

Abstract
Purpose
Conditionally Replicative Adenovirus (CRAd) has been previously demonstrated to augment the activity of radiation, resulting in synergy of cell kill. However, previous models combining radiation with CRAd have not focused on the methods of radiation delivery.
Materials and methods
We model the combination of a novel prostate-specific CRAd, Ad5 PSE/PBN E1A-AR (Ad5: adenovirus 5; PSE: prostate-specific enhancer; PBN: rat probasin promoter; E1A: early region 1A; AR: androgen receptor), with radiation delivered both acutely and continuously, in an effort to better mimic the potential clinical modes of prostate cancer radiotherapy.
Results
We demonstrate that pre-treatment of cells with acute single high dose rate (HDR) radiation 24 hours prior to viral infection results in significantly enhanced viral replication and virus-mediated cell death. In addition, this combination causes increased level of γ-H2AX (Phosphorylated histone protein H2AX on serine 139), a marker of double-stranded DNA damage and an indirect measure of nuclear fragmentation. In contrast, continuous low dose rate (LDR) radiation immediately following infection of the same CRAd results in no enhancement of viral replication, and only additive effects in virus-mediated cell death.
Conclusions
These data provide the first direct assessment of the real-time impact of radiation on viral replication and the first comparison of the effect of radiation delivery on the efficacy of CRAd virotherapy. Our data demonstrate substantial differences in CRAd efficacy based on the mode of radiation delivery.
Keywords: adenocarcinoma of prostate, radiosensitisation, oncolytic viral therapy, prostate-specific conditionally replicative adenovirus, acute single high dose rate radiation, continuous low dose rate radiation
Introduction
Radiation therapy is a common treatment for adenocarcinoma of the prostate (PCa); however, despite significant improvements in delivery technologies, many patients develop recurrence after treatment with curative intent (Nguyen and Zietman 2007). In its most simplistic terms, radiation can be delivered from an external source (i.e., external beam radiation therapy or EBRT) or from an internal source, such as a radioactive seed. When the radiation is delivered externally, it is typically fractionated into multiple high dose rate (HDR) fractions. When the radiation is delivered through the permanent instillation of radioactive seeds, the radiation exposure is continuous but at a low dose rate (LDR brachytherapy). Because of the ease of treatment and recovery, LDR brachytherapy has been gaining popularity as the CaPSURE (Cancer of the Prostate Strategic Urologic Research Endeavor) database has reported an increase in the use of LDR brachytherapy from 3.1–12.0% in the past decade (Cooperberg et al. 2004).
The total dose of radiation given by these strategies is limited by the potential for damage to surrounding tissues (Teh et al. 2004). Despite continuously improving technology, some prostate cancer cells survive. This radiation-resistance may be due to the unusually slow growth rate of prostate cancers, compared with other cancers, or to alterations in cellular pathways such as DNA damage repair, cell cycle, or apoptosis. Recently, there have been significant advances in circumventing this radiation-resistance by combining radiation therapy with radiation sensitisers, such as certain chemotherapies (Maggiorella et al. 2003, Lebedeva et al. 2007, Shewach and Lawrence 2007). While successful, one of the important limitations has been the concomitant radiation sensitisation of non-cancerous tissues. It would be more advantageous to establish approaches that provide tissue-specific radiation sensitisation. Adenoviral gene therapy has this potential because it can be highly tailored to prostate tissues through tissue-specific promoters. We have previously described prostate-specific CRAd gene therapy, using the promoter of Prostate-Specific Antigen (PSA) to control the E1A adenoviral gene (Rodriguez et al. 1997). The persistently active status of the PSA promoter in most advanced prostate cancers, evident by the presence of continuously rising serum PSA, makes the use of the PSA promoter an attractive strategy for all stages of PCa (Wu et al. 2001, Zegarra-Moro et al. 2002, Chen et al. 2004). The original vector (CN706, also known as CG7060) and the subsequent vector (CV787, also known as CG7870, in which E1A and E1B (early region 1B) are under the control of two separate prostate-specific promoters) have been applied to several prostate cancer clinical trials, in which they were each used alone for treatment of locally-recurrent PCa following radiation or meta-static PCa through intravenous delivery (DeWeese et al. 2001, Small et al. 2006). In all trials, there has been compelling evidence of viral mediated therapeutic effect. We have recently modified the prostate-specific CRAd concept by inserting a fusion of the viral E1A gene with the androgen receptor (Hoti et al. 2007). The E1A-AR chimera overcomes a mutual inhibition effect of viral E1A and host AR, makes the virus more androgen responsive, tissue-specific, and therapeutically effective. We utilise this newest generation of prostate-specific CRAd virotherapy (Ad5 PSE/PBN E1A-AR) in combination with two different models of radiation therapy to assess the oncolytic activity in vitro.
Several studies have demonstrated that oncolytic adenoviral gene therapy can enhance the therapeutic effect of radiation. However, there have only been a few reports on targeting this combination therapy to a specific tissue. Moreover, all of these have been with EBRT (Chen et al. 2001, Advani et al. 2006, Idema et al. 2007). To date there have been no studies to evaluate the efficacy of combining oncolytic gene therapy with continuous LDR radiation. Since LDR brachytherapy is minimally invasive and well tolerated, we reasoned this might be a superior method for combination with CRAd therapy, as both radioactive seeds and CRAd could be delivered directly into the prostate under a single anesthetic. Here we compare the effects of combining a prostate-specific CRAd, Ad5 PSE/PBN E1A-AR, with both acute HDR radiation and continuous LDR radiation. Our results indicate that these two radiation strategies do not provide equal benefit when combined with CRAd gene therapy; the combination with acute HDR radiation appears to be most efficacious. Nevertheless, both combination strategies do result in a significant benefit.
Materials and methods
Cell culture
LNCaP, PC3, and DU145, from American Type Culture Collection (ATCC, Manassas, VA, USA), and LAPC4 (obtained from Dr John Isaac laboratory at Johns Hopkins University School of Medicine, MD, USA) were cultured in RPMI-1640 medium (Roswell Park Memorial Institute-1640 medium) (Cellgro Mediatech, Herndon, VA, USA) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-Products, West Sacramento, CA, USA), and maintained at passages 50–60. LNCaP (ATCC® Number: CRL-1740™) and LAPC4, human PCa cell lines that produce PSA and possess a mutated but functional androgen receptor or a wild-type androgen receptor respectively, were used as the target cell lines. PC3 (ATCC® Number: CRL-2698™) and DU145 (ATCC® Number: HTB-81™), human PCa cell lines that lack the androgen receptor and do not produce PSA, were used as control cell lines for AR responsiveness. HT29 (ATCC® Number: HTB-38™), a colon cancer cell line, and U2OS (ATCC® Number: HTB-96™), an osteosarcoma cell line, were used as control cell lines for tissue specificity. Both were maintained according to the manufacture’s protocols. Virus packaging line DPL-S11 was maintained in Dulbecco’s Modified Eagle Medium (DMEM, Cellgro Mediatech, Herndon, VA, USA) with 5% FBS and 200 μg/ml G418 (Mediatech, Manassas, VA, USA). It was derived from helper cell line Per.C6 (Fallaux et al. 1998) by selection for diphtheria toxin resistance, and expresses a previously characterised membrane-bound anti-fiber single chain antibody as a pseudo-receptor (van Beusechem et al. 2002). The 293HEK cell line (Quantum Biotechnologies, Laval, Quebec, Canada) was used for viral titration. It was maintained in DMEM with 10% FBS. All media were supplemented with 1% Penicillin-Streptomycin (Invitrogen Corporation, Carlsbad, CA, USA). All cells were maintained at 37°C in an atmosphere containing 5% CO2.
Viruses and their dosage
Ad5 PSE/PBN E1A-AR (Hoti et al. 2007) was generated in the AdEasy system (a gift from Dr Bert Vogelstein laboratory at Johns Hopkins University School of Medicine) through recombination of line-arised shuttle plasmid RpS-PSE-PBN-E1A-AR (RpS: Rodriguez plasmid shuttle) with pAdEasy-1 in DPL-S11 cells. The E1A-AR chimera includes wild-type AR. Adeno-X-LacZ, a recombinant, ΔE1/ΔE3 adenovirus that encodes β-galactosidase (Clontech, Mountain View, CA, USA) was used as a control virus. Both viruses were employed at doses ranging from 0.5–5 multiplicities of infection (MOI). FFIG (Ad5 Fiber IRES GFP) (a gift from Dr Gary Ketner laboratory at Johns Hopkins University School of Public Health), a replication-defective reporter virus that links green fluorescent protein (GFP) expression to fiber gene (which is part of the major late transcription unit) production through the use of an internal ribosome entry site (IRES) (Hoti et al. 2007), was used at 30 MOI. Viral infection was performed on 48-well plates with 2 × 104 cells per well.
Large-scale viral purification was performed using CsCl2 (Invitrogen) gradient ultracentrifuge and stored in dialysis buffer containing 15 mM Tris (pH 7.8), 2 mM MgCl2 and 5% sucrose. The titer of the viral stocks was determined using the Adeno-X™ Rapid Titer Kit and 293HEK cells. All viral stocks were tested for wild-type replication competent adenovirus (RCA) background generated by homologous recombination, using quantitative PCR with primers against wild-type E1A promoter. The RCA content of all the viruses was at undetectable level.
In vitro irradiation: Timing and dosage
Acute single HDR radiation was performed 24 h prior to viral infection at doses of 2 Gy or 6 Gy (0.67 Gy/min) (Gammacell 40 137Cs irradiator, Atomic Energy Commission of Canada, Ltd, Mississauga, Ontario, Canada); un-irradiated control cells were seeded and infected at the same time. Continuous LDR radiation (25 cGy/h in an incubated 137Cs irradiator, custom-built) was performed as described (DeWeese et al. 1998). Cell culture plates were placed in a sealed chamber filled with 5% CO2 and maintained at 37°C in the LDR irradiator for 24 h (up to 6 Gy) or 8 h (up to 2 Gy) immediately after viral infection; un-irradiated control cells were seeded and infected at the same time.
Cell viability
Cell viability was measured using MTT ((3-(4,5-Dimethylthiazol-2-yl)- 2,5- diphenyltetrazolium bromide, a tetrazole) Cell Proliferation Assay kit (ATCC) five, seven, nine or 12 days post-treatment. Growth media was removed and replaced with MTT solution 10-fold diluted with fresh media; after incubation for 3 h at 37°C, detergent reagent provided in the kit was added to dissolve the purple precipitates. Absorbance was recorded at 570 nm using Spectramax M2e fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA). At each time point, percentage of cell survival after respective treatment was calculated by comparison to the growth of untreated cells, i.e., optical density (OD) of treated cells/OD of untreated cells.
Real time viral replication influenced by radiation
To observe the reciprocal effect of radiation on viral replication, FFIG was co-infected with Ad5 PSE/PBN E1A-AR. GFP expression of FFIG is a direct measure of CRAd replication (Hoti et al. 2007). The level of GFP at five, seven and nine days post-treatment was recorded using Spectramax M2e fluorescence plate reader (Molecular Devices) (excitation at 485 nm, and emission at 535 nm). Considering the significant cell death after treatment, the GFP reading was normalised to percentage of surviving cells (GFP reading/cell). The data was confirmed with viral output/input assay to avoid bias in data collection.
Viral output/input assay
LNCaP and LAPC4 cells infected with Ad5 PSE/PBN E1A-AR virus (2 MOI) in the presence and absence of radiation were pelleted and lysed to release viruses at the end of seven days post-treatment. The amount of released viruses was measured by titration using 293HEK cells. The ‘amplification ratio’ of a virus produced from an infected cell (Output) to the amount originally used to infect the cells (Input) were determined and plotted as viral output/input ratio.
Viral uptake influenced by radiation
To observe the effect of radiation on viral uptake, AdTrack virus (a gift from Dr Bert Vogelstein laboratory at Johns Hopkins University School of Medicine) that is driven by CMV (Cytomegalovirus) promoter and expresses GFP (He et al. 1998) was employed. LNCaP cells irradiated with 6 Gy of acute single HDR radiation were infected with AdTrack 24 h after irradiation. The level of GFP 24, 48, and 72 h post-infection was recorded using the above mentioned fluorescence plate reader. The GFP data are plotted as GFP fold increase relative to the mock-infected cells (signal/background).
Western blot analysis
LNCaP cells were infected with Ad5 PSE/PBN E1A-AR virus (2 MOI) on 100 mm culture dishes. Cells were washed with 1 × PBS, scraped, lysed on ice using RIPA buffer (buffer for radio immunoprecipitation assay) (Pierce Biotechnology, Rockford, IL, USA) supplemented with fresh protease inhibitor cocktail (Complete, EDTA-free, Roche Applied Science, Indianapolis, IN, USA), sonicated and centrifuged to remove the cell debris five and seven days post-treatment. Protein concentration was assessed using BCA (bicinchoninic acid) Protein Assay Kit (Pierce Biotechnology). 10–20 μg of protein was heated at 95°C and loaded on 4–15% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gels (Bio-Rad, Hercules, CA, USA). Gels were run for 2 h until the loading dyes reached to the bottom, and transferred to PVDF (Polyvinylidene Fluoride) (Bio-Rad) membrane. Pre-stained molecular weight markers (Rainbow Marker; Bio-Rad) were run in the same gels for comparison of molecular weight and estimation of transfer efficiency. The membranes were blocked with 1 × TBS (Tris-Buffered Saline) including 0.01% Tween 20 and 5% milk for 1 h and then probed with primary antibodies, i.e., a rabbit polyclonal antibody against γ-H2AX (working concentration 0.1 μg/ml) (Upstate Biotechnology, Billerica, MA, USA) or a rabbit monoclonal antibody against β-actin (1:5000 dilution) (Sigma, St Louis, MO, USA) overnight at 4°C. Secondary mouse anti-rabbit antibodies labeled with horseradish peroxidase were subsequently applied. Films were developed after incubation for 1–5 min with the horseradish peroxidase substrate provided in the enhanced chemiluminescence kit (Amersham, Piscataway, NJ, USA).
Statistical analysis
Statistical analysis was performed on Graph Pad Prism 5.0, running on an IBM compatible computer, using the Windows XP operating system. Comparisons for paired data were analysed using the Student’s t-test. Statistical significance was defined as a p-value <0.05 and was denoted in each of the figures by an asterisk. Expected total cell survival in Figure 1 was calculated as (1 − cell death by radiation × cell death by CRAd).
Figure 1.

In vitro cytotoxicity of CRAd in combination with two types of radiation. LNCaP cells were infected with 2 MOI of Ad5 PSE/PBN E1A-AR on 48-well plates for both combinations. In (a), acute single HDR radiation (6 Gy) was performed 24 hours prior to infection. In (b), continuous LDR radiation (6 Gy) was performed immediately after infection. MTT assay was used to assess cell viability. The enhanced cytotoxicity of the CRAd monotherapy in the LDR experiment, in comparison to the HDR experiment, is due to the difference in shorter time gap (24 hours vs. 48 hours) between cell plating and viral infection. Error bars indicate the standard error of the mean (SEM) for three independent experiments.
Results
Acute single HDR radiation versus continuous LDR radiation combined with oncolytic adenovirus
To evaluate the effect of combining CRAd virotherapy with radiation delivery, two in vitro radiation models were established. The first model is acute administration of radiation, followed by virotherapy. This model is most analogous to clinical HDR radiation. The model is intended to provide information most relevant for either EBRT, where total radiation therapy is fractionated at HDRs, or HDR brachytherapy, where a HDR radioactive source is instilled via catheters into the prostate for short intervals. In contrast, we also performed combination of CRAd virotherapy with a continuous LDR mode of radiation. In this case the model is most analogous to LDR brachytherapy, where radioactive seeds and viruses would be placed/or injected simultaneously. In this set of experiments we applied a continuous LDR radiation source immediately after infection using a custom made 137Cs irradiator at 25 cGy/h. Cell viability for the individual therapies as well as the two combinations was assessed five, seven and nine days post-treatment using MTT assay.
For the acute HDR studies, an acute single HDR radiation (6 Gy or 2 Gy at a rate of 0.67 Gy/min) was delivered to cells 24 h prior to viral infection. This timing was chosen based on our comparison of resulting combined cytotoxicity from experiments where radiation was delivered 24 h prior to infection versus irradiation 72 h post-infection (data not shown) and previously published reports (Qian et al. 2005, Egami et al. 2008). Ad5 PSE/PBN E1A-AR was infected at doses ranging from 0.5–5 MOI. For LNCaP cells, as shown in Figure 1a, 6 Gy of acute single HDR radiation resulted in 45% cell death at Day 9, and virotherapy (Ad5 PSE/PBN E1A-AR) at an MOI of 2 resulted in 10% cell death. However, when radiation and virotherapy were combined at these doses, cell death on Day 9 increased significantly to 90% (p <0.05). This observed value of cell death is much greater than the calculated additive death rate of 50%. In this experiment, even though virus alone killed only 10% cells on Day 9, at the end of 12 days, it killed 90% of cells (Figure 1a). Combinations of other doses of CRAd (0.5 MOI and 5 MOI) and radiation (2 Gy) showed similar results (data not shown).
On the other hand, in Figure 1b, continuous LDR radiation at the same dose of 6 Gy resulted in 46% and 66% cell death seven and nine days post-radiation, whereas virotherapy with an MOI of 2 resulted in 29% and 87% death of LNCaP cells seeded at the same time with the radiation-treated cells over the same period of time. (It is important to note that the enhanced cytotoxicity of the CRAd monotherapy in the LDR experiment, in comparison to the HDR experiment, is due to the shorter time gap between cell plating and viral infection). However, when radiation and CRAd were combined at these doses, cell death increased significantly to 63% (p <0.05) and 92%, respectively, at the end of the same period post-radiation. In this case, the combined therapies resulted in an additive cytotoxic effect. These data demonstrate that the combination of CRAd with acute single HDR radiation, performed 24 h prior to viral infection, was superior to continuous LDR radiation performed immediately after viral infection. However, over time both combination therapies achieved ~90% cell kill in these in vitro models. Combinations of other doses of CRAd (0.5 MOI and 5 MOI) and radiation (2 Gy) showed similar results (data not shown). Another AR positive PCa cell line, LAPC4, was also tested for the combination of radiation and CRAd virotherapy, and similar data were obtained (Supplementary Figure S1, online version only).
Reciprocal effect of radiation on viral replication
To interrogate the differences in cytotoxicity between the two combination therapies, we studied the effects of radiation dose rate and the timing of radiation delivery on viral replication. A special replication-incompetent reporter virus (FFIG) was used to assess viral replication (Hoti et al. 2007). In this virus, the major late promoter (MLP) is used to drive GFP expression. Since the MLP is only active during the final stages of viral replication, this reporter virus provides an indirect readout of viral replication when combined with a replication-competent virus such as Ad5 PSE/PBN E1A-AR.
FFIG was co-infected with the Ad5 PSE/PBN E1A-AR to evaluate viral replication in real time. For these studies, GFP reading was normalised to the percentage of living cells as determined using MTT assay. As shown in Figure 2a, MLP induction increased 11-fold in LNCaP cells pre-treated with acute single HDR radiation 24 h prior to infection, whereas normal viral replication in un-irradiated LNCaP cells showed only a two-fold induction of the MLP in the same period of time. The difference in viral replication was statistically significant (p <0.05). This increase in MLP induction at a later stage of the time course coincided with the sudden increase in cell death shown in Figure 1a, indicating that there was a significant viral outburst on Day 9.
Figure 2.

Reciprocal effect of radiation on viral replication. In (a) and (c), LNCaP cells were infected with 2 MOI of Ad5 PSE/PBN E1A-AR together with 30 MOI of FFIG on 48-well plates. GFP level was measured using a fluorescent plate reader and the data was normalised to the percentage of surviving cells. In (b) and (d), LNCaP cells were infected with 2 MOI of Ad5 PSE/PBN E1A-AR on 100 mm dishes; the amount of viral amplification seven days after treatment was titered using antibody against viral hexon. In (a) & (b): acute single HDR radiation (6 Gy) was performed 24 hours prior to infection. In (c) & (d): continuous LDR radiation (6 Gy) was performed immediately after infection. Error bars indicate the SEM for three independent experiments.
To confirm the result from FFIG reporter assay, a second experiment was performed to evaluate viral output titers (a direct measurement of replication), under these same conditions, over seven days. As shown in Figure 2b, we confirmed a two-fold greater viral titer in the irradiated cells compared to un-irradiated LNCaP cells. This increase was statistically significant (p <0.05) and is consistent with the findings from the FFIG reporter studies at Day 7 (Figure 2a).
In similar studies with LDR radiation, there did not appear to be a radiation-induced enhancement of viral replication using the FFIG reporter assay (Figure 2c). In identical viral output titration experiments, it was surprising to find that significantly less virus (p <0.05) was produced after continuous LDR radiation treatment when compared to untreated control (Figure 2d). Under these conditions, where there was not a significant increase in viral number, it appears that CRAd oncolysis contributed less to total host cell death when combined with LDR radiation (Figure 1b).
LAPC4 was also employed for the comparison of the reciprocal effect of the two radiation models on viral replication, and similar data were obtained (Supplementary Figure S2, online version only).
These results indicate that the mode of radiation delivery can affect viral replication, and that the difference in replication may account for the disparity in the efficacy of acute HDR radiation versus LDR radiation in this experimental system.
It appeared that CRAd replicated to a greater extent and caused more significant cytotoxicity in the LDR experimental system compared to in HDR experiments when it was used as monotherapy (Figures 1 and 2). This can be explained by the difference in the experimental settings, i.e., there was a shorter time gap (24 h) between cell seeding and viral infection in LDR experiments compared to that in the HDR experiments (48 h). Indeed it seems that physiological status of cells at the time of viral infection may affect viral entry. It is reported that oncolytic Herpes Simplex Virus has better penetration when cancer cells are induced to go through apoptosis (Nagano et al. 2008). In our study, freshly seeded cells in LDR experiments might be more prone to viral penetration, which led to increased replication and subsequent cytotoxicity.
Viral uptake is not upregulated by acute single HDR radiation
To investigate whether the increase in viral replication following acute single HDR radiation was due to enhanced viral uptake, a replication-deficient reporter virus AdTrack, which constitutively expresses GFP, was employed. LNCaP was infected with 2 MOI of AdTrack 24 h following 2 and 6 Gy of acute single HDR radiation and the GFP expression was assessed 24, 48, and 72 h after infection using the fluorescent plate reader. As shown in Figure 3, GFP level of AdTrack in cells pre-treated with radiation at a dose of 6 Gy was not significantly different from the one of un-irradiated cells 24, 48, and 72 h after infection. We have carefully counted the number of cells expressing GFP in every time points (data not shown), and have drawn the same conclusion from that set of data. The same was true when cells were treated with 2 Gy acute single HDR radiation (data not shown). This result demonstrated that acute single HDR radiation treatment 24 h prior to infection did not significantly affect viral uptake.
Figure 3.
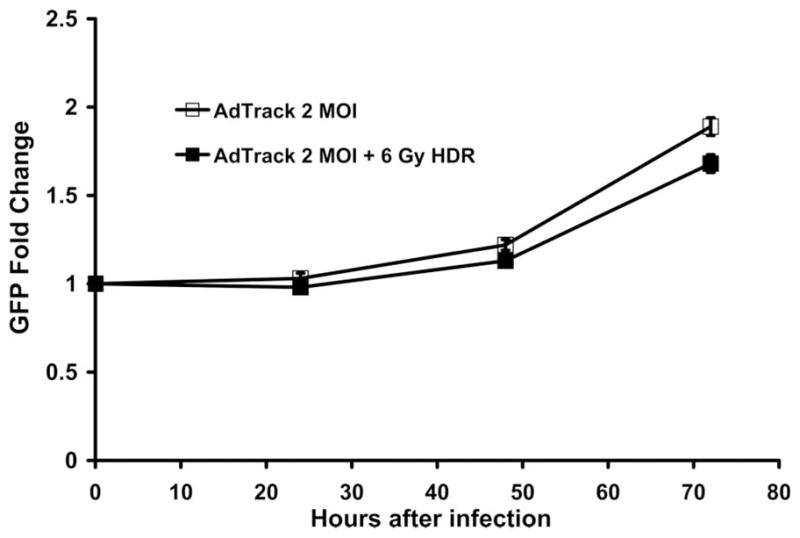
Viral uptake is not upregulated by acute single HDR radiation performed 24 hours prior to infection. LNCaP cells were infected with 2 MOI of GFP expressing AdTrack 24 hours following acute single HDR radiation. GFP level was measured using a fluorescent plate reader. Error bars indicate the SEM for three independent experiments.
Viral/acute single HDR radiation combination results in increased expression of γ-H2AX
To further investigate the mechanism contributing to the difference in the cytotoxicity between the two combination therapies, we evaluated their distinctive response to DNA damage. We chose to measure the expression level of γ-H2AX, which is a marker of double-stranded DNA injury and also an indirect reflection of nuclear fragmentation and cell death. As shown in Figure 4, combination treatment of LNCaP with 2 MOI of Ad5 PSE/PBN E1A-AR 24 h after 6 Gy acute single HDR radiation resulted in increased expression of γ-H2AX compared to the virus and radiation treatments individually. This effect was seen on Day 7 (Figure 4), but not Day 5 (data not shown), following respective treatment, suggesting that these differences occur later during viral amplification cycles. We hypothesise that the concurrence of enhanced DNA damage was probably the result of viral oncolysis, which led to increased nuclear DNA fragmentation and subsequently increased phosphorylation of histone H2AX. On the contrary, combination treatment of the same MOI of virus with 6 Gy of continuous LDR radiation did not cause a significant increase in γ-H2AX expression relative to individual treatments alone on Day 7 (Figure 4) and Day 5 (data not shown).
Figure 4.

Viral/acute single HDR radiation combination causes increased expression of double-stranded DNA damage marker γ-H2AX. LNCaP cells were infected with 2 MOI of Ad5 PSE/PBN E1A-AR on 100 mm dishes either 24 hours following acute single HDR radiation (6 Gy) (as control, un-irradiated cells were seeded and infected at the same time, denoted as a) or immediately followed by continuous LDR radiation (6 Gy) (as control, un-irradiated cells were seeded and infected at the same time, denoted as b). Whole cell extracts were collected seven days post-treatment. For Western blotting, 10–20 μg of whole cell extract was loaded on 4–15% acrylamide gels.
Discussion
The application of tissue-selective oncolytic adenoviral gene therapy for the purpose of sensitising cancer cells to radiation is an emerging and promising strategy (O’Shea et al. 2004, Freytag et al. 2007). Several studies have confirmed that adenovirus sensitises cells to radiation through a number of pathways, primarily through interfering with DNA repair. The viral protein E4orf6 (orf: open reading frame) plays an important role in blocking DNA double-strand repair genes. Viral E4orf6 interacts with DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (Boyer et al. 1999) and cooperates with E1B 55K to target MRE11 (Meiotic Recombination 11) and p53 for ubiquitin-mediated degradation, thus interfering with non-homologous end joining (NHEJ) and double strand break (DSB) repair (Querido et al. 2001a, 2001b, Hart et al. 2005). Inhibition of DNA-PKcs alone is sufficient for enhancing radiation therapy (Collis et al. 2003). E4orf6 has also been shown to prolong the signaling of DNA damage by inhibiting PP2A, a phosphatase responsible for dephosphorylating γ-H2AX, leading to prolonged H2AX phosphorylation that initiates caspase 3-independent and AIF (Apoptosis-Inducing Factor)-dependent apoptosis (Hart et al. 2007). In addition to radiosensitising cells through inhibition of DNA repair processes, prostate-specific CRAds also have the advantage of being able to replicate in specific cancer cells causing cell death through oncolysis (Rajecki et al. 2007).
To propagate virotherapy as a radiosensitiser, in contrast to prior studies which concentrated on the effect of adenoviral proteins on DNA repair pathways, here we focused on how viral replication and virus-mediated oncolysis were affected by different forms of radiation delivery. Acute HDR radiation appeared to be most efficacious in promoting viral replication and hence virus-mediated cytotoxicity. Continuous LDR radiation, although superior as a means of monotherapy for prostate cancer, appeared to have less favorable impact on viral replication as demonstrated by FFIG reporter assay and viral titration assay (Figure 2c, 2d, and Supplementary Figure S2b, online version only). While we would prefer to confirm these findings with in vivo models in tumour xenografts, the LDR continuous treatment has proven difficult to mimic in animals. Despite this, we feel that our in vitro data provide valuable insight into the mechanisms by which CRAd augment radiation-induced cytotoxity.
We hypothesise that viral oncolysis contributes to the supra-additive cytotoxicity in the CRAd/HDR radiation combination, but it does not play a role in the CRAd/LDR combination. One supporting evidence was that in the former combination the increase in expression of γ-H2AX coincided with up-surge of viral replication and cytotoxicity at late stage of viral amplification cycle, but in the latter combination, there was neither increase in γ-H2AX expression nor a sudden rise of viral replication and cytotoxicity. While γ-H2AX is a marker of double-stranded DNA injury, it can indirectly reflect the degree of nuclear fragmentation; nuclear fragmentation can be the result of many cellular insults, but its concurrence with up-surge of viral replication and cytotoxicity suggest that oncolysis has occurred.
Previous studies have also reported that pre-treating cells with radiation causes up-regulation of host cell membrane receptor dynamin 2, leading to increased viral uptake, and subsequently increased viral cytotoxicity (Qian et al. 2005, Egami et al. 2008). However, our studies with the replication deficient adenovirus, AdTrack, failed to find increases in viral infection following acute radiation. This discrepancy might be explained by the differences in the viral vectors, assays and timing employed for observation of viral entry. Nonetheless, it remains to be determined whether radiation-enhanced replication is due to up-regulation of viral entry, activation of viral proteins, or alteration of host cellular environment.
To confirm that the enhanced cytotoxicity demonstrated in the combination of CRAd virotherapy and acute radiation was specifically mediated by tissue-specific viral replication, we compared the cell kill caused by the CRAd virotherapy and a control virus Adeno-X-LacZ in LNCaP, LAPC4, PC3, DU145, HT29, and U2OS cells following 6 Gy of HDR radiation treatment performed 24 h prior to infection. As shown in Supplementary Figure S3 (online version only), we found that it was our prostate-specific CRAd (Ad5 PSE/PBN E1A-AR), but not Adeno-X-LacZ, that caused enhanced cell death in LNCaP and LAPC4, and there was no significantly CRAd-mediated cell death in AR-negative PCa cells (PC3 and DU145) and non-prostate cancer cells (HT29 and U2OS).
In summary, our study demonstrates that prostate-specific CRAd is capable of enhancing the therapeutic effects of radiation for the treatment of AR positive PCa. In this experimental system, radiation appeared to enhance viral replication, but only when the radiation was given at a HDR 24 h prior to infection. This enhancement of viral replication appears to be the dominant mechanism contributing to the supra-additive effect of combination therapy with acute single HDR radiation, compared to combination therapy with LDR radiation. To our knowledge, this is the first demonstration of a direct effect on viral replication by radiation and also the first comparison of LDR radiation to HDR radiation, when combined with CRAd virotherpy. These data lay a clear foundation for the translation of combination CRAd virotherapy with radiation therapy, suggesting that clinical protocols utilising the combination are best done with either EBRT or HDR brachytherapy.
Supplementary Material
Acknowledgments
The authors would like to thank Marikki Laiho, MD, PhD, Danny Song, MD, and Gary Ketner, PhD (The Johns Hopkins University School of Medicine) for helpful discussions and advices, and Tarana Kudrolli, MS, and Nasir Hoti, PhD (The Johns Hopkins University School of Medicine) for technical assistance during the course of these studies. This project has been supported in whole or in part through the Prostate Cancer SPORE (5 P50 CA58236), DOD, Prostate Cancer Consortium (DAMD17-03-2-033), and Prostate Cancer Foundation (private philanthropy).
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Advani SJ, Mezhir JJ, Roizman B, Weichselbaum RR. ReVOLT: Radiation-enhanced viral oncolytic therapy. International Journal of Radiation Oncology Biology Physics. 2006;66:637–646. doi: 10.1016/j.ijrobp.2006.06.034. [DOI] [PubMed] [Google Scholar]
- Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263:307–312. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen Y, DeWeese T, Dilley J, Zhang Y, Li Y, Ramesh N, Lee J, Pennathur-Das R, Radzyminski J, Wypych J, Brignetti D, Scott S, Stephens J, Karpf DB, Henderson DR, Yu DC. CV706, a prostate cancer-specific adenovirus variant, in combination with radiotherapy produces synergistic antitumor efficacy without increasing toxicity. Cancer Research. 2001;61:5453–5460. [PubMed] [Google Scholar]
- Collis SJ, Swartz MJ, Nelson WG, DeWeese TL. Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors. Cancer Research. 2003;63:1550–1554. [PubMed] [Google Scholar]
- Cooperberg MR, Lubeck DP, Meng MV, Mehta SS, Carroll PR. The changing face of low-risk prostate cancer: Trends in clinical presentation and primary management. Journal of Clinical Oncology. 2004;22:2141–2149. doi: 10.1200/JCO.2004.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWeese TL, Shipman JM, Dillehay LE, Nelson WG. Sensitivity of human prostatic carcinoma cell lines to low dose rate radiation exposure. Journal of Urology. 1998;159:591–598. doi: 10.1016/s0022-5347(01)63990-9. [DOI] [PubMed] [Google Scholar]
- DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, Hamper U, DeJong R, Detorie N, Rodriguez R, Haulk T, DeMarzo AM, Piantadosi S, Yu DC, Chen Y, Henderson DR, Carducci MA, Nelson WG, Simons JW. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Research. 2001;61:7464–7472. [PubMed] [Google Scholar]
- Egami T, Ohuchida K, Mizumoto K, Onimaru M, Toma H, Nishio S, Nagai E, Matsumoto K, Nakamura T, Tanaka M. Radiation enhances adenoviral gene therapy in pancreatic cancer via activation of cytomegalovirus promoter and increased adenovirus uptake. Clinical Cancer Research. 2008;14:1859–1867. doi: 10.1158/1078-0432.CCR-07-0933. [DOI] [PubMed] [Google Scholar]
- Fallaux FJ, Bout A, van der Velde I, van den Wollenberg DJ, Hehir KM, Keegan J, Auger C, Cramer SJ, van Ormondt H, van der Eb AJ, Valerio D, Hoeben RC. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Human Gene Therapy. 1998;9:1909–1917. doi: 10.1089/hum.1998.9.13-1909. [DOI] [PubMed] [Google Scholar]
- Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J, Zhang Y, Barton KN, Brown SL, Lu M, Savera A, Kim JH. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Molecular Therapy. 2007;15:1016–1023. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- Hart LS, Ornelles D, Koumenis C. The adenoviral E4orf6 protein induces atypical apoptosis in response to DNA damage. Journal of Biological Chemistry. 2007;282:6061–6067. doi: 10.1074/jbc.M610405200. [DOI] [PubMed] [Google Scholar]
- Hart LS, Yannone SM, Naczki C, Orlando JS, Waters SB, Akman SA, Chen DJ, Ornelles D, Koumenis C. The adenovirus E4orf6 protein inhibits DNA double strand break repair and radiosensitizes human tumor cells in an E1B-55K-independent manner. Journal of Biological Chemistry. 2005;280:1474–1481. doi: 10.1074/jbc.M409934200. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proceedings of the National Academy of Sciences of the USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoti N, Li Y, Chen CL, Chowdhury WH, Johns DC, Xia Q, Kabul A, Hsieh JT, Berg M, Ketner G, Lupold SE, Rodriguez R. Androgen receptor attenuation of Ad5 replication: Implications for the development of conditionally replication competent adenoviruses. Molecular Therapy. 2007;15:1495–1503. doi: 10.1038/sj.mt.6300223. [DOI] [PubMed] [Google Scholar]
- Idema S, Geldof AA, Dirven CM, van der Jagt M, Gerritsen WR, Vandertop WP, Lamfers ML. Evaluation of adenoviral oncolytic effect on glioma spheroids by 18F-DG positron-emission tomography. Oncology Research. 2007;16:471–477. doi: 10.3727/096504007783338304. [DOI] [PubMed] [Google Scholar]
- Lebedeva IV, Emdad L, Su ZZ, Gupta P, Sauane M, Sarkar D, Staudt MR, Liu SJ, Taher MM, Xiao R, Barral P, Lee SG, Wang D, Vozhilla N, Park ES, Chatman L, Boukerche H, Ramesh R, Inoue S, Chada S, Li R, De Pass AL, Mahasreshti PJ, Dmitriev IP, Curiel DT, Yacoub A, Grant S, Dent P, Senzer N, Nemunaitis JJ, Fisher PB. mda-7/IL-24, novel anticancer cytokine: Focus on bystander antitumor, radiosensitization and antiangiogenic properties and overview of the phase I clinical experience (Review) International Journal of Oncology. 2007;31:985–1007. [PubMed] [Google Scholar]
- Maggiorella L, Deutsch E, Frascogna V, Chavaudra N, Jeanson L, Milliat F, Eschwege F, Bourhis J. Enhancement of radiation response by roscovitine in human breast carcinoma in vitro and in vivo. Cancer Research. 2003;63:2513–2517. [PubMed] [Google Scholar]
- Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Research. 2008;68:3795–3802. doi: 10.1158/0008-5472.CAN-07-6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PL, Zietman AL. High-dose external beam radiation for localized prostate cancer: Current status and future challenges. Cancer Journal. 2007;13:295–301. doi: 10.1097/PPO.0b013e318156dbe3. [DOI] [PubMed] [Google Scholar]
- O’Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, Shen Y, Habets G, Ginzinger D, McCormick F. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Qian J, Yang J, Dragovic AF, Abu-Isa E, Lawrence TS, Zhang M. Ionizing radiation-induced adenovirus infection is mediated by Dynamin 2. Cancer Research. 2005;65:5493–5497. doi: 10.1158/0008-5472.CAN-04-4526. [DOI] [PubMed] [Google Scholar]
- Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes & Development. 2001a;15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E, Morrison MR, Chu-Pham-Dang H, Thirlwell SW, Boivin D, Branton PE. Identification of three functions of the adenovirus e4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. Journal of Virology. 2001b;75:699–709. doi: 10.1128/JVI.75.2.699-709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajecki M, Kanerva A, Stenman UH, Tenhunen M, Kangasniemi L, Sarkioja M, Ala-Opas MY, Alfthan H, Sankila A, Rintala E, Desmond RA, Hakkarainen T, Hemminki A. Treatment of prostate cancer with Ad5/3Delta24hCG allows non-invasive detection of the magnitude and persistence of virus replication in vivo. Molecular Cancer Therapeutics. 2007;6:742–751. doi: 10.1158/1535-7163.MCT-06-0403. [DOI] [PubMed] [Google Scholar]
- Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Research. 1997;57:2559–2563. [PubMed] [Google Scholar]
- Shewach DS, Lawrence TS. Antimetabolite radiosensitizers. Journal of Clinical Oncology. 2007;25:4043–4050. doi: 10.1200/JCO.2007.11.5287. [DOI] [PubMed] [Google Scholar]
- Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, Yu DC, Aimi J, Ando D, Working P, Kirn D, Wilding G. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Molecular Therapy. 2006;14:107–117. doi: 10.1016/j.ymthe.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Teh BS, Amosson CM, Mai WY, McGary J, Grant WH, 3rd, Butler EB. Intensity modulated radiation therapy (IMRT) in the management of prostate cancer. Cancer Investigation. 2004;22:913–924. doi: 10.1081/cnv-200039674. [DOI] [PubMed] [Google Scholar]
- van Beusechem VW, Grill J, Mastenbroek DC, Wickham TJ, Roelvink PW, Haisma HJ, Lamfers ML, Dirven CM, Pinedo HM, Gerritsen WR. Efficient and selective gene transfer into primary human brain tumors by using single-chain antibody-targeted adenoviral vectors with native tropism abolished. Journal of Virology. 2002;76:2753–2762. doi: 10.1128/JVI.76.6.2753-2762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Matherly J, Smallwood A, Adams JY, Billick E, Belldegrun A, Carey M. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Therapy. 2001;8:1416–1426. doi: 10.1038/sj.gt.3301549. [DOI] [PubMed] [Google Scholar]
- Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Research. 2002;62:1008–1013. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.