin-Creuza V., Marden M. (2004) IUBMB Life 56, 709–719 [DOI] [PubMed] [Google Scholar]
- 9. Giuffrè A., Moschetti T., Vallone B., Brunori M. (2008) Biochem. Biophys. Res. Commun. 367, 893–898 [DOI] [PubMed] [Google Scholar]
- 10. Burmester T., Hankeln T. (2009) J. Exp. Biol. 212, 1423–1428 [DOI] [PubMed] [Google Scholar]
- 11. Greenberg D. A., Jin K., Khan A. A. (2008) Curr. Opin. Pharmacol. 8, 20–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan A. A., Wang Y., Sun Y., Mao X. O., Xie L., Miles E., Graboski J., Chen S., Ellerby L. M., Jin K., Greenberg D. A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 17944–17948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X., Liu J., Zhu H., Tejima E., Tsuji K., Murata Y., Atochin D. N., Huang P. L., Zhang C., Lo E. H. (2008) Stroke 39, 1869–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun Y., Jin K., Mao X. O., Zhu Y., Greenberg D. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 15306–15311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiland T. R., Kundu S., Trent J. T., 3rd, Hoy J. A., Hargrove M. S. (2004) J. Am. Chem. Soc. 126, 11930–11935 [DOI] [PubMed] [Google Scholar]
- 16. Nadra A. D., Martí M. A., Pesce A., Bolognesi M., Estrin D. A. (2008) Proteins 71, 695–705 [DOI] [PubMed] [Google Scholar]
- 17. Garrocho-Villegas V., Gopalasubramaniam S. K., Arredondo-Peter R. (2007) Gene 398, 78–85 [DOI] [PubMed] [Google Scholar]
- 18. Huang Z., Shiva S., Kim-Shapiro D. B., Patel R. P., Ringwood L. A., Irby C. E., Huang K. T., Ho C., Hogg N., Schechter A. N., Gladwin M. T. (2005) J. Clin. Invest. 115, 2099–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shiva S., Huang Z., Grubina R., Sun J., Ringwood L. A., MacArthur P. H., Xu X., Murphy E., Darley-Usmar V. M., Gladwin M. T. (2007) Circ. Res. 100, 654–661 [DOI] [PubMed] [Google Scholar]
- 20. Hendgen-Cotta U. B., Merx M. W., Shiva S., Schmitz J., Becher S., Klare J. P., Steinhoff H. J., Goedecke A., Schrader J., Gladwin M. T., Kelm M., Rassaf T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10256–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Basu S., Azarova N. A., Font M. D., King S. B., Hogg N., Gladwin M. T., Shiva S., Kim-Shapiro D. B. (2008) J. Biol. Chem. 283, 32590–32597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamdane D., Kiger L., Dewilde S., Green B. N., Pesce A., Uzan J., Burmester T., Hankeln T., Bolognesi M., Moens L., Marden M. C. (2003) J. Biol. Chem. 278, 51713–51721 [DOI] [PubMed] [Google Scholar]
- 23. Hamdane D., Kiger L., Dewilde S., Green B. N., Pesce A., Uzan J., Burmester T., Hankeln T., Bolognesi M., Moens L., Marden M. C. (2004) Micron 35, 59–62 [DOI] [PubMed] [Google Scholar]
- 24. Nicolis S., Monzani E., Ciaccio C., Ascenzi P., Moens L., Casella L. (2007) Biochem. J. 407, 89–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grubina R., Huang Z., Shiva S., Joshi M. S., Azarov I., Basu S., Ringwood L. A., Jiang A., Hogg N., Kim-Shapiro D. B., Gladwin M. T. (2007) J. Biol. Chem. 282, 12916–12927 [DOI] [PubMed] [Google Scholar]
- 26. Burmester T., Weich B., Reinhardt S., Hankeln T. (2000) Nature 407, 520–523 [DOI] [PubMed] [Google Scholar]
- 27. Grassetti D. R., Murray J. F., Jr. (1967) Arch. Biochem. Biophys. 119, 41–49 [DOI] [PubMed] [Google Scholar]
- 28. Pesce A., Dewilde S., Nardini M., Moens L., Ascenzi P., Hankeln T., Burmester T., Bolognesi M. (2003) Structure 11, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 29. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi L., Jenkins P. M., Leichert L. I., Jakob U., Martens J. R., Ragsdale S. W. (2009) J. Biol. Chem. 284, 20556–20561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azarov I., Huang K. T., Basu S., Gladwin M. T., Hogg N., Kim-Shapiro D. B. (2005) J. Biol. Chem. 280, 39024–39032 [DOI] [PubMed] [Google Scholar]
- 32. Van Doorslaer S., Dewilde S., Kiger L., Nistor S. V., Goovaerts E., Marden M. C., Moens L. (2003) J. Biol. Chem. 278, 4919–4925 [DOI] [PubMed] [Google Scholar]
- 33. Petersen M. G., Dewilde S., Fago A. (2008) J. Inorg. Biochem. 102, 1777–1782 [DOI] [PubMed] [Google Scholar]
- 34. Grubina R., Basu S., Tiso M., Kim-Shapiro D. B., Gladwin M. T. (2008) J. Biol. Chem. 283, 3628–3638 [DOI] [PubMed] [Google Scholar]
- 35. Salhany J. M. (2008) Biochemistry 47, 6059–6072 [DOI] [PubMed] [Google Scholar]
- 36. Wakasugi K., Nakano T., Morishima I. (2003) J. Biol. Chem. 278, 36505–36512 [DOI] [PubMed] [Google Scholar]
- 37. Schafer F. Q., Buettner G. R. (2001) Free Radic. Biol. Med. 30, 1191–1212 [DOI] [PubMed] [Google Scholar]
- 38. Du W., Syvitski R., Dewilde S., Moens L., La Mar G. N. (2003) J. Am. Chem. Soc. 125, 8080–8081 [DOI] [PubMed] [Google Scholar]
- 39. Xu J., Li L., Yin G., Li H., Du W. (2009) J. Inorg. Biochem. 103, 1693–1701 [DOI] [PubMed] [Google Scholar]
- 40. Crawford J. H., Isbell T. S., Huang Z., Shiva S., Chacko B. K., Schechter A. N., Darley-Usmar V. M., Kerby J. D., Lang J. D., Jr., Kraus D., Ho C., Gladwin M. T., Patel R. P. (2006) Blood 107, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nienhaus K., Kriegl J. M., Nienhaus G. U. (2004) J. Biol. Chem. 279, 22944–22952 [DOI] [PubMed] [Google Scholar]
- 42. Ikeda-Saito M., Hori H., Andersson L. A., Prince R. C., Pickering I. J., George G. N., Sanders C. R., 2nd, Lutz R. S., McKelvey E. J., Mattera R. (1992) J. Biol. Chem. 267, 22843–22852 [PubMed] [Google Scholar]
- 43. Biram D., Garratt C. J., Hester R. E. (1993) Biochim. Biophys. Acta 1163, 67–74 [DOI] [PubMed] [Google Scholar]
- 44. Gladwin M. T., Grubina R., Doyle M. P. (2009) Acc. Chem. Res. 42, 157–167 [DOI] [PubMed] [Google Scholar]
- 45. Yi J., Safo M. K., Richter-Addo G. B. (2008) Biochemistry 47, 8247–8249 [DOI] [PubMed] [Google Scholar]
- 46. Basu S., Grubina R., Huang J., Conradie J., Huang Z., Jeffers A., Jiang A., He X., Azarov I., Seibert R., Mehta A., Patel R., King S. B., Hogg N., Ghosh A., Gladwin M. T., Kim-Shapiro D. B. (2007) Nat. Chem. Biol. 3, 785–794 [DOI] [PubMed] [Google Scholar]
- 47. Copeland D. M., Soares A. S., West A. H., Richter-Addo G. B. (2006) J. Inorg. Biochem. 100, 1413–1425 [DOI] [PubMed] [Google Scholar]
- 48. Hoshino M., Maeda M., Konishi R., Seki H., Ford P. C. (1996) J. Am. Chem. Soc. 118, 5702–5707 [Google Scholar]
- 49. Brancaccio A., Cutruzzolá F., Allocatelli C. T., Brunori M., Smerdon S. J., Wilkinson A. J., Dou Y., Keenan D., Ikeda-Saito M., Brantley R. E., Jr. (1994) J. Biol. Chem. 269, 13843–13853 [PubMed] [Google Scholar]
- 50. Doyle M. P., Pickering R. A., DeWeert T. M., Hoekstra J. W., Pater D. (1981) J. Biol. Chem. 256, 12393–12398 [PubMed] [Google Scholar]
- 51. Jung K. H., Chu K., Lee S. T., Sunwoo J. S., Park D. K., Kim J. H., Kim S., Lee S. K., Kim M., Roh J. K. (2010) Biochem. Biophys. Res. Commun. 403, 66–72 [DOI] [PubMed] [Google Scholar]
- 52. Raychaudhuri S., Skommer J., Henty K., Birch N., Brittain T. (2010) Apoptosis 15, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu J., Yu Z., Guo S., Lee S. R., Xing C., Zhang C., Gao Y., Nicholls D. G., Lo E. H., Wang X. (2009) J. Neurosci. Res. 87, 164–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brunori M., Giuffrè A., Forte E., Mastronicola D., Barone M. C., Sarti P. (2004) Biochim. Biophys. Acta 1655, 365–371 [DOI] [PubMed] [Google Scholar]
- 55. Mason M. G., Nicholls P., Wilson M. T., Cooper C. E. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 708–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Presley T. D., Morgan A. R., Bechtold E., Clodfelter W., Dove R. W., Jennings J. M., Kraft R. A., King S. B., Laurienti P. J., Rejeski W. J., Burdette J. H., Kim-Shapiro D. B., Miller G. D. (2011) Nitric Oxide 24, 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.