

Abstract
The etiology of autoimmune diseases remains largely unknown. Concordance rates in monozygotic twins are lower than 50% while genome-wide association studies propose numerous significant associations representing only a minority of patients. These lines of evidence strongly support other complementary mechanisms involved in the regulation of genes expression ultimately causing overt autoimmunity. Alterations in the post-translational modification of histones and DNA methylation are the two major epigenetic mechanisms that may potentially cause a breakdown of immune tolerance and the perpetuation of autoimmune diseases. In recent years, several studies both in clinical settings and experimental models proposed that the epigenome may hold the key to a better understanding of autoimmunity initiation and perpetuation. More specifically, data support the impact of epigenetic changes in systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis and other autoimmune diseases, in some cases based on mechanistical observations. We herein discuss what we currently know and what we expect will come in the next future. Ultimately, epigenetic treatments already being used in oncology may soon prove beneficial also in autoimmune diseases.
Keywords: DNA methylation, histone modification, microRNA, tolerance breakdown
Why epigenetics?
Autoimmune diseases are generally considered as complex (and/or multifactorial) diseases. Genetic background confers susceptibility to or protection from disease onset, but it is neither sufficient nor causative for disease development. While numerous similarities between conditions are being identified,1 the etiology of the majority of autoimmune diseases remains largely unknown. Although strong genetic bases have been found by recent genome-wide association studies,2 these studies fail to demonstrate the presence of a unique genetic mechanism underlying immune tolerance breakdown and, moreover, the significant genetic associations identified are found only in relative small proportion of patients.
The largely incomplete concordance rates of autoimmune diseases in monozygotic (MZ) twins (Table 1) strongly support other complementary mechanisms involved in gene expression regulation ultimately causing overt autoimmunity. Based on these observations, the use of novel strategies focusing on the analysis of histone modifications and DNA methylation supports the notion that epigenetic alterations may play a crucial role in triggering autoimmunity. Epigenetics (from the Greek επί (epi) over and γενετικός (genetics)) studies mechanisms that determine and/or perpetuate heritable genomic functions without changes in DNA sequence.
Table 1. Pairwise CRs of autoimmune diseases in MZ and DZ twin sets were calculated as n of concordant sets/n of studied sets.
| MZ twins CR | DZ twins CR | |
|---|---|---|
| Systemic lupus erithematosus | 0.24 | 0.02 |
| Sjögren's syndrome | Concordant pair reported | — |
| Type I diabetes mellitus | 0.21–0.70a | 0.00–0.13 |
| Rheumatoid arthritis | 12.3–15.4 | 3.50–3.60 |
| Primary biliary cirrhosis | 0.63 | 0.00 |
| Primary sclerosing cholangitis | Concordant pair reported | — |
| Graves' disease | 0.17–0.29 | 0.00–0.02 |
| Multiple sclerosis | 0.25–0.31a | 0.03–4.7 |
| Celiac disease | 0.75–0.83 | 0.11 |
Abbreviations: CR, concordance rate; DZ, dizygotic; MZ, monozygotic.
7.5 years of observation.
Epigenome and/or epigenotype is, thus, considered as a cell-specific and stable pattern of gene expression induced by such epigenetic mechanisms. Functionally, epigenetic mechanisms are, indeed, crucial for cell type development and differentiation, being able to induce stable expression or repression of genes. Epigenetic mechanisms are, also, able to confer a metabolic plasticity to cell, thus allowing the cell to adapt itself to environmental changes.
The potential role of epigenetics in environmental/genetic interactions, where environmental changes produce modifications in gene expression, has been suggested by some intriguing experimental studies.
Firstly, a seminal study investigated the use of a specific dietary regimen, i.e. foods rich in methyl donors, in order to modify coat color in agouti pregnant rodents. Such regimen led the offspring to manifest a specific coat color compared to mothers fed with a standard diet. This observation has been explained by an altered DNA methylation process that is the most thoroughly studied epigenetic mechanism. Such process silences the intracisternal A particle retroviral insertional element, ultimately limiting the appearance of agouti alleles. A second major example came from Dutch individuals who were exposed to famine during intrauterine life and childhood during the World War II. The DNA methylation analysis of the region regulating the insulin-like growth factor 2 (IGF2) expression constitutes a major example of epigenetic imprinting by demonstrating subjects a well-conserved hypomethylation status in exposed compared to non-exposed subjects.3
Recent observational studies have shown association of DNA methylation profiles with several environmental factors including exposure to prenatal tobacco smoke,4 alcohol consumption,5 and environmental pollutants.6, 7 Based on these observations, it is becoming clear how epigenetic mechanisms should be considered as the new frontier in the interaction between genome and environment, thus well conjugating the adagio stating that complex diseases ensue from ‘bad genes and bad luck'. This was strongly supported by the experimental data proposed by Dr Fraga and colleagues who demonstrated how epigenetics may well explain the discordance of autoimmune diseases in MZ twins.3 Phenotypic differences significantly increased along with age of the twins in a trend coined as ‘epigenetic drift', which occurs during life according with the different expositions to environmental stressors.8
The present article will first illustrate the major epigenetic mechanisms under investigation and then will discuss available data in the field of autoimmune diseases.
Epigenetic mechanisms
All epigenetic mechanisms share the feature of not altering DNA sequence, but only the possibility that gene sequences are transcribed under specific conditions. Comparing prokaryotic and eukaryotic organisms may help our understanding of how epigenetic mechanisms can regulate gene expression. Prokaryotes do not have nucleus and have a limited number of genes, whose transcription is regulated by factors binding directly to DNA promoter elements.
In contrast, eukaryotes have a significantly larger number of genes that have to be compacted in order to fit within the cell nucleus. In order to do so, genes are packed together with specific proteins, i.e. histones, in an ultra-structure known as chromatin. This DNA functional packaging system has regulatory functions, as chromatin can undergo steric changes, thus being able to interfere with proteins involved in gene transcription. Similarly, transcription regulation is derived from the methylation of cytosine DNA residues (particularly within gene promoters) which inhibits the transcription of the downstream genetic sequence. DNA methylation and histone modifications constitute the major epigenetic changes known so far, and therefore their underlying processes will be discussed below in further details. Moreover, the newest field of microRNA (miRNA) will be briefly illustrated as an additional gene regulatory mechanism.
Histone modifications
As mentioned before, histones are highly conserved proteins that reside within nuclei of eukaryotic cells. They can be classified into two main groups: (i) core histones (H2A, H2B, H3 and H4) that are part of the nucleosome core, the basic unit of DNA packaging in eukaryotics; and (ii) linker histones (H1 and H5). Two of each of the core histones assemble to form an octameric nucleosome core particle by wrapping about 147 base pairs of DNA around the protein spool in a 1.7 left-handed super-helical turn9 (Figure 1), thus providing DNA condensation and organization in the nucleus, as well as modulating DNA accessibility to the transcription machinery. This latter process could be represented as a drawer that can be opened or closed following specific stimuli. In fact, each histone subtype can be modified by different chemical modification at defined amino acids leading to transcription modulation and, therefore, cell cycle regulation, development and differentiation.
Figure 1.
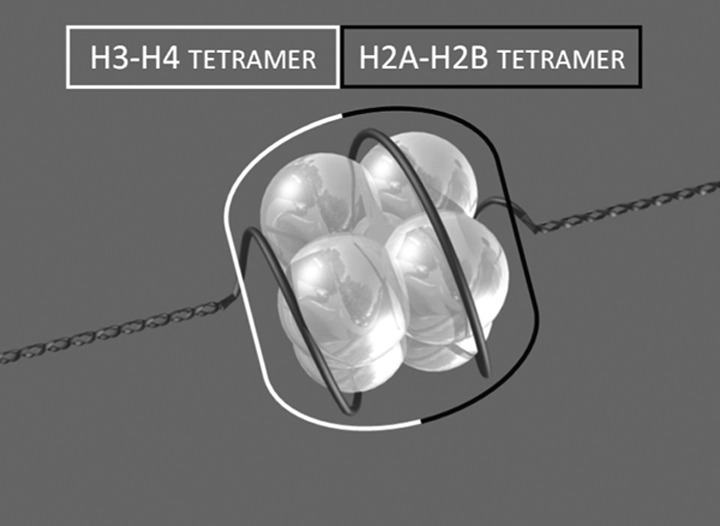
The structure of the nucleosome. The figure depicts the histone composed of two tetramers with DNA wrapped around the proteins.
Each of the four core histones shares the same folding structure known as histone fold domain, which consists of three α-helices (α1, α2 and α3) separated by two loops (L1 and L2).10 The histone fold domains fold together in antiparallel pairs (H3 with H4 and H2A with H2B) to constitute tetramers. The subsequent assembly of two tetramers forms the octameric core structure (H3/H4-H2A/H2B1) of the nucleosome.11 The N-terminal regions of histones protrude outside the nucleosome core and are prone to post-translational modifications, which are important in chromatin compaction and gene regulation. Histone post-translational modifications concur to determine the pattern defined as ‘histone code' and will be summarized below. All these histone modifications are caused by specific enzymes which recognize histone tails and can work to add or remove functional groups which are in turn recognized by nuclear factors. Specific proteins have affinity for modified amino acid residues (for instance bromodomains bind acetylated lysines or chromodomains methylated lysines) and promote specific changes in chromatin determining respectively the activation or the silencing of gene transcription (Figure 2).
Figure 2.
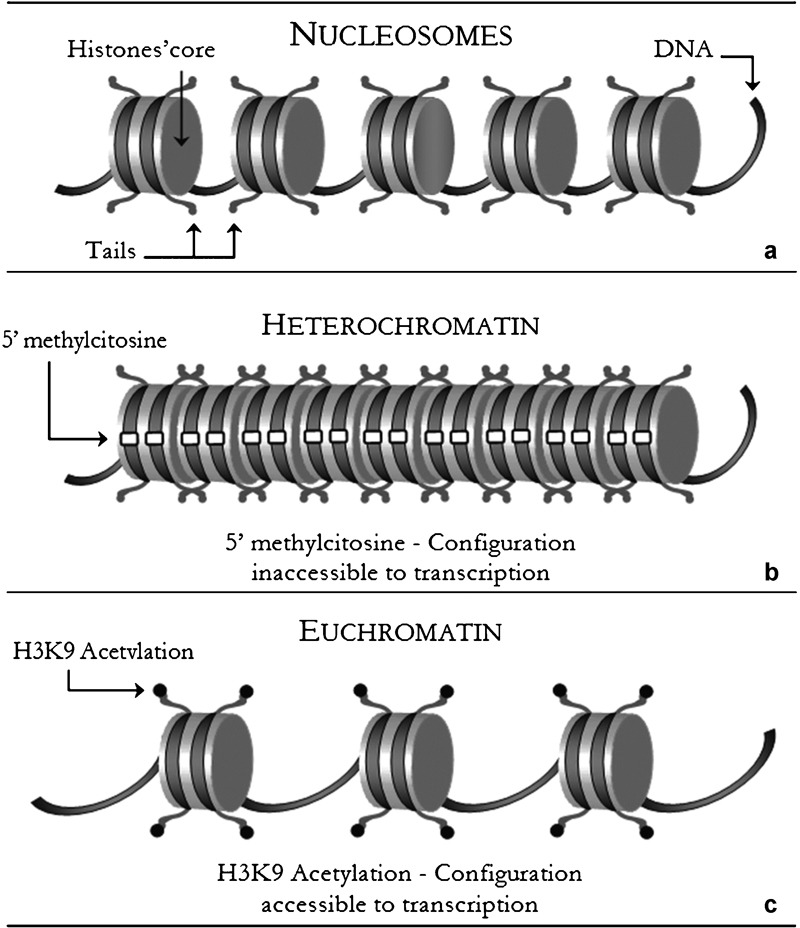
(a) Nucleosomes: the interactions between eight histone proteins determine a quaternary structure which leads to a double wrapping of DNA molecule. (b) Heterochromatin: histone deacetylation with the association of other histone modifications confers a dense configuration to DNA molecules. (c) Euchromatin: the epigenetic process of histones' tails acetylation is usually associated with an active configuration.
Among histone modifications, acetylation and deacetylation are one of the most important gene expression regulatory mechanisms. These processes involve selected lysine residues in the tails of nucleosomal histones and are induced by histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes, respectively.12, 13, 14, 15 HAT enzymes share the ability to promote gene expression by transferring acetyl groups to lysine16, 17, 18 while HDACs remove acetyl groups and generally associate with gene repression.19, 20, 21 A second mechanism involves histone methylation and its effects depend on the position of the modified lysine residue within the histone tail and on the number of methyl groups added to such residues. As an example, the presence of three methyl groups on lysine 4 residue on histone H3 (Me-H3K4), has been associated with transcriptional activation whereas the triple methylation of residues 9 or 27 determines repression.3, 22, 23, 24, 25, 26
As a third mechanism, arginine can also be methylated/demethylated by specific enzymes and play a critical role in the dynamic regulation of gene expression.27 Methylation of arginine residue 3 on histone H4 (H4R3) and arginine 17 on histone H3 (H3R17) have been shown to induce gene activation.23, 28, 29, 30 Finally, ubiquitin is a 76 amino acid protein that is involved in specific protein labeling. Ubiquitinated proteins are committed to proteosomal degradation and ubiquitination thus controlling the stability and intracellular localization of numerous proteins. Ubiquitination ultimately influences the status of histones methylation or acetylation31 to modulate gene expression, as in the case of the nuclear factor-κB pathway.32
DNA methylation
DNA methylation consists of the addition of a methyl group to the fifth carbon of cytosine residues, converting these to 5-methylcytosines. This reaction involves specific enzymes called DNA methyltransferases (DNMTs) and a methyl group donor, S-adenosylmethionine (Figure 3).
Figure 3.
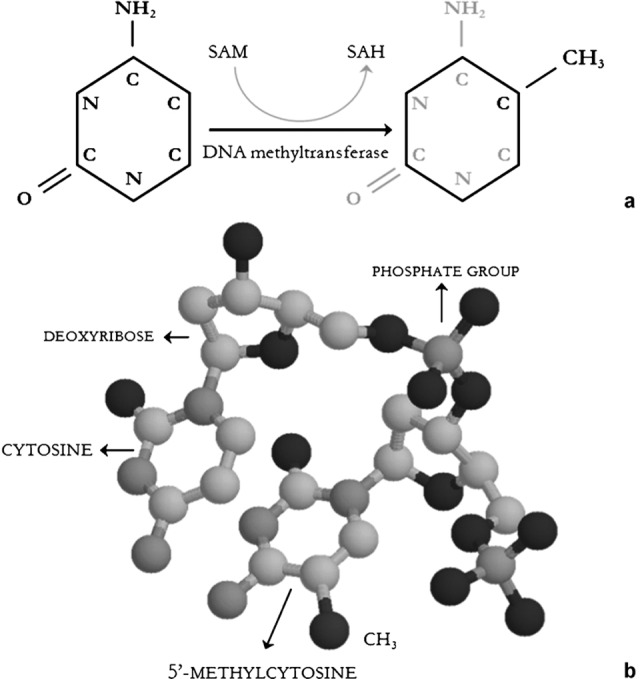
Panel a: Biochemistry of DNA methylation. DNA methyltransferase catalyze the reaction. The enzyme shifts the methyl group from SAM to the fifth carbon of cytosine. The reaction produces 5′-methylcytosine and SAH. Cytosine bases are integrant part of DNA filament and cannot be found as free molecules. Panel b: Cytosine and 5′-methylcytosine molecular structure in a DNA fragment. The brown atom represents the methyl group -CH3. Grey: carbons; blue: azotes; red: oxygens; yellow: phosphates. SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Among DNMTs, DNMT3A and DNMT3B are responsible for de novo methylation, while DNMT1 maintains epigenetic covalent modifications during cell replication. In mammalian genome, DNA methylation occurs mostly at CpG islands that are regions exceeding 500 base pairs with a CG content higher than 55%.33, 34 CpG islands have key regulatory functions, and can be found in promoter regions of about half of all genes.35 Altered CpG island methylation may, indeed, change chromatin structure, being typically able to modulate the finely promoter–transcription factor interactions within the transcription machinery.36
As a consequence, in most cases, both acquisition and somatic maintenance of such methylation state induce gene repression. Moreover, CpG-methylated sites can also interact with specific proteins containing a domain called methyl-CpG-binding domain. Although their specific contribution to different sites in different cell contexts is still unclear, methyl-CpG-binding domain proteins have been proposed to induce gene repression by binding to methylated DNA, a process that would lead to chromatin modification and remodeling complexes.
The importance of DNA methylation process in gene expression is well represented by sex chromosome balance, where females are characterized by partial transcriptional suppression of X chromosome obtained through methylation of specific genetic sequences.37 Mammalian genome contains dispersed clusters of genes, whose specific allele expression is determined by inheritance of parental methylation status. These genome regions that are rich in CpG and by which an organism inherits a state of expressed or unexpressed genomic sequence from parents, are usually referred as imprinting centers. Therefore, errors in methylation of such imprinting centers can induce an inherited altered gene expression.
From a clinical standpoint, the importance of DNA methylation, and in particular, of an impaired methylation, is suggested by the two rare congenital diseases known as Silver–Russel and Beckwith–Wiedemann syndromes. Both conditions derive from errors in methylation of a specific imprinting center located on chromosome 11 between genes encoding IGF2 and imprinted maternally expressed transcript (H19). In physiological condition this center is methylated only on the paternal allele. Beckwith–Wiedemann syndrome is characterized by an overexpression of IGF2 due to methylation of both parental alleles, while, in a complementary fashion, the double loss of imprinting centre methylation suppresses IGF2 locus and increases H19 expression causing Silver–Russel syndrome.
miRNAs
miRNAs are a group of post-transcriptional regulators involved in many biological processes including development, differentiation, proliferation and apoptosis.38 miRNAs are about 22 nucleotide-long non-coding RNAs that suppress translation by binding to complementary target mRNA species and causing the degradation of the target.39 miRNAs are genome encoded and transcribed by RNA polymerase II, similar to ordinary protein-coding RNAs40 and were recently investigated in autoimmune and chronic inflammatory conditions.41
One of the best known examples of epigenetic regulation due to miRNA–DNA interactions is represented by X-chromosome inactivation in women. Indeed, one of the two X chromosomes in female organisms is silenced by epigenetic mechanisms leading to dosage compensation of X-chromosome products. This balance is obtained with the transcription of X-inactive-specific transcript gene from the so called X-inactivation center and it leads to adapt gene expression in female sex with male organisms.42 X-inactive-specific transcript codes for a non-coding mRNA that coats one X chromosome, so this process silences the expression of a great part of genetic sequences on the future inactivated X chromosome.43 Silencing is stabilized by the addiction of repressive histone marks and DNA methylation. For these reasons it has been assumed that errors in epigenetic X-chromosome silencing could be involved in the pathogenesis of several diseases, including autoimmune diseases. This is indeed an attractive hypothesis that could explain the noticeable female predominance in autoimmune related disorders.44
Autoimmune diseases and epigenetic modifications
The impact of epigenetics is rapidly increasing in all complex diseases and is soon expected to gain a more prominent role in our future understanding of medicine. In general terms, we may well hypothesize that epigenetics will fill the gap between genomics and environmental factors in the pathogenesis of this type of conditions.
In the case of autoimmunity, presence of specific impairments in the regulation of epigenetic processes in immune cells would be responsible for immune-tolerance breakdown through both hypomethylation of genes or involvement of transcription repressors.45, 46, 47
In autoimmune diseases, epigenetics is mostly studied using peripheral blood mononuclear cells in humans or using animal models. Both in vitro and in vivo experimental models have shown that variation of the epigenome may lead to the onset of autoreactive T-cell clones. Specific epigenetic defects have been associated with autoimmune disorders.
As an example, the differentiation of T helper cells to Th1 subsets producing interferon-γ (IFN-γ) and acting against bacteria or Th2 subpopulations producing interleukin-4 (IL-4) and IL-13 cytokines is epigenetically regulated. Th1 cells present an IFN-γ demethylated promoter but have repressive epigenetic histone modifications at IL-4-13 locus, opposite to what is observed in the Th2 subsets.48 This is an example of the interactions between immune system dysregulation and epigenetics but the loss of immune tolerance is a complex process difficult to investigate particularly for non-traditional antigens.49 The study of specific models may help to understand the mechanisms involved in tolerance breakdown; one major example is represented by a unique human model: MZ twins.50
MZ twins are an ideal model to study environmental and epigenetic influences which could contribute to the autoimmune process, as proposed by Francis Galton, an English scientist of the nineteenth century. It was Galton's work published in 1874 in his book ‘English man of science: their nature and nurture' that constitute the basis for current epigenetics.3, 51 Over a century later, Fraga et al.8, 52 demonstrated the presence of an epigenetic drift in several MZ twin pairs and highlighted the importance of epigenetic modifications in the development of differences between twins. From a clinical standpoint, the concordance rates for autoimmune diseases in MZ twins are a powerful tool to determine the impact of genetics and the environment in determining disease onset. Concordance rates among MZ twins vary widely but, with only two exceptions, are well below 50% (Table 1), thus making this model an ideal target to investigate the role of epigenetics. Recent years have witnessed an increasing number of studies investigating epigenetics in specific autoimmune diseases and will be discussed in the sections below and are summarized in Table 2.
Table 2. Available evidence on the epigenetics involvement in specific autoimmune diseases.
| Systemic lupus erithematosus | T- and B-cell global DNA hypomethylation60, 61 with decreased DNMT1 transcription64 |
| miR-146a probably involved in disease onset82 | |
| CD4+ T-cell changes: | |
| •CD70 demethylation;65, 67 | |
| •CD40L demethylation (in women);68 | |
| •hypoacetylation of histone proteins H3 and H4;81 | |
| •H3 acetylation negatively correlates with disease activity.81 | |
| B-cell changes: | |
| •CD70 demethylation;67, 79 | |
| •perforin demethylation.67, 79 | |
| Rheumatoid arthritis | RASF changes: |
| •global DNA hypomethylation;92 | |
| •hypomethylation of CpG islands in LINE-1 promoter;92, 93 | |
| •hypomethylation of DR-3 promoter;95 | |
| •unmethylated CpG in IL-6 promoter;94 | |
| •miR-155,100 and miR-146 (Ref. 102) upregulated. | |
| Systemic sclerosis | Methylation of CpG islands in FL1 promoter with reduced expression107, 109, 111 |
| Sjögren's syndrome | Upregulated miR-574-3p and -768-3p in salivary glands114 |
| Upregulated miR-150 and -146 in salivary glands and lymphocytes115 | |
| Type 1 diabetes mellitus | Global hypermethylation activity caused by altered metabolism: |
| •glucose and insulin levels increase methylation by altering homocysteine metabolism;130, 179, 180, 181 | |
| •low protein diet decreases islet mass and vascularity.134, 135 | |
| Multiple sclerosis | PAD2 hypomethylation in white matter cells150, 151 |
| Effects of trichostatin A (histone deacetylase inhibitor) in murine models157 |
Abbreviations: DR-3, death receptor 3; DNMT, DNA methyltransferase; RASF, rheumatoid arthritis synovial fibroblast; PAD2, peptidyl arginine deiminase, type II
Systemic lupus erithematosous (SLE)
SLE is a systemic multiorgan autoimmune disease with different immunological and clinical manifestations characterized by an autoantibody response to nuclear and/or cytoplasmic antigens.
Most recent genome-wide association studies demonstrated that genomics significantly predispose to SLE onset,53, 54, 55, 56, 57 but the incomplete disease concordance between identical twins suggests a role for other complementary factors.52, 58 In this undefined scenario, experimental data indicate that epigenetic mechanisms, and in particular impaired T- and B-cell DNA methylation, may constitute one of these factors.59
Several studies have uncovered the importance of DNA hypomethylation in SLE etiology,60, 61 and in particular it has been suggested that this phenomenon may affect the structure of T-cell chromatin, resulting in cellular hyperactivity. Changes in DNA methylation are regulated by the extracellular signal-regulated kinase signaling pathway62 and this pathway is reduced in murine T cells causing a decreased expression of DNMT1 and an overexpression of methylation-sensitive autoimmunity genes, similar to T cells in human SLE.63 Human data further confirmed these views as T cells from patients with active SLE manifest decreased total deoxymethylcytosine content and decreased DNMT1 transcripts64 leading to the hyperexpression of several genes. Epigenetic similarities between patient lymphocyte and experimentally demethylated T cells were also demonstrated with SLE cells capable of stimulating antibody production by autologous B cells.65, 66 Another line of evidence came from the elevated CD70 expression in SLE cells similar to what is observed in vitro stimulating CD4+ T cells with the epigenetically active molecules procainamide and hydralazine, as both drugs cause CD70 demethylation.65, 67 Further, cells from women with SLE overexpress CD40L and manifest a demethylation of the corresponding gene on the inactivated X chromosome. Since cells from male patients do not overexpress CD40L,68 this finding has been advocated to explain SLE female predominance. The common trait of these observations could reside in the epigenetically-mediated downregulation of the transcription factor RFX1 in CD4+ T cells.69
The study of 5-azacytidine to understand SLE pathogenesis was prompted by the clinical evidence of a lupus-like syndrome in patients treated with procainamide and hydralazine. It has been found that CD4+ T cells treated with an inhibitor of DNMT1, such as 5-azacitidyne, become autoreactive, and the process is reversible after the drug is discontinued.61 The study of these patients demonstrated that only a group of treated subjects develop the syndrome, thus suggesting the presence of an idiosyncratic reaction which remains one of the most studied phenomena in modern pharmacology. These rare adverse reactions arise in a restricted subset of people. This group of patients is difficult to treat. Moreover, it is impossible to predict what element of the human population could develop the reaction. The most important drugs involved in induction of a lupus-like disease are procainamide and hydralazine, even though both cause antinuclear antibodies, ANA in a majority of people.70 The development of a systemic involvement and clinical manifestation probably require the presence of lupus susceptibility genes.71 Interestingly, both drugs are DNA methylation inhibitors but procainamide is a competitive inhibitor of DNMT1 enzymatic activity72 and hydralazine inhibits T- and B-cell signal-regulated kinase pathways.73, 74 The observation of an increased expression of adhesion molecules on lupus drug-induced lymphocytes proves the epigenetic mechanisms and their role in the induction of autoreactivity.75 Similarly, lupus CD4+ T cells have an abnormal interaction with major histocompatibility complex (MHC) molecules as supported by the experimental evidence of an abnormal self-antigen response following treatment with 5-azacytidine76 which demethylates sequences encoding costimulatory molecules like CD11a.77, 78 These molecules take part in CD4+ T-cell activation and their hyperexpression influences lymphocytes interaction with self-antigens. Furthermore, 5-azacytidine demethylates the cytotoxic molecule perforin and the B-cell costimulatory molecule CD70 causing their overexpression, both phenomena being observed also in patients' T cells.67, 79 The increased perforin and CD70 expression levels contribute to autoreactive macrophage killing capability which can generate a source of antigenic apoptotic nucleosomes,79 and antibody overproduction,65 respectively. One recent study compared DNA methylation in genome-wide loci in a cohort of MZ twins discordant for SLE, rheumatoid arthritis (RA) and dermatomyositis.80 MZ twins discordant for SLE manifested DNA methylation and expression changes in genes relevant to SLE pathogenesis and a global decrease in the methylation content.
Histone modifications have been studied in both lupus mouse models and human lupus. Global acetylation of histones H3 and H4 in active SLE CD4+ T cells was found to be decreased and H3 acetylation negatively correlated with disease activity.81 A recent study also demonstrated that a negative regulator of the IFN pathway, miR-146a, may contribute to disease onset.82 Both H3 and H4 histones are hypoacetylated in spleen-isolated cells from lupus-prone mice compared with controls.83 As observed in DNA methylation, the use of the HDAC inhibitors such as trichostatin A or suberoylanilide hydroxamic acid demonstrated improvement in glomerulonephritis and splenomegaly commonly observed in SLE.84, 85 These results are further supported by the in vitro use of HDAC inhibitors leading to increased histones H3 and H4 acetylation and reduced production of IL-12, IFN-γ, IL-6 and IL-10.84 A murine strain carrying a HAT mutation develops a severe lupus-like disease with serum anti-dsDNA autoantibodies, glomerulonephritis and premature death.86
RA
RA is a chronic systemic inflammatory disease that primarily affects peripheral joints. As observed for SLE, the clinical onset RA requires a combination of genetic susceptibility factors, deregulated immunomodulation and environmental influences.87, 88, 89 We may, then, hypothesize that only a genetic predisposition in concert with specific epigenetic alterations leads to the RA-associated immune system dysregulation. The typical joint localization of RA can be explained with the presence of local and environmental factors which remain unknown but may well include epigenetic changes.
In recent years, the epigenetics of RA have been widely investigated.90 It has been proposed that RA synovial fibroblasts (RASFs) have a major role in the initiation and perpetuation of RA,91 possibly via decreased global DNA methylation92 or hypomethylation of CpG islands in LINE-1 promoter.92, 93
Unmethylated CpG islands within IL-6 promoter gene in monocytes have been associated with a local hyperactivation of the inflammation circuit.94 RA monocyte cells also manifest a change in methylation status of CpG islands within the promoter of death receptor 3 (DR-3), which is, then, downregulated inducing resistance to apoptosis.95
RA synovial tissues are characterized by a drift of the balance between HAT and HDAC activity toward the former96 as supported by the proposed benefits induced by HDAC inhibitors97 such as FK228 which inhibits joint swelling, synovial inflammation and joint destruction in murine RA models.98 Furthermore, FK228 suppresses the production of vascular endothelial growth factor in vivo and blocks angiogenesis in synovial tissue in collagen antibody-induced arthritis.99 Conversely, a twofold lower HDAC activity was reported in synovial extracts from RA patients compared to osteoarthritis patients.96
Lastly, it has also been suggested that specific expression and function of miRNA, in particular miR-155 and miR-146, might be involved in RA pathogenesis100 as these are highly expressed in RASFs but not in osteoarthritis synovial fibroblast.100, 101 Tumor necrosis factor-α and IL-1β enhance miR-155 expression, which manifests a repressive effect on metalloproteinases expression in RASFs.100 miR-146 is upregulated by proinflammatory molecules in RA synovial tissues and has a negative regulatory function of the nuclear factor-κB pathway of RA patients' monocytes.102
Systemic sclerosis (SSc)
SSc or scleroderma is a rare condition of unknown etiology characterized by excessive collagen deposition in skin and other tissues with progressive vasculopathy. The presence of autoantibodies against nuclear autoantigens in patients with SSc, female predominance and frequent autoimmune comorbidity are considered signs of autoimmunity.103, 104 Aberrant fibroblast activation and collagen deposition ultimately lead to fibrosis with a gradual but progressive alteration of involved tissues and organs, a process characterized by an inbalance of stimuli favouring pro-collagen and a defective production of metalloproteinases.105, 106 This is particularly evident for the skin where the increased collagen pool confers anelasticity to the derma causing the pathognomonic hard and thick appearance.
Cultured SSc fibroblasts manifest typical cellular abnormalities for multiple generations and maintain the profibrotic phenotype when transferred outside the disease environment,107 thus suggesting the presence of an imprinted profibrotic cell phenotype. This phenotype is determined by an increase in production of a defined cytokines pool including TGF-β and other growth factors in association with reduced synthesis of matrix metalloproteinases 1 and 3. The clonal selection of profibrotic fibroblasts108 is one of the proposed pathogenetic mechanisms but there are insufficient data to confirm this hypothesis and the causative mechanism could be represented by epigenetics. This hypothesis is primarily supported by Wang and colleagues, who reported an epigenetic influence on collagen gene expression by the addiction of DNMT and HDACs inhibitors in cultured SSc fibroblasts. Most recent studies identified a reduced expression of FL1,107 a transcription factor that inhibits collagen production109 with an inverse correlation between FL1 expression and type I collagen production in cultured fibroblasts.110 An epigenetic regulation of FL1 is indirectly suggested by the presence of CpG islands in FL1 promoter that can be methylated and bound to specific regulatory proteins.111 Epigenetic FL1 changes lead to increased collagen synthesis which is not balanced by metalloproteinase activity ultimately leading to collagen accumulation and fibrosis. This finding raises the possibility that aberrant DNA methylation within fibroblasts may contribute to the development of the disorder,107 but genome-wide studies on DNA methylation and possibly histone modifications are awaited.
Sjögren's syndrome (SjS)
SjS affects salivary and lacrimal glands, resulting in dry mouth and/or dry eye conditions in patients as a consequence of autoimmune responses to self-antigens.112 Despite extensive investigations into the etiology of SjS focusing on genetic, environmental and immune factors, neither the triggering nor the disease-initiating events have been identified.113 Nevertheless, most recent data reported the overexpression of two miRNAs (namely, miR-574-3p and -768-3p) in the salivary glands of SjS patients,114 while the study of non-obese diabetic mice with associated SjS demonstrated the upregulation of other miRNAs (miR-150 and -146) in target tissues and in peripheral lymphocytes.115 The overexpression of miR-146 was confirmed in the salivary glands and peripheral lymphocytes of patients with SjS.115
Type 1 diabetes (T1D)
T1D is a T cell-mediated autoimmune disease116 that develops in genetically susceptible individuals. In fact, predisposing genetic polymorphisms have been identified in T1D patients such as those in MHC class II (DR and DQ), insulin, PTPN22, CTLA4 and IL-rRA.117 The disease incidence has been increasing over the past decades, as well represented by the data from Finland, where T1D yearly incidence has increased from 12 to 63 per 100 000,118, 119 somehow in conflict with MHC data.120, 121 Several studies focus on environmental exposures to dietary antigens and infectious agents, but evidence is limited.122, 123
Epigenetic studies on MZ twins concordant for T1D demonstrated significant differences in the epigenome, particularly for DNA methylation content and histone modifications through a trend coined ‘epigenetic drift'.8 The presence of a primitive pancreatic damage can initiate the autoimmune attack and lead to the activation of repair mechanisms like cellular proliferation124, 125 and ultimately influence the integrity of the epigenome. Another mechanism by which epigenetics may play an important role in T1D is by modulating lymphocyte maturation and cytokine gene expression.126 An example is the differentiation of subtype T helper cells, one of the most complex immune process, which is ruled by epigenetic controls.127, 128, 129 The epigenetic modifications are also important in pancreatic islet cells through the influence of repair mechanisms. Interestingly, glucose and insulin levels are major determinants on the methylation processes that take place in the cell via elevated homocysteine and homocysteine remethylation with a concurrent reduced capacity to eliminate homocysteine by transsulfuration. Homocysteine can be thus remethylated to form methionine and then converted to S-adenosylmethionine, the major methyl group donor in cellular methylation reactions.130
The establishment and maintenance of methylation patterns of CpG dinucleotides in DNA and histones depend on cellular methyl group metabolism, which is dependent on various nutrients, as in the case of folate.130 These relations between food and epigenetic mechanisms acquire importance during embryogenesis, intrauterine and perinatal life as demonstrated in animal models131 and human studies.132 These changes may well affect the offspring pancreas133 as in the case of a low-protein diet decreasing islet mass and vascularity.134, 135
Multiple sclerosis (MS)
MS is an inflammatory chronic disease characterized by myelin destruction followed by a progressive grade of neurodegeneration in multifocal loci called plaque.136, 137 The etiopathogenesis of MS remains largely unknown but the current hypothesis encompasses an immune-mediated damage determined by the activation of immune cells types against self white matter epitopes to develop diffuse plaques in the central nervous system with resulting inflammation. Genetic linkage studies and genome-wide profiling arrays have enabled the identification of several genes significantly associated with MS susceptibility,138, 139, 140, 141 as in the case of MHC.142 However, the 20–30% concordance rate for MS among MZ twins143, 144 emphasizes the importance of environmental factors in MS pathogenesis possibly via epigenetic mechanisms.142 Tissue damage implies the activation of developmental pathways,145, 146 but in patients with MS these appear to be unregulated in the presence of repair events.147, 148
There is limited data on epigenetics of MS, but a 30% reduction was reported in the methylation rate of cytosines in CpG islands was found in the white matter of affected central nervous tissue compared to controls.149 Further evidence on the role of hypomethylation was found at the promoter region of peptidyl arginine deiminase, type II, which is overexpressed in MS and is involved in the citrullination process of myelin basic protein (MBP).150, 151 These data are in agreement with the observation of an increased demethylase enzyme activity in MS.152 The citrullination of MBP by peptidyl arginine deiminase determines important biologic effects such as to promote protein autocleavage153 and a resulting increased probability to create new epitopes.154 Several studies support the importance of citrullination of MBP in modulating the immune response in MS155 via two mechanisms. In fact, citrullination increases the production of immunodominant peptides, due to increased autocleavage of the protein.153 This process leads to irreversible changes in the biological properties of MBP which becomes more prone to proteolytic digestion and causes myelin instability.154 These MBP alterations during the early stages of MS may contribute to the sensitization of T cells also by enhancing the autoimmune response153 leading through a chronic inflammatory response. Most recently, the comparison of multiple epigenetic readouts in CD4+ T cells from MZ twins discordant for MS failed to identify consistent associations,156 thus possibly suggesting that broad approaches may not constitute the ideal approach to complex conditions. These negative findings also applied to HLA haplotypes, confirmed MS susceptibility polymorphisms, copy number variations, mRNA and genomic single nucleotide polymorphism and insertion/deletion genotypes, or the expression of approximately 19 000 genes.
The importance of determining the epigenetic bases of MS, similar to other complex conditions, is of particular importance in the search for therapeutic implications, as well represented by the beneficial effects of trichostatin A, a HDAC inhibitor, in MS murine models.157
Primary biliary cirrhosis (PBC)
PBC is a chronic immune-mediated cholestatic liver disease characterized by the destruction of the small interlobular bile ducts, leading to portal inflammation, fibrosis and/or cirrhosis.158
Similar to most autoimmune diseases, PBC affects primarily women (female/male ratio estimated as 10∶1) with a peak of incidence in the fifth decade of life. Enhanced awareness of the condition and increased availability of diagnostic tools, in particular serological testing, have led to a more frequent and earlier diagnosis of PBC,159, 160 more commonly at asymptomatic stages.161
PBC etiopathogenesis recognizes an important role for genomics, possibly stronger than in other autoimmune disorders.162, 163 Indeed, PBC concordance rate in MZ twins is 63% the highest among autoimmune diseases with the exception of celiac disease.164
New approaches are being sought to identify the presence of epigenetic marks which can participate to PBC susceptibility. In fact not only genetic polymorphisms in the first genome-wide studies but also epigenetic impaired mechanism could be involved in the break of self-tolerance. Mitchell and colleagues most recently described the differential expression of two X-linked genes (PIN4 and CLIC2) that were differentially methylated in discordant MZ twins.165 This is of particular importance based on our previous report of a possible X-chromosome haploinsufficiency.166
Where we are and what is next for the epigenetics of autoimmunity
Most recently, there have been numerous studies to support the importance of epigenetics in the initiation and perpetuation of autoimmunity in specific conditions. In some cases, findings were recapitulated in different conditions, thus supporting the theory of a common theme for the autoimmunologist1, 167 and providing fascinating bases for the geographical pattern of autoimmunity epidemiology (i.e. geoepidemiology).89, 168, 169, 170 Over the past years, there has been an enormous development of genome-wide mapping for DNA methylation171 and histone modifications26 with novel issues arising172, 173 particularly in terms of environmental epigenetics.174 Every cell process is permeated by epigenetic regulation, from cancer175 to autoimmune diseases.176 The understanding of these mechanisms and the identification of target molecules are expected to lead to new classes of therapeutical molecules, coined ‘epigenetic therapies'.177, 178 We foresee that only a common effort between researchers involved in human and experimental autoimmunity and the use of powerful tools such as MZ twins will soon provide fascinating developments in the relatively young field of epigenomics. As an example, epidemiology and basic epigenetics should work together to provide solid associations between environmental factors and DNA methylation or histone changes in patients with autoimmune diseases.
Acknowledgments
This work was supported by the American Liver Foundation (CS) and NIH R21DK075400 (CS). AB receives salary support from New Investigator funding from the HSPH-NIEHS Center for Environmental Health (ES000002). The authors are grateful to Dr. Esteban Ballestar for the valuable discussion and contribution to the manuscript.
References
- Shoenfeld Y, Selmi C, Zimlichman E, Gershwin ME. The autoimmunologist: geoepidemiology, a new center of gravity, and prime time for autoimmunity. J Autoimmun. 2008;31:325–330. doi: 10.1016/j.jaut.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Hardy J, Traynor BJ, Houlden H. Towards a complete resolution of the genetic architecture of disease. Trends Genet. 2010;26:438–442. doi: 10.1016/j.tig.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestar E. Epigenetics lessons from twins: prospects for autoimmune disease. Clin Rev Allergy Immunol. 2010;39:30–41. doi: 10.1007/s12016-009-8168-4. [DOI] [PubMed] [Google Scholar]
- Breton CV, Salam MT, Vora H, Gauderman WJ, Gilliland FD. Genetic variation in the glutathione synthesis pathway, air pollution, and children's lung function growth. Am J Respir Crit Care Med. 2010. [DOI] [PMC free article] [PubMed]
- Zhang H, Zhu Z, Meadows GG. Chronic alcohol consumption decreases the percentage and number of NK cells in the peripheral lymph nodes and exacerbates B16BL6 melanoma metastasis into the draining lymph nodes. Cell Immunol. 2010. [DOI] [PMC free article] [PubMed]
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179:572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect. 2009;117:217–222. doi: 10.1289/ehp.11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci USA. 1995;92:11170–11174. doi: 10.1073/pnas.92.24.11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, Bishop TC, Victor JM, Jackson V, van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Matsuyama A, Komatsu Y, Nishino N. From discovery to the coming generation of histone deacetylase inhibitors. Curr Med Chem. 2003;10:2351–2358. doi: 10.2174/0929867033456602. [DOI] [PubMed] [Google Scholar]
- Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PD, Wagner K, Horz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265:195–202. doi: 10.1006/excr.2001.5187. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann NY Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T. Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep. 2002;3:39–44. doi: 10.1093/embo-reports/kvf013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006;11:344–355. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, et al. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- Osley MA, Fleming AB, Kao CF. Histone ubiquitylation and the regulation of transcription. Results Probl Cell Differ. 2006;41:47–75. doi: 10.1007/400_006. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP. CpG islands—‘a rough guide'. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- Chow J, Heard E. X inactivation and the complexities of silencing a sex chromosome. Curr Opin Cell Biol. 2009;21:359–366. doi: 10.1016/j.ceb.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight. Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett. 2009;285:116–126. doi: 10.1016/j.canlet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Strietholt S, Maurer B, Peters MA, Pap T, Gay S. Epigenetic modifications in rheumatoid arthritis. Arthritis Res Ther. 2008;10:219. doi: 10.1186/ar2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra M, Bernuzzi F, Invernizzi P, Danese S. MicroRNAs in autoimmunity and inflammatory bowel disease: crucial regulators in immune response. Autoimmun Rev. 2010. [DOI] [PubMed]
- Sidhu SK, Minks J, Chang SC, Cotton AM, Brown CJ. X chromosome inactivation: heterogeneity of heterochromatin. Biochem Cell Biol. 2008;86:370–379. doi: 10.1139/o08-100. [DOI] [PubMed] [Google Scholar]
- Wutz A. Xist function: bridging chromatin and stem cells. Trends Genet. 2007;23:457–464. doi: 10.1016/j.tig.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Ozcelik T. X chromosome inactivation and female predisposition to autoimmunity. Clin Rev Allergy Immunol. 2008;34:348–351. doi: 10.1007/s12016-007-8051-0. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136:1122–1135. doi: 10.1016/j.cell.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–357. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Baguet A, Bix M. Chromatin landscape dynamics of the IL4–IL13 locus during T helper 1 and 2 development. Proc Natl Acad Sci USA. 2004;101:11410–11415. doi: 10.1073/pnas.0403334101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay IR. Autoimmunity since the 1957 clonal selection theory: a little acorn to a large oak. Immunol Cell Biol. 2008;86:67–71. doi: 10.1038/sj.icb.7100135. [DOI] [PubMed] [Google Scholar]
- Ballestar E. Epigenetics lessons from twins: prospects for autoimmune disease. Clin Rev Allerg Immu. 2010;39:30–41. doi: 10.1007/s12016-009-8168-4. [DOI] [PubMed] [Google Scholar]
- Haque FN, Gottesman II, Wong AH. Not really identical: epigenetic differences in monozygotic twins and implications for twin studies in psychiatry. Am J Med Genet C Semin Med Genet. 2009;151C:136–141. doi: 10.1002/ajmg.c.30206. [DOI] [PubMed] [Google Scholar]
- Jarvinen P, Aho K. Twin studies in rheumatic diseases. Semin Arthritis Rheum. 1994;24:19–28. doi: 10.1016/0049-0172(94)90096-5. [DOI] [PubMed] [Google Scholar]
- Clancy RM, Marion MC, Kaufman KM, Ramos PS, Adler A, Harley JB, et al. Genome-wide association study of cardiac manifestations of neonatal lupus identifies candidate loci at 6p21 and 21q22. Arthritis Rheum. 2010. [DOI] [PMC free article] [PubMed]
- Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R151. doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- Cunninghame Graham DS. Genome-wide association studies in systemic lupus erythematosus: a perspective. Arthritis Res Ther. 2009;11:119. doi: 10.1186/ar2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Knapp AM, Nehme-Schuster H, Soulas-Sprauel P, Poindron V, Pasquali JL, et al. Peripheral B cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory B cells and membrane CD19 expression. J Autoimmun. 2010;34:426–434. doi: 10.1016/j.jaut.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Balada E, Ordi-Ros J, Vilardell-Tarres M. DNA methylation and systemic lupus erythematosus. Ann NY Acad Sci. 2007;1108:27–136. doi: 10.1196/annals.1422.015. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lu Q. DNA methylation in T cells from idiopathic lupus and drug-induced lupus patients. Autoimmun Rev. 2008;7:376–383. doi: 10.1016/j.autrev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Gorelik G, Richardson B. Aberrant T cell ERK pathway signaling and chromatin structure in lupus. Autoimmun Rev. 2009;8:196–198. doi: 10.1016/j.autrev.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–378. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M, et al. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–1673. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, et al. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum. 2004;50:1850–1860. doi: 10.1002/art.20255. [DOI] [PubMed] [Google Scholar]
- Crow MK, Kirou KA. Regulation of CD40 ligand expression in systemic lupus erythematosus. Curr Opin Rheumatol. 2001;13:361–369. doi: 10.1097/00002281-200109000-00004. [DOI] [PubMed] [Google Scholar]
- Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005;174:6212–6219. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007;179:6352–6358. doi: 10.4049/jimmunol.179.9.6352. [DOI] [PubMed] [Google Scholar]
- Zhao M, Sun Y, Gao F, Wu X, Tang J, Yin H, et al. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J Autoimmun. 2010;35:58–69. doi: 10.1016/j.jaut.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Yung RL, Richardson BC. Drug-induced lupus. Rheum Dis Clin North Am. 1994;20:61–86. [PubMed] [Google Scholar]
- Batchelor JR, Welsh KI, Tinoco RM, Dollery CT, Hughes GR, Bernstein R, et al. Hydralazine-induced systemic lupus erythematosus: influence of HLA-DR and sex on susceptibility. Lancet. 1980;1:1107–1109. doi: 10.1016/s0140-6736(80)91554-8. [DOI] [PubMed] [Google Scholar]
- Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005;280:40749–40756. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003;48:746–756. doi: 10.1002/art.10833. [DOI] [PubMed] [Google Scholar]
- Mazari L, Ouarzane M, Zouali M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc Natl Acad Sci USA. 2007;104:6317–6322. doi: 10.1073/pnas.0610434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, et al. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Richardson B. Effect of an inhibitor of DNA methylation on T cells. II. 5-azacytidine induces self-reactivity in antigen-specific T4+ cells. Hum Immunol. 1986;17:456–470. doi: 10.1016/0198-8859(86)90304-6. [DOI] [PubMed] [Google Scholar]
- Alcolado JC, Laji K, Gill-Randall R. Maternal transmission of diabetes. Diabet Med. 2002;19:89–98. doi: 10.1046/j.1464-5491.2002.00675.x. [DOI] [PubMed] [Google Scholar]
- Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- Kaplan MJ, Lu Q, Wu A, Attwood J, Richardson B. Demethylation of promoter regulatory elements contributes to perforin overexpression in CD4+ lupus T cells. J Immunol. 2004;172:3652–3661. doi: 10.4049/jimmunol.172.6.3652. [DOI] [PubMed] [Google Scholar]
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N, Qiu X, Luo Y, Yuan J, Li Y, Lei W, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol. 2008;35:804–810. [PubMed] [Google Scholar]
- Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- Garcia BA, Busby SA, Shabanowitz J, Hunt DF, Mishra N. Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition. J Proteome Res. 2005;4:2032–2042. doi: 10.1021/pr050188r. [DOI] [PubMed] [Google Scholar]
- Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111:539–552. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly CM, Mishra N, Miller JM, Joshi D, Ruiz P, Richon VM, et al. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. 2004;173:4171–4178. doi: 10.4049/jimmunol.173.6.4171. [DOI] [PubMed] [Google Scholar]
- Forster N, Gallinat S, Jablonska J, Weiss S, Elsasser HP, Lutz W. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol. 2007;178:6941–6948. doi: 10.4049/jimmunol.178.11.6941. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet. 2009;41:1313–1318. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobon GJ, Youinou P, Saraux A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J Autoimmun. 2010;35:10–14. doi: 10.1016/j.jaut.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Maciejewska-Rodrigues H, Karouzakis E, Strietholt S, Hemmatazad H, Neidhart M, Ospelt C, et al. Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J Autoimmun. 2010;35:15–22. doi: 10.1016/j.jaut.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Meinecke I, Rutkauskaite E, Gay S, Pap T. The role of synovial fibroblasts in mediating joint destruction in rheumatoid arthritis. Curr Pharm Des. 2005;11:563–568. doi: 10.2174/1381612053381945. [DOI] [PubMed] [Google Scholar]
- Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:3613–3622. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- Neidhart M, Rethage J, Kuchen S, Kunzler P, Crowl RM, Billingham ME, et al. Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression. Arthritis Rheum. 2000;43:2634–2647. doi: 10.1002/1529-0131(200012)43:12<2634::AID-ANR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58:2686–2693. doi: 10.1002/art.23758. [DOI] [PubMed] [Google Scholar]
- Takami N, Osawa K, Miura Y, Komai K, Taniguchi M, Shiraishi M, et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum. 2006;54:779–787. doi: 10.1002/art.21637. [DOI] [PubMed] [Google Scholar]
- Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. 2007;56:1087–1093. doi: 10.1002/art.22512. [DOI] [PubMed] [Google Scholar]
- Grabiec AM, Tak PP, Reedquist KA. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on. Arthritis Res Ther. 2008;10:226. doi: 10.1186/ar2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 2004;50:3365–3376. doi: 10.1002/art.20709. [DOI] [PubMed] [Google Scholar]
- Manabe H, Nasu Y, Komiyama T, Furumatsu T, Kitamura A, Miyazawa S, et al. Inhibition of histone deacetylase down-regulates the expression of hypoxia-induced vascular endothelial growth factor by rheumatoid synovial fibroblasts. Inflamm Res. 2008;57:4–10. doi: 10.1007/s00011-007-7036-z. [DOI] [PubMed] [Google Scholar]
- Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–1009. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58:1284–1292. doi: 10.1002/art.23429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora-Singh RK, Assassi S, del Junco DJ, Arnett FC, Perry M, Irfan U, et al. Autoimmune diseases and autoantibodies in the first degree relatives of patients with systemic sclerosis. J Autoimmun. 2010;35:52–57. doi: 10.1016/j.jaut.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourh P, Agarwal SK, Martin E, Divecha D, Rueda B, Bunting H, et al. Association of the C8orf13-BLK region with systemic sclerosis in North-American and European populations. J Autoimmun. 2010;34:155–162. doi: 10.1016/j.jaut.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest. 1974;54:880–889. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derk CT, Jimenez SA. Systemic sclerosis: current views of its pathogenesis. Autoimmun Rev. 2003;2:181–191. doi: 10.1016/s1568-9972(03)00005-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54:2271–2279. doi: 10.1002/art.21948. [DOI] [PubMed] [Google Scholar]
- Maxwell DB, Grotendorst CA, Grotendorst GR, LeRoy EC. Fibroblast heterogeneity in scleroderma: Clq studies. J Rheumatol. 1987;14:756–759. [PubMed] [Google Scholar]
- Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem. 2001;276:20839–20848. doi: 10.1074/jbc.M010133200. [DOI] [PubMed] [Google Scholar]
- Kubo M, Czuwara-Ladykowska J, Moussa O, Markiewicz M, Smith E, Silver RM, et al. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163:571–581. doi: 10.1016/S0002-9440(10)63685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nat Genet. 1994;6:236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- Chiorini JA, Cihakova D, Ouellette CE, Caturegli P. Sjogren syndrome: advances in the pathogenesis from animal models. J Autoimmun. 2009;33:190–196. doi: 10.1016/j.jaut.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez P, Anaya JM, Aguilera S, Urzua U, Munroe D, Molina C, et al. Gene expression and chromosomal location for susceptibility to Sjogren's syndrome. J Autoimmun. 2009;33:99–108. doi: 10.1016/j.jaut.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Alevizos I, Illei GG. MicroRNAs in Sjogren's syndrome as a prototypic autoimmune disease. Autoimmun Rev. 2010;9:618–621. doi: 10.1016/j.autrev.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulosan M, Pauley KM, Yo K, Chan EK, Katz J, Peck AB, et al. Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjogren's syndrome before disease onset. Immunol Cell Biol. 2009;87:81–90. doi: 10.1038/icb.2008.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knip M, Siljander H. Autoimmune mechanisms in type 1 diabetes. Autoimmun Rev. 2008;7:550–557. doi: 10.1016/j.autrev.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet. 2009;373:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, et al. The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care. 2008;31:1546–1549. doi: 10.2337/dc08-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie KM, Bain SC, Barnett AH, Bingley PJ, Christie MR, Gill GV, et al. The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet. 2004;364:1699–1700. doi: 10.1016/S0140-6736(04)17357-1. [DOI] [PubMed] [Google Scholar]
- Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O, Akerblom HK. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005;54 Suppl 2:S125–136. doi: 10.2337/diabetes.54.suppl_2.s125. [DOI] [PubMed] [Google Scholar]
- Lefebvre DE, Powell KL, Strom A, Scott FW. Dietary proteins as environmental modifiers of type 1 diabetes mellitus. Annu Rev Nutr. 2006;26:175–202. doi: 10.1146/annurev.nutr.26.061505.111206. [DOI] [PubMed] [Google Scholar]
- Kauri LM, Wang GS, Patrick C, Bareggi M, Hill DJ, Scott FW, et al. Increased islet neogenesis without increased islet mass precedes autoimmune attack in diabetes-prone rats. Lab Invest. 2007;87:1240–1251. doi: 10.1038/labinvest.3700687. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C, Meierhoff G, Kolb H, Schloot NC. Association of T-cell reactivity with beta-cell function in recent onset type 1 diabetes patients. J Autoimmun. 2010;34:127–135. doi: 10.1016/j.jaut.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- Sawalha AH. Epigenetics and T-cell immunity. Autoimmunity. 2008;41:245–252. doi: 10.1080/08916930802024145. [DOI] [PubMed] [Google Scholar]
- Aune TM, Collins PL, Chang S. Epigenetics and T helper 1 differentiation. Immunology. 2009;126:299–305. doi: 10.1111/j.1365-2567.2008.03026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JT, Stover PJ. Folate-mediated one-carbon metabolism. Vitam Horm. 2008;79:1–44. doi: 10.1016/S0083-6729(08)00401-9. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempainen J, Vaarala O, Makela M, Veijola R, Simell O, Knip M, et al. Interplay between PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun. 2009;33:155–164. doi: 10.1016/j.jaut.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Chamson-Reig A, Arany EJ, Summers K, Hill DJ. A low protein diet in early life delays the onset of diabetes in the non-obese diabetic mouse. J Endocrinol. 2009;201:231–239. doi: 10.1677/JOE-09-0002. [DOI] [PubMed] [Google Scholar]
- Boujendar S, Arany E, Hill D, Remacle C, Reusens B. Taurine supplementation of a low protein diet fed to rat dams normalizes the vascularization of the fetal endocrine pancreas. J Nutr. 2003;133:2820–2825. doi: 10.1093/jn/133.9.2820. [DOI] [PubMed] [Google Scholar]
- Arany E, Strutt B, Romanus P, Remacle C, Reusens B, Hill DJ. Taurine supplement in early life altered islet morphology, decreased insulitis and delayed the onset of diabetes in non-obese diabetic mice. Diabetologia. 2004;47:1831–1837. doi: 10.1007/s00125-004-1535-z. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Akkad DA, Hoffjan S, Petrasch-Parwez E, Beygo J, Gold R, Epplen JT. Variation in the IL7RA and IL2RA genes in German multiple sclerosis patients. J Autoimmun. 2009;32:110–115. doi: 10.1016/j.jaut.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet. 2008;9:516–526. doi: 10.1038/nrg2395. [DOI] [PubMed] [Google Scholar]
- Brynedal B, Duvefelt K, Jonasdottir G, Roos IM, Akesson E, Palmgren J, et al. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PLoS One. 2007;2:e664. doi: 10.1371/journal.pone.0000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark F, Duvefelt K, Iacobaeus E, Kockum I, Wallstrom E, Khademi M, et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat Genet. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- Zivadinov R, Uxa L, Bratina A, Bosco A, Srinivasaraghavan B, Minagar A, et al. HLA-DRB1*1501, -DQB1*0301, -DQB1*0302, -DQB1*0602, and -DQB1*0603 alleles are associated with more severe disease outcome on MRI in patients with multiple sclerosis. Int Rev Neurobiol. 2007;79:521–535. doi: 10.1016/S0074-7742(07)79023-2. [DOI] [PubMed] [Google Scholar]
- Chao MJ, Ramagopalan SV, Herrera BM, Lincoln MR, Dyment DA, Sadovnick AD, et al. Epigenetics in multiple sclerosis susceptibility: difference in transgenerational risk localizes to the major histocompatibility complex. Hum Mol Genet. 2009;18:261–266. doi: 10.1093/hmg/ddn353. [DOI] [PubMed] [Google Scholar]
- Hansen T, Skytthe A, Stenager E, Petersen HC, Bronnum-Hansen H, Kyvik KO. Concordance for multiple sclerosis in Danish twins: an update of a nationwide study. Mult Scler. 2005;11:504–510. doi: 10.1191/1352458505ms1220oa. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:12877–12882. doi: 10.1073/pnas.1932604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Herranz E, Pardo LA, Bunt G, Gold R, Stuhmer W, Linker RA. Re-expression of a developmentally restricted potassium channel in autoimmune demyelination: Kv1.4 is implicated in oligodendroglial proliferation. Am J Pathol. 2007;171:589–598. doi: 10.2353/ajpath.2007.061241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Shankar SL, Shafit-Zagardo B, Massimi A, Lee SC, Raine CS, et al. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A. Limiting and repairing the damage in multiple sclerosis. Schweiz Med Wochenschr. 1993;123:1145–1152. [PubMed] [Google Scholar]
- Mastronardi FG, Noor A, Wood DD, Paton T, Moscarello MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. 2007;85:2006–2016. doi: 10.1002/jnr.21329. [DOI] [PubMed] [Google Scholar]
- Moscarello MA, Brady GW, Fein DB, Wood DD, Cruz TF. The role of charge microheterogeneity of basic protein in the formation and maintenance of the multilayered structure of myelin: a possible role in multiple sclerosis. J Neurosci Res. 1986;15:87–99. doi: 10.1002/jnr.490150109. [DOI] [PubMed] [Google Scholar]
- Moscarello MA, Wood DD, Ackerley C, Boulias C. Myelin in multiple sclerosis is developmentally immature. J Clin Invest. 1994;94:146–154. doi: 10.1172/JCI117300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronardi FG, Tsui H, Winer S, Wood DD, Selvanantham T, Galligan C, et al. Synergy between paclitaxel plus an exogenous methyl donor in the suppression of murine demyelinating diseases. Mult Scler. 2007;13:596–609. doi: 10.1177/1352458506072167. [DOI] [PubMed] [Google Scholar]
- D'Souza CA, Wood DD, She YM, Moscarello MA. Autocatalytic cleavage of myelin basic protein: an alternative to molecular mimicry. Biochemistry. 2005;44:12905–12913. doi: 10.1021/bi051152f. [DOI] [PubMed] [Google Scholar]
- Musse AA, Boggs JM, Harauz G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc Natl Acad Sci USA. 2006;103:4422–4427. doi: 10.1073/pnas.0509158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranquill LR, Cao L, Ling NC, Kalbacher H, Martin RM, Whitaker JN. Enhanced T cell responsiveness to citrulline-containing myelin basic protein in multiple sclerosis patients. Mult Scler. 2000;6:220–225. doi: 10.1177/135245850000600402. [DOI] [PubMed] [Google Scholar]
- Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Hohenester S, Oude-Elferink RP, Beuers U. Primary biliary cirrhosis. Semin Immunopathol. 2009;31:283–307. doi: 10.1007/s00281-009-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kD mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. 1987;138:3525–3531. [PubMed] [Google Scholar]
- Bruggraber SF, Leung PS, Amano K, Quan C, Kurth MJ, Nantz MH, et al. Autoreactivity to lipoate and a conjugated form of lipoate in primary biliary cirrhosis. Gastroenterology. 2003;125:1705–1713. doi: 10.1053/j.gastro.2003.09.034. [DOI] [PubMed] [Google Scholar]
- Nakano T, Inoue K, Hirohara J, Arita S, Higuchi K, Omata M, et al. Long-term prognosis of primary biliary cirrhosis (PBC) in Japan and analysis of the factors of stage progression in asymptomatic PBC (a-PBC) Hepatol Res. 2002;22:250–260. doi: 10.1016/s1386-6346(01)00148-6. [DOI] [PubMed] [Google Scholar]
- Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, et al. Genome-wide meta-analyses identifies three loci associated with primary biliary cirrhosis Nat Genet 2010in press. [DOI] [PMC free article] [PubMed]
- Invernizzi P, Selmi C, Mackay IR, Podda M, Gershwin ME. From bases to basis: linking genetics to causation in primary biliary cirrhosis. Clin Gastroenterol Hepatol. 2005;3:401–410. doi: 10.1016/s1542-3565(04)00678-0. [DOI] [PubMed] [Google Scholar]
- Selmi C, Invernizzi P, Miozzo M, Podda M, Gershwin ME. Primary biliary cirrhosis: does X mark the spot. Autoimmun Rev. 2004;3:493–499. doi: 10.1016/j.autrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Mitchell MM, Lleo A, Zammataro L, Mayo MJ, Invernizzi P, Bach N, et al. Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics. 2011;6 doi: 10.4161/epi.6.1.13405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invernizzi P, Miozzo M, Battezzati PM, Bianchi I, Grati FR, Simoni G, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet. 2004;363:533–535. doi: 10.1016/S0140-6736(04)15541-4. [DOI] [PubMed] [Google Scholar]
- Matthias T, Shoenfeld Y. Challenges for the autoimmunologist. Clin Rev Allergy Immunol. 2010;38:75–76. doi: 10.1007/s12016-009-8141-2. [DOI] [PubMed] [Google Scholar]
- Invernizzi P. Geoepidemiology of autoimmune liver diseases. J Autoimmun. 2010;34:J300–J306. doi: 10.1016/j.jaut.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Shapira Y, Agmon-Levin N, Shoenfeld Y. Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun. 2010;34:J168–J177. doi: 10.1016/j.jaut.2009.11.018. [DOI] [PubMed] [Google Scholar]
- Youinou P, Pers JO, Gershwin ME, Shoenfeld Y. Geo-epidemiology and autoimmunity. J Autoimmun. 2010;34:J163–J167. doi: 10.1016/j.jaut.2009.12.005. [DOI] [PubMed] [Google Scholar]
- van Steensel B. Mapping of genetic and epigenetic regulatory networks using microarrays. Nat Genet. 2005;37 Suppl:S18–S24. doi: 10.1038/ng1559. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- Brooks WH, Le Dantec C, Pers JO, Youinou P, Renaudineau Y. Epigenetics and autoimmunity. J Autoimmun. 2010;34:J207–219. doi: 10.1016/j.jaut.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Cang S, Lu Q, Ma Y, Liu D. Clinical advances in hypomethylating agents targeting epigenetic pathways. Curr Cancer Drug Targets. 2010;10:539–545. doi: 10.2174/156800910791517217. [DOI] [PubMed] [Google Scholar]
- Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Lebdeh HS, Barazzoni R, Meek SE, Bigelow ML, Persson XM, Nair KS. Effects of insulin deprivation and treatment on homocysteine metabolism in people with type 1 diabetes. J Clin Endocrinol Metab. 2006;91:3344–3348. doi: 10.1210/jc.2006-0018. [DOI] [PubMed] [Google Scholar]
- Chiang EP, Wang YC, Chen WW, Tang FY. Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J Clin Endocrinol Metab. 2009;94:1017–1025. doi: 10.1210/jc.2008-2038. [DOI] [PubMed] [Google Scholar]
- Fonseca V, Dicker-Brown A, Ranganathan S, Song W, Barnard RJ, Fink L, et al. Effects of a high-fat-sucrose diet on enzymes in homocysteine metabolism in the rat. Metabolism. 2000;49:736–741. doi: 10.1053/meta.2000.6256. [DOI] [PubMed] [Google Scholar]