

Abstract
More than half of the primordial follicles that are formed by Day 6 of postnatal life in the mouse will be eliminated from the ovary by the time of puberty. Apoptosis, a form of programmed cell death, is one mechanism by which these follicles could be actively lost. To investigate whether apoptosis is responsible for the loss of primordial follicles, follicular atresia was examined during the prepubertal period, when follicles die and are cleared from the ovary at an extremely high rate. Four hallmarks of classical apoptosis were measured in follicles present in prepubertal ovaries. The primordial follicle cohort was not positively associated with nuclear condensation or cell shrinkage, activation of caspase 3, cleavage of poly(ADP ribose) polymerase 1 (PARP1), or fragmentation of DNA. These data are consistent with a nonapoptotic pathway that is responsible for small follicle death.
Keywords: apoptosis, atresia, follicular development, prepubertal, primordial follicle
Primordial follicle atresia during the prepubertal period in mice is not due to classical apoptosis.
INTRODUCTION
In the ovary, each germ cell is either selected to grow and ovulate or is lost to an atretic pathway; the female reproductive capacity requires a balance between these fates. Germ cells can be lost to death processes before or after they are surrounded by somatic cells to form follicles; the death of oocytes and germ cells preceding follicle formation is termed attrition, whereas the degenerative process of follicle death once germ cells are encapsulated by somatic cells is termed atresia [1]. More than 99% of germ cells in the ovary are lost through attrition and atresia; only very few ever reach ovulation. The precise mechanisms governing germ cell attrition rely mainly on apoptosis and are well documented [2–8], but less is understood about the biological mechanism of follicular atresia. Both biological processes are crucial for the selection and maturation of an adequate number of good-quality oocytes that will last throughout the reproductive life of the organism.
The mechanism of cell death and follicular atresia in the ovary has been widely assumed to be apoptosis [9–11]. Indeed, apoptosis does appear to be the major mechanism of germ cell attrition during fetal development. Atretic primordial germ cells exhibit morphological signs of apoptosis, including nuclear condensation, membrane blebbing, and cell shrinkage, with eventual fragmentation [2, 5, 6]. Additionally, the germ cells lost during this period exhibit DNA fragmentation and laddering characteristic of apoptosis [7, 8]. Germ cells also exhibit signs of apoptosis during the period of germ cell nest breakdown that immediately precedes primordial follicle formation. Both cleaved PARP1 and DNA fragmentation, as evidenced by TUNEL staining, are apparent in germ cells during this time of primordial follicle assembly in both the mouse and human [3, 4, 12, 13]. In fact, hormonally controlled apoptosis of germ cells is thought to be necessary for proper completion of nest breakdown and follicle assembly in the mouse [12]. However, even during this period, alternative forms of germ cell death or loss have been documented. For example, the process of shedding, where germ cells squeeze through the ovarian surface and are shed into the intrabursal space, also has been reported during this time period, but the exact contribution to attrition is unknown [14]. Additionally, there are reports of nonclassical apoptotic phenotypes (those that do not exhibit hallmarks of apoptosis, like DNA fragmentation or cellular and nuclear condensation) that resemble meiotic arrest, necrotic cell death, or autophagic cell death [15, 16]. The biological processes responsible for all follicular atresia are not as well studied as those responsible for earlier attrition. Follicular atresia itself is defined only by a measured quantitative loss of follicles; it is distinct from any explanation of the biological mechanism responsible for cell death. The dynamics of follicular atresia are difficult to pinpoint because they depend on the age of the animal; different follicle cohorts are present before and after puberty. An additional complicating factor is that follicles may be lost from the initial follicle pool by atresia or merely by the healthy transition to the next phase of follicular growth; the decision between these two fates differs greatly based on the follicle class involved [17–20]. After germ nest breakdown and accompanying germ cell attrition [4], primordial follicles are formed that consist of a central germ cell with surrounding squamous granulosa cells. These quiescent, nongrowing follicles are the class that experiences the most significant loss due to atresia during the prepubertal period [17, 18]. Upon activation, which involves actions of the phosphatidylinositol 3-kinase pathway [21], the transition from primordial to primary is associated with morphological and proliferative changes in the granulosa cells and growth of the oocyte. Further proliferation of granulosa cells is indicative of multilaminar secondary follicles. Contrary to primordial follicles, primary and small secondary follicles are thought to transition mostly to more mature follicles and have a negligible rate of atresia [17–19]. After stimulation by follicle-stimulating hormone, the granulosa cells begin a rapid proliferation stage coincident with reorganization around a fluid-filled space called the antrum. These proliferative and differentiation changes mark the final growth stage before ovulation. Follicles at each of these later stages that are not selected for further growth also undergo atresia [17–20, 22–24].
Like earlier attrition, the death of large follicles in the adult ovary involves apoptosis although, importantly, these signs of apoptosis are detected primarily in the somatic granulosa cells, not the oocyte itself [8, 25–29]. Apoptotic markers have been detected in the granulosa cells of large secondary and antral (preovulatory) atretic follicles of human, rodent, bovine, porcine, and avian ovaries [20, 27, 29–35]. However, these studies did not report similar signs of apoptosis in smaller follicles in the adult ovary [8, 20, 29]. Human primordial and primary follicles in the adult do not exhibit morphological signs of apoptosis or the proteolytic cleavage of caspase 3, a highly conserved effector of apoptosis [27]. Indeed, caspase 3 and DNA fragmentation were only identified in older follicles in which morphological signs of apoptosis existed, and all three identifiers of apoptosis were absent in the primordial follicles of human adult ovary biopsies [36].
Unlike adult ovaries, the prepubertal ovary undergoes a significant loss of primordial follicles from the initial follicle pool that is formed after nest breakdown. In mice, primordial follicles decrease in number from more than 10 000 at Postnatal Day 6 to fewer than 3000 by Day 45 [17]. This massive atresia is mirrored in the human ovary, which has approximately 2 million follicles present at birth but only about 300 000 by age 7 yr; even more follicles are lost in the years preceding menarche [37]. The mechanisms underlying the loss of follicles prior to puberty establish the “ovarian reserve,” or those follicles available to the adult animal to ensure normal fertility and the endocrine hormones necessary to the overall health of the organism. Loss of too many or too few follicles would change the ovarian dynamics of the adult. Indeed, premature loss of ovarian follicles is associated with premature ovarian insufficiency, and the mechanisms governing prepubertal follicle loss may provide a window of understanding on this largely idiopathic disease.
The current study investigates whether apoptosis is the mechanism of cell death and follicular atresia in the prepubertal ovary.
MATERIALS AND METHODS
Animals
CD1 mice were maintained in accordance with the policies of the Northwestern University's Animal Care and Use Committee. Mice were housed and bred in a controlled barrier facility within Northwestern University's Center of Comparative Medicine (Chicago and Evanston, IL) and were provided with food and water ad libitum. Temperature, humidity, and photoperiod (12L:12D) were kept constant. Animals were fed Teklad Global (Madison, WI) irradiated 2919 or 2916 chow, which does not contain soybean or alfalfa meal and contains minimal phytoestrogens.
Tissue Processing and Histological Staining
Mouse ovaries were placed in 4% formaldehyde (Sigma, St. Louis, MO) fixative for 8 to 24 h, depending on size. The tissue was then dehydrated and paraffin embedded. Five-micrometer sections were cut with a microtome and mounted on Superfrost-Plus slides (Vector Laboratories Inc., Burlingame, CA) or Surgipath Snowcoat Extra slides (Surgipath, Richmond, IL). Hematoxylin and eosin staining were performed using a Leica Autostainer XL (Leica Microsystems, Wetzlar, Germany). All tissue processing was performed by the Northwestern University Center for Reproductive Sciences Histology Core.
Antibodies
DEAD (Asp-Glu-Ala-Asp) box polypeptide 4 (DDX4; or mouse vasa homolog, MVH), cleaved caspase 3, and cleaved poly(ADP ribose) polymerase 1 (PARP1) were detected by immunohistochemistry or immunofluorescence (IMF). The antibodies used were rabbit polyclonal anti-DDX4 (graciously provided by Dr. Toshiaki Noce, Mitsubishi Kagaku Institute of Life Sciences, Tokyo, Japan), rabbit polyclonal anti-cleaved caspase 3 (catalog no. 9661; Cell Signaling Technology Inc., Danvers, MA), and rabbit polyclonal anti-cleaved PARP1 (catalog no. 9544; Cell Signaling Technology). Primary antibodies against DDX4, cleaved caspase 3, and cleaved PARP1 were diluted 1:4000, 1:25, and 1:75, respectively. The secondary antibody used was biotinylated goat anti-rabbit, diluted 1:200 except for anti-cleaved caspase 3 staining, when the secondary was diluted 1:75. Primary antibody was omitted for all negative controls.
Immunohistochemistry and IMF
Immunohistochemistry and IMF for cleaved PARP1 were performed and visualized as described previously [38], except that antigen retrieval was performed by microwaving on high for 2 min and low for 7 min.
A modified protocol for cleaved caspase 3 staining (courtesy of Dr. P.J. Devine, University of Quebec, Pointe-Claire, QC, Canada) was used. Peroxide quenching, Tris-buffered saline with tween (TBS-T) permeabilization, and ABC kit steps were omitted, 5% bovine serum albumin-PBS was used as block, and all antibodies were diluted in PBS. After secondary antibody incubation, the slides were rinsed in TBS-T and incubated in streptavidin-conjugated Alexa Fluor 568 (1:100 dilution) for 1 h (Molecular Probes, Eugene, OR).
In Situ DNA Fragmentation Detection
TUNEL staining was performed using the DeadEnd Fluorometric TUNEL System Kit following the manufacturer's protocol (Promega Corp., Madison, WI) as described previously [38]. DNAse pretreatment was done according to manufacturer's protocol with a DNAseI (Worthington Biochemical Corp., Lakewood, NJ) concentration of 20 U/ml.
Preparation of Ovarian Lysates
Ovaries were placed in lysis buffer (50 mM Tris-HCl, pH 7.5; 10% glycerol; 5 mM ethylenediaminetetraacetic acid; 150 mM NaCl; and 0.5% Nonidet P-40) with freshly added protease inhibitors (one tablet per 10 ml; catalog no. 11836153001; Roche Laboratories, Indianapolis, IN) and phosphatase inhibitors (diluted 1:100; catalog no. P2850; Sigma-Aldrich, St. Louis, MO) and homogenized with a handheld motorized pestle (catalog no. 749540-0000; Kontes Glass Company, Vineland, NJ). Lysates were centrifuged 5 min, and supernatant was quantified for protein content using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE) and placed at −80°C until use.
Caspase 3/7 Activity Assay and Cleaved PARP1 Western Blotting
Caspase 3/7 activity was assayed using the Caspase Glo 3/7 Assay (catalog no. G8093; Promega) as described previously [39], using 5 μg of protein.
Western blotting was performed as described previously [40], with the following modifications: between 35 and 60 μg of protein was loaded for each sample, blocking was done in 5% milk in TBS-T, and primary antibodies were incubated overnight at 4°C. Dilutions for anti-cleaved PARP1, anti-beta actin (catalog no. A-2066; Sigma-Aldrich), and horseradish peroxidase-conjugated goat anti-rabbit secondary (catalog no. 816120; Zymed Laboratories, San Francisco, CA) were 1:500, 1:3000, and 1:3000, respectively. Expression was determined as a ratio of cleaved-PARP1 to actin signal.
For both quantitative measures, activity/expression was represented on a per-ovary basis after determining the wet weights of ovaries using C-31 Microbalance (Cahn Instruments, Cerritos, CA).
Mathematical Calculation of Primordial Follicle Death
Using the models developed by Bristol-Gould et al. [17, 38], equations 1–5 were derived to determine the number of primordial follicles that underwent atresia (A) or transitioned to primary stage (T) as a function of time (in days). In equation 1, P represents the number of primordial follicles at time t after the initial time point (i.e., Day 6). Calculations used Day 6 as t = 0 because all germ cells are encapsulated into follicles by this time, and germ cell loss can therefore be attributed to follicle atresia rather than germ cell attrition. The initial number of primordial follicles is denoted as Po, the kinetic constant kL represents the rate of loss for primordial follicles by atresia, and kT is the kinetic constant describing the rate of loss for primordial follicles by transition to the primary stage (Fig. 1A). equation 1 can be plugged into equation 2 or equation 4 to provide a solvable form for number of follicles lost to atresia (A) or transition (T), respectively. equation 3 is the solution to equation 2, and equation 5 is the solution to equation 4. Models from Bristol-Gould et al. [17, 38] did not pool primordial and primary follicles into a single group.
FIG. 1.

Mathematical model of primordial follicle dynamics. Modeling based on empirical counts provides predictions for (A) loss dynamics and (B) total remaining primordial follicle number. C) Primordial follicles can leave the initial follicle pool by dying (L) or through transition to the primary stage (T).
Follicle Counting
Except in the case of cortical follicle counts for examination of follicle shedding, follicles were classified as either primordial/primary if they had only one surrounding granulosa cell layer, or secondary/antral if they had multiple granulosa cell layers [23]. Follicles with either an oocyte or single positive granulosa cell stained positive by IMF or TUNEL, or those that were deemed pyknotic were considered apoptotic follicles. All counts were conducted beginning at the first section of ovarian tissue on every 10th section of serially sectioned tissue and were normalized to the entire ovary as described previously, with the modification that all secondary follicles were counted even if they did not appear morphologically healthy [38]. An average of at least three sections was counted for each ovary; counting serial sections at regular intervals allows the same relative amount of each ovary to be counted. Counting multiple sections has been shown to be important for accuracy of germ cell counts [41]. For examination of follicle shedding, only primordial follicles (distinct from primary follicles) were counted; these were defined as described previously [38]. All counts were performed by three independent investigators. Follicle counting procedure was tested with current counters and was found to be similar to results obtained by counters in Bristol-Gould et al. [17, 38].
Although all results are reported in terms of primordial follicles, primordial and primary follicles were quantified together in this study (not including modeling derived from Bristol-Gould et al. [17, 38] or counts of cortical follicles for follicle shedding) to distinguish them from multilayered follicles. Despite this pooling, the results are stated in terms of primordial follicles alone. We have done this because several studies from our lab and others have shown that primary follicles do not undergo atresia to an appreciable degree [17–20]. Therefore, although our results do not show apoptosis occurring in primary follicles, this is likely because of the fact that these follicles are simply not dying, rather than the conclusion that follicles are dying but are not doing so by apoptosis. Because primary follicles are not dying to any significant degree, they do not contribute to counts of apoptotic marker-positive follicles. Because counts are presented in terms of positive follicles per section, not percent of total follicles, primary follicles do not influence counts, and results can therefore be stated in terms of primordial follicles alone.
Statistics
Data were analyzed by one-way ANOVA between groups, followed by a Tukey test of significance. Significance was determined for P < 0.05. All statistical analysis was performed using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA).
RESULTS
Mathematical and Empirical Assessment of the Number of Atretic Primordial Follicles Lost per Day in the Prepubertal Ovary
An estimated number of primordial follicles that are lost to atresia between Days 6 and 19 was determined by mathematical modeling of empirically derived follicle numbers counted in the ovaries of CD1 mice (equations 1–3). A model derived from follicle counts performed on time points of Days 6, 10, 19, and 45 and Months 4.5, 6, and 12 (follicle counts were performed and published in Bristol-Gould et al. [17]) indicated an initial follicle pool of approximately 10 300 primordial follicles (Postnatal Day 6), from which approximately 5100 primordial follicles are lost by Day 19 [17, 38] (approximately 2600 primordial follicles per ovary; Fig. 1A). This loss of primordial follicles from the initial follicle pool is only relevant to a discussion of follicle death if follicles are lost to atresia rather than a transition to the primary stage; follicle counts from Bristol-Gould et al. [17] did not group primordial and primary follicles, allowing the calculation of the number of follicles lost to each endpoint. A formula was derived to calculate this from the original model [17, 38]. We calculate a transition of 81 ± 3.8 follicles per ovary per day to primary follicles, with an approximately 2-fold greater loss of primordial follicles to atresia (155 ± 7.4 primordial follicles per ovary per day; Fig. 1, B and C). Thus, atresia accounts for the majority of loss from the initial primordial follicle pool.
Classic Apoptotic “Death Effectors” Remain Inactive in Atretic Primordial Follicles
Caspase 3, which requires proteolytic cleavage for activation, is a key mediator of apoptosis and is partially or totally responsible for cleaving many of the proteins that are degraded to complete apoptosis; caspase 7 is a family member of caspase 3 that can act in its place in some apoptotic processes [42]. The presence of active caspase 3 or 7 was investigated across the prepubertal period using a highly sensitive assay. Very little caspase activity was found until Postnatal Day 16, with significant increases by Day 19 (Fig. 2A). To determine where the caspase activity was localized, the presence of cleaved caspase 3 was measured in ovarian tissue from Postnatal Days 7, 10, 13, 16, 19, 22, and 26 using immunofluorescent assessment. Cleaved caspase 3 was detected in the cytoplasm of granulosa cells in secondary and antral follicles at all days in which they were present; the number of positive follicles increased significantly by Postnatal Day 16 (Fig. 2, B and C). Cytoplasmic, nonnucleated bodies present in the antral space, thought to be cell fragments from cells undergoing apoptosis (i.e., apoptotic bodies) were positive for cleaved caspase 3 (Fig. 2C), as were some stromal cells. In contrast, granulosa cells from primordial and primary follicles were negative for cleaved caspase 3 (Fig. 2D). Oocytes from all follicular stages were negative for cleaved caspase 3. Interestingly, in the negligible number of primordial follicles deemed positive for cleaved caspase 3, staining was typically in the oocyte rather than the granulosa cells, a reversal of the localization monitored in larger follicles.
FIG. 2.

Apoptotic pathway effectors are not activated in primordial or primary follicles (P). A) Assay of activity shows significant increase of active caspase 3 or 7 only at Day 19. B) An antibody against the cleavage product of caspase 3 localizes to granulosa cells of secondary (S) and antral (A) follicles across the entire prepubertal period. Different lowercase letters indicate a significant difference between groups. Symbol indicates that the group marked is not significantly different from any unmarked groups. Error bars are + SEM. Representative (C) secondary/antral and (D) primordial follicles (yellow asterisks) are shown. Bars = 100 μm (C) and 25 μm (D).
Characteristic Apoptotic Pathway Substrates Remain Uncleaved in Primordial Follicles
Downstream of the activation of caspases or any compensating protease, the degradation of certain substrates is considered a hallmark of apoptosis. For example, internucleosomal DNA fragmentation has been the best-studied molecular indicator of apoptosis. It can be detected as DNA laddering when by gel electrophoresis, or studied in situ by labeling DNA strand breaks (TUNEL method) [43–45]. A second substrate targeted for degradation in apoptosis is PARP1, which is responsible for repairing DNA strand breaks. Cleavage of PARP1 allows the accumulation of genotoxic assaults [46]. To rule out the possibility that a compensatory protease was acting in place of caspase 3 or 7, cleavage of PARP1 and DNA fragmentation were examined in tissue from Postnatal Days 7, 10, 13, 16, 19, 22, and 26. The results corroborate the results of caspase 3; both DNA (Fig. 3, A–E) and PARP1 (Fig. 3, F–I) were significantly degraded in secondary and antral follicles only, and only in the granulosa cells of these follicles. In addition to measuring fragmentation in these follicles known to undergo apoptosis, the positive control of DNAse pretreatment in the TUNEL protocol clearly labeled all cells, with oocytes appearing to be to be labeled or visualized more readily than somatic cells when DNA fragmentation is induced (Fig. 3C). Across all time points, oocytes were negative for both DNA fragmentation and PARP1 cleavage. Positive granulosa cells were identified throughout secondary follicles and in the cumulus and mural granulosa cell layers of antral follicles, most commonly proximal to the antrum (Fig. 3, A and F). Labeling of fragmented DNA is clearly isolated to the nucleus, whereas cleaved PARP1 is distributed throughout the cytoplasm. Primordial and primary follicles, both oocytes and granulosa cells, were almost completely negative for both markers (Fig. 3, B and G). Quantification of PARP1 from Western blotting supports this pattern; PARP1 expression is increased by Day 16, and significantly so by Day 19 (Fig. 3I). Cleaved PARP1 seems to be the most sensitive apoptotic marker studied, because it detected a greater number of secondary and antral follicles as apoptotic than any other method. As with cleaved caspase 3, the very few primordial follicles ever deemed reactive for cleaved PARP1 immunostaining were positive within the oocyte, not the granulosa cells.
FIG. 3.
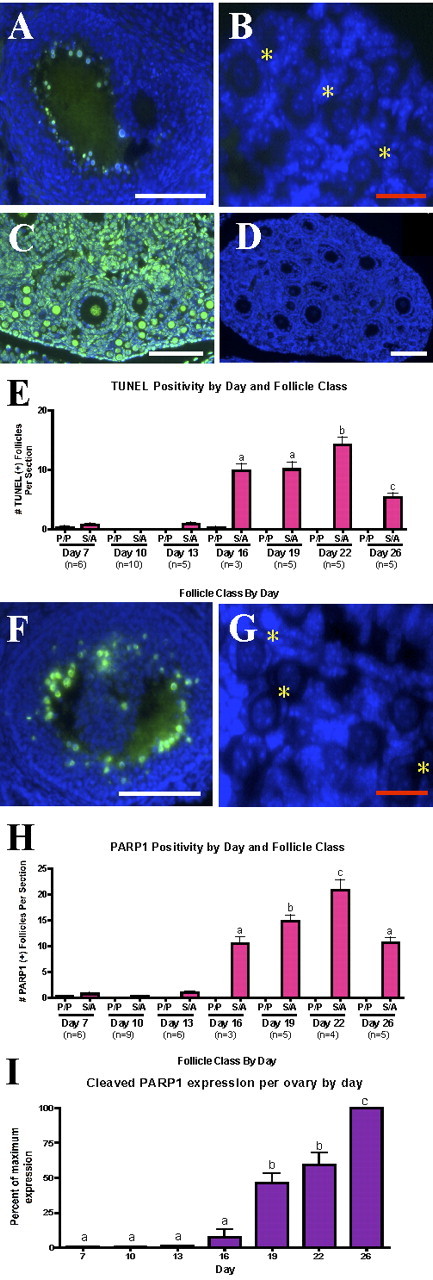
Primordial and primary follicles do not exhibit classic apoptotic pathway endpoints. DNA fragmentation as indicated by TUNEL staining can be recognized in antral follicles (A) from Day 26, but not in primordial follicles (B). DNAse treatment serves as a clear positive control (C), and the image of an entire Day 10 ovary demonstrates global absence of DNA fragmentation at this day (D). Trend shown in A and B is true across the period (C). The same results can be seen for the cleavage of PARP1 (F–H). Western blots show that cleaved PARP1 increases significantly at Day 19 (I). Different lowercase letters indicate a significant difference between groups. Yellow asterisks indicate representative primordial follicles. Error bars are + SEM. P, primordial or primary follicles; S, secondary follicles; A, antral follicles. Bars = 100 μm (A, C, D, F) and 25 μm (B, G).
Morphological Endpoints of Apoptosis Are Absent in Primordial Follicles
Pyknosis—the shrinkage of the cell and condensation of the nucleus that can be observed by light microscopy—is the classical morphological signature of apoptosis [47]. To confirm results from apoptotic pathway component localization, ovarian tissue collected from mice 7, 10, 13, 16, 19, 22, and 26 days old was stained with hematoxylin, and every 10th section of serially sectioned tissue was counted to quantify pyknosis. Pyknotic nuclei were restricted to the granulosa cells of large-growing secondary and antral follicles in tissue from later days, where such developed follicles were present. These pyknotic nuclei appeared throughout the granulosa cell layers in preantral follicles but were more commonly found surrounding the antral space after antrum formation (Fig. 4A). Granulosa cells with pyknotic nuclei often also exhibited cell fragmentation. Pyknotic stromal cell nuclei also were detected but were not quantified. No pyknotic nuclei were identified in oocytes of any follicle size at any day examined. Negligible numbers of primordial or primary follicles with pyknotic cells were identified on any day (Fig. 4, B and C).
FIG. 4.
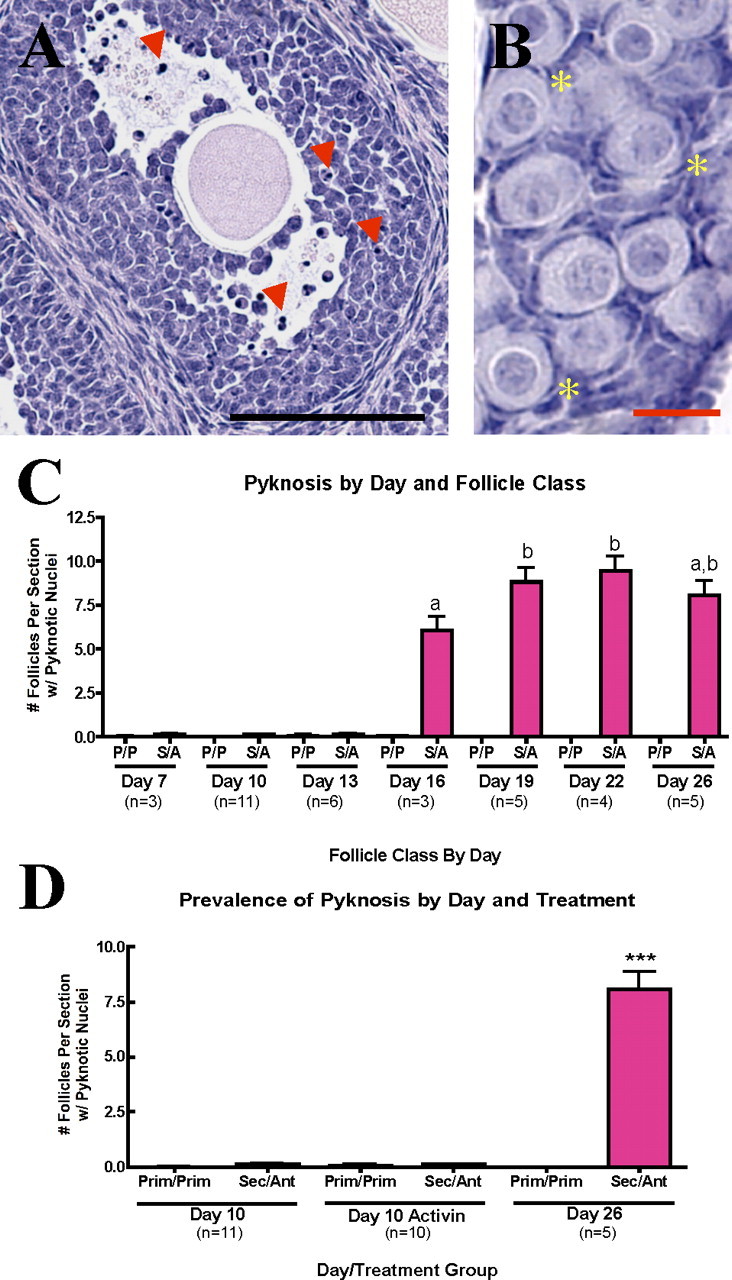
Morphological endpoints of apoptosis are absent in primordial and primary follicles. A) Large secondary and antral follicles show extensive nuclear condensation and cell shrinkage (pyknosis; red arrowheads) in granulosa cells. Bar = 100 μm. B) Pyknosis is not detected in primordial or primary follicles (representatives marked with yellow asterisks). Bar = 20 μm. Quantification is shown in C. D) Animal model with supraphysiological follicle loss exhibits no significant increase in pyknosis in primordial follicles. Different lowercase letters and asterisks (***) indicate a significant difference between groups. Error bars are + SEM. Prim, primary; Sec, secondary; Ant, antral.
Apoptosis Is Not Initiated in Primordial Follicles During Supraphysiological Follicle Atresia
All observations to this point have used untreated animals to examine the normal physiological loss of follicles in the prepubertal period. There are, however, several models in which exogenous hormone or drug treatment can increase the number of primordial follicles formed, which is accompanied by a greater than normal loss of follicles prior to the establishment of the ovarian reserve at puberty. Previous data from our lab showed that activin increases the number of perinatal primordial follicles by 27%. However, by Day 19, activin-treated animals have a similar number of primordial follicles as the vehicle-treated animals. Thus, between Days 6 and 19, activin-treated animals undergo an increased rate of primordial follicle atresia [38]. This model allows us to determine whether the ovary initiates apoptosis to discard the excess follicles. To investigate this possibility, pyknotic primordial and primary follicles in ovarian tissue collected from 10-day-old, activin-treated animals [38] were counted.
Although activin-treated ovaries have a higher rate of primordial follicle loss (approximately 8000 primordial follicles lost per animal between Days 6 and 19 compared with 5000 in untreated controls), no significant increase in apoptosis in primordial (or primary) follicles in activin-treated ovaries was measured. Similar to primordial follicles from untreated ovaries, neither oocytes nor granulosa cells of primordial follicles from activin-treated animals appeared pyknotic (Fig. 4D).
Lack of Apoptosis Visualization Cannot Be Explained by Rapid Clearance
Rapid turnover and clearance of apoptotic cells by resident phagocytes has been suggested as a reason that counts of germ cells fail to label a large percentage of those expected to be apoptotic [4]. To address this possibility, cleaved PARP1-positive primordial and primary follicles were counted for eight time points (every 3 h) across the 24-h time period of Postnatal Day 10. Day 10 was selected because it represents the midpoint in the period of maximum follicle loss. The counting methodology assumes that the period between the onset of PARP1 cleavage and complete clearance of all cells in the primordial follicle is less than 3 h (an ultraconservative time frame for most apoptotic cells). By selecting 3 h, it is possible that we could overestimate the number of atretic follicles if their clearance is slower than this time span. Only 37 primordial or primary follicles were marked positive for cleaved PARP1 across the entire 24-h period, far fewer than the estimated 155 follicles that are being eliminated on this day (Fig. 5).
FIG. 5.

Lack of apoptosis visualization is not due to rapid clearance. Using an antibody against cleaved PARP1, positive primordial and primary follicles were counted across a 24-h period on Postnatal Day 10 (D10). Error bars are + SEM.
Follicle Shedding Does Not Explain Primordial Follicle Loss
As an alternative to an apoptotic mechanism, the hypothesis that primordial follicles are shed from the ovary was investigated by counting cortical follicles from every 10th section of Day 10 ovarian tissue. Cortical follicles were designated as “normal” if the oocyte was separated from the intrabursal space by at least two cell layers (hypothetically, the granulosa and ovarian surface epithelial layers; Fig. 6A). Follicles were identified as subepithelial if only a single cell layer separated the germ cell cytoplasm from the intrabursal space (Fig. 6B) and intraepithelial if the oocyte cytoplasm was in direct contact with the intrabursal region (Fig. 6C). Finally, follicles were classified as intrabursal if the oocyte or follicle was completely outside of the ovary. The bursa was maintained intact through dissection, processing, and sectioning for all ovaries counted.
FIG. 6.

Follicle shedding does not explain prepubertal primordial follicle loss. Cortical follicles were categorized as either normal (A), subepithelial (B), intraepithelial (C), or intrabursal. Isolated cells were seen in the intrabursal space (D). To ascertain whether these cells were actually degenerated oocytes, DDX4 antibody was used to label oocytes. E) Intrabursal cells and stained oocytes in primordial follicles (yellow asterisks). Black arrows indicate ovarian surface epithelium, and red arrowheads indicate the bursa. Quantification of above results is shown in F. Error bars are + SEM. Bar = 2 μm.
Approximately 90% of cortical follicles from Day 10 tissue were classified as normal (Fig. 6F). Although significantly less than the normal population, nearly 10% of the follicles were designated as subepithelial. An insignificant number of follicles transitioned further toward the surface; a negligible number of follicles were deemed intraepithelial. A primordial follicle was never found in the intrabursal space in all 11 ovaries examined. However, many intrabursal cells were detected (Fig. 6D), which confirmed that cells were not lost from the bursal sac during tissue preparation. Although these cells were not intact follicles, they could be oocytes that were shed from the ovary without accompanying granulosa cells. To address this possibility, every 10th section of Day 10 ovaries was stained for DDX4, an oocyte marker [48]. Although oocytes from all primordial follicles (and larger-growing follicles) stained positively for DDX4, no intrabursal cells were determined to be positive (Fig. 6E), verifying that these cells were not shed follicles.
DISCUSSION
Thousands of primordial follicles are lost during the prepubertal period in mice. We have demonstrated that during this period, primordial and primary follicles have a near-complete absence of all the standard hallmarks of classical apoptosis, such as nuclear condensation, activation of caspase 3, degradation of PARP1, and DNA fragmentation, all of which were identified in granulosa cells of larger atretic follicles. Furthermore, we found that the activin-treated ovary does not resort to apoptosis to eliminate supraphysiological numbers of primordial follicles. Finally, the phenomenon of epithelial shedding that has been described in germ cells during nest breakdown does occur during primordial follicle loss in the prepubertal ovary.
Failure to detect the morphologic and biochemical markers of apoptosis may have been due to technical limitations or utilization of nonclassical apoptotic pathway components. Single methods of identifying apoptosis are limited in their ability to mark all apoptotic cells; only a subpopulation of germ cells lost during follicle assembly, when germ cells are known to undergo apoptosis to some degree, can be identified (by TUNEL or cleaved PARP1 antibody staining) as apoptotic [4, 27, 38]. Second, nonclassical apoptotic pathways may be involved in follicle atresia, such as germ cells' utilization of caspase 2 instead of caspase 3 [49]. This study addresses these challenges by simultaneously performing multiple apoptosis detection techniques and performing quantification during the prepubertal period, as well as performing 24-h counts to exclude the possibility of rapid clearance preventing the complete visualization of markers.
Follicle count results are presented here using pooled counts from primordial and primary follicles (except in the case of follicle shedding counts, where only primordial follicles are counted). Although we have found that primary follicles do not exhibit characteristics of apoptosis, such as DNA fragmentation, pyknosis, PARP1 cleavage, or caspase 3 activation, similar to results found in primordial follicles, it is very important that we not extend the same conclusions regarding follicle death to this group. The initial primordial follicle pool loses 155 follicles per ovary per day to atresia (death); however, the rate of primary follicle loss to atresia is negligible [17–20]. Therefore, the conclusions presented here regarding primordial follicles—namely, that apoptosis is not the mechanism of their death and that a nonapoptotic death mechanism must exist in these follicles—is not applicable to primary follicles.
We conclude, therefore, that primordial follicles are not lost by classical apoptosis or follicle shedding and can furthermore report that the mechanism of germ cell elimination depends on the developmental stage or local follicular environment of the cell. In addition, apoptosis as a mechanism of cell death is limited by cell type within each follicle during the prepubertal period, and a nonapoptotic mechanism is likely responsible for atresia of primordial follicles in the ovary.
The Mechanism of Germ Cell Elimination Depends on Developmental Stage
Our results demonstrate that the mechanism of elimination for germ cells and follicles depends on the developmental stage at the time of loss. Although it has been reported that germ cells can be shed from the ovary surface during nest breakdown, follicle shedding from the ovary is not the mechanism by which primordial follicles are lost during the prepubertal period. These findings are in accordance with the original report of germ cell shedding in mice, which indicated that the phenomenon was extremely rare at 2 and 3 wk of age [14] but contrary to a report that suggests approximately 25% of primordial follicles are extruded from the ovary on Postnatal Days 7 and 12 [50]. This contradiction could be due to strain differences or differences in classification of cortical follicles. It is, however, difficult to postulate that one fourth of all primordial follicles could be extruded from the ovary at separate time points, yet none of these follicles would be found in the intrabursal region. Thus, the conclusion most consonant with the data is that follicle shedding is a rare event in the postpubertal animal and does not account for the loss of ovarian follicles in the time leading to puberty.
Similarly, the current study demonstrates that atresia via apoptosis is limited to specific developmental stages. Germ cells during fetal development and nest breakdown and granulosa cells of larger follicles have all been reported to exhibit characteristics of apoptosis; however, our results suggest that neither granulosa cells nor oocytes of primordial follicles undergo apoptosis. These results are consistent with previous studies, including two detailed studies of the morphology of dying primordial follicles. de Bruin et al. [51] described that human primordial oocytes die in an organized fashion characterized by the accumulation of vacuoles and late mitochondrial and nuclear rupture. Only 1 in 182 follicles contained any sign of pyknosis, leading the authors to conclude that the method of oocyte degeneration was necrotic rather than apoptotic [51]. In a similar study in rats, Devine et al. [25] demonstrated a high degree of cytoplasmic vacuolization and a complete lack of nuclear condensation and other morphological hallmarks in atretic primordial follicles. In contrast to the de Bruin study, this study reported no nuclear rupture, suggesting that neither necrosis nor apoptosis could explain oocyte death. Additionally, cleaved caspase 3 was not detected in primordial follicles of a different mouse strain [26]. Moreover, DNA fragmentation was not detected in whole-ovary preparations from Postnatal Day 3 or 7 mice. In the time following primordial follicle formation, DNA fragmentation was not detected until Day 14 [7]. Similar studies in rats found that nuclear pyknosis of granulosa cells is not detected until Day 13 [52]. Both of these time points coincide with the appearance of atretic secondary follicles in the ovary; the large loss of primordial follicles prior to this time is unaccompanied by either apoptotic hallmark. A recently published study performed in rats is the sole study to find expression of apoptotic markers, such as TUNEL and cleaved caspase 3, in primordial follicles, and indeed in oocytes across the period [53]. Surprisingly, the study found expression of these markers in 25%–30% of oocytes at every day examined across the prepubertal period, a number far larger than the numbers of follicles thought to be lost at any one moment. In the context of the previous literature mentioned and our own study, we are unable to explain this finding, although species differences are possible. Nevertheless, even this study agrees with the current conclusion that primordial follicles are not lost by classical apoptosis.
Apoptosis Is Limited by Cell Type Within Formed Follicles
Apoptosis is not only limited by developmental stage, because this study demonstrates that it is also limited to certain cells within each follicle. Previous work reported that cell death in smaller follicles begins with death of the oocyte, whereas death of larger follicles can exhibit massive granulosa cell loss while maintaining an intact oocyte [8, 25, 51, 54]. Therefore, larger-follicle atresia is apparently dependent on granulosa cell death, whereas primordial follicle atresia is not. Because of the nonapoptotic state of the granulosa cells, the mechanism of cell death in primordial follicular oocytes becomes the defining factor in atresia. The current study demonstrates, however, that oocytes of primordial follicles (as well as the granulosa cells) exhibit negligible amounts of apoptotic death.
Genetic models that manipulate the classic apoptotic pathway components provide conflicting results regarding the role of apoptosis in the death of oocytes of primordial follicles. Two studies involving overexpression or ablation of the antiapoptotic factor Bcl2 demonstrated a significant change in surviving primordial follicles [7, 55], but one of these studies suggested that the difference is due to a change in germ cell attrition that is then carried over to primordial follicle number [55]. One study concluded that there was a direct effect on primordial follicle number, but the authors did not perform early follicle counts [7]. It is possible that such counts would have measured changes in germ cell number preceding follicle formation. Similar to the Bcl2 studies, a study using a Bax knockout mouse model reported an increase in primordial follicle number at a late time point but demonstrated that this increase was already present at Postnatal Days 7 and 4, and that even embryonic germ cell number was higher in the transgenic animal [56]. Conversely, a second group using a Bax knockout mouse model performed early germ cell counts, and their results suggested an actual effect on primordial follicle loss [57]. Bax knockout animals in the study had three times as many primordial follicles as controls at Day 42, although they contained comparable numbers at Postnatal Day 4. These data suggest that Bax deficiency renders primordial follicles more resistant to apoptosis, preventing them from undergoing the normal physiological loss. To date, this study represents the best support for the presence of apoptosis in primordial follicles. Unfortunately, the study used morphology to identify primordial follicles as atretic; morphology alone has been reported to poorly identify oocytes as apoptotic [25, 51]. Therefore, although atresia was used synonymously with apoptosis, this was not actually demonstrated. This is especially problematic because alternate forms of cell death can also be regulated by Bcl2 family members [58]. Also, other work involving Bax has been highly suggestive of a role for the protein in primordial germ cell survival [59], but this stage was surprisingly unaffected in the study by Perez et al. [57].
Therefore, the literature regarding the death of follicle-enclosed oocytes by apoptosis is controversial; within this context, our results indicate that there is no evidence of detectable oocyte apoptosis occurring at any stage of follicle development after the completion of follicle formation. Because germ cells have been reported to undergo apoptosis in germ cell nests and later when ovulated and denuded of cumulus cells [60], it seems possible that the environment of the intact follicle, when the oocyte is surrounded by the somatic granulosa cells, supports an alternative pathway of germ cell death. Indeed, gap junctions begin to form as soon as oocytes and granulosa cells come into contact [61]. We postulate that oocytes of the earliest primordial follicles may be exposed to new signaling pathways that were previously unavailable within the germ cell nest, and that these new connections may alter the mechanism of oocyte cell death.
Alternate Mechanisms of Follicle Atresia in the Prepubertal Ovary Must Exist
Several studies already suggest that apoptosis is not the only mechanism of cell death in the ovary. Indeed, two studies demonstrated an interesting “meiotic arrest” phenotype in which germ cells die subsequent to arresting after their last mitotic division and never truly enter meiotic prophase I [16, 41]. The ensuing death of the cell did not exhibit the morphological signs of apoptosis: the chromatin did not seem to condense in the nucleus, and characteristic DNA degradation did not occur (the cells remain TUNEL negative) [16]. The authors suggested that this nonapoptotic cell death may be responsible for a significant portion of the death of germ cells during folliculogenesis and could therefore be extended to the death of oocytes within follicles. Likewise, alternative forms of cell death have been described in the adult ovary of various animals. A necrosislike form of programmed cell death has been observed in atretic follicles of the adult quail and goose [30, 32]. This form of cell death does not result from cellular injury or trigger an immunological response, and it does not follow the traditional apoptotic pathway. Finally, recent studies have reported that an alternate form of programmed cell death called autophagy occurs in the atretic follicles of quail [30], can be detected in human granulosa cells cultured in vitro [62], and may account for some portion of cell death in cultured fetal germ cells [63]. Literally translated as the cell “eating itself,” autophagy refers to the nonreversible degradation of substrates, including whole organelles, inside the cell's own lysosomal compartments. An extreme version of this normal housekeeping function can result in the death of the cell [64]. Autophagic cell death can be regulated by Bax, Bcl2l1, and Bcl2, indicating the dual role of the Bcl2 family members in autophagy and apoptosis [58]. Therefore, genetic manipulations thought to affect follicular atresia through apoptosis could actually be acting through mechanisms of autophagy. It seems evident, therefore, that apoptosis is not a “universal pathway of cellular suicide” responsible for the controlled deletion of ovarian cell populations, as was believed previously [10].
We have demonstrated that the mechanism of cell death in the ovary is dependent on both the developmental stage and cell type involved. Specifically, primordial follicles do not die by classical apoptosis; we hypothesize that all oocytes encapsulated in follicles may similarly eschew apoptosis. Future studies will investigate other possible mechanisms of follicle atresia to determine which pathways are used and what factors influence the mechanism of atresia chosen. Better insight into how follicles are eliminated by the ovary will dramatically increase basic understanding of normal physiological follicle loss and help shape clinical applications for women with aberrant follicle management leading to infertility.
Acknowledgments
The authors gratefully acknowledge the assistance of Cristina Thomas of Northwestern University and Jessina Thomas and Brian Chang of the Illinois Math and Science Academy (Aurora, IL) in performing extensive data quantification; the donation of DDX4 antibody by Dr. Toshiaki Noce; assistance with the cleaved caspase 3 IMF protocol from Dr. P.J. Devine; assistance from Alison Kim in figure preparation; and Jen Jozevic for animal care concerns. We are indebted to the P01-funded Northwestern University Histology Core (Tyler Wellington, director) for all tissue processing.
Footnotes
1Supported by National Institutes of Health grant P01 HD 21921.
REFERENCES
- Hsueh AJ, Billig H, Tsafriri A. Ovarian follicle atresia: a hormonally controlled apoptotic process. Endocr Rev 1994; 15: 707 724 [DOI] [PubMed] [Google Scholar]
- Coucouvanis EC, Sherwood SW, Carswell-Crumpton C, Spack EG, Jones PP. Evidence that the mechanism of prenatal germ cell death in the mouse is apoptosis. Exp Cell Res 1993; 209: 238 247 [DOI] [PubMed] [Google Scholar]
- Ghafari F, Gutierrez CG, Hartshorne GM. Apoptosis in mouse fetal and neonatal oocytes during meiotic prophase one. BMC Dev Biol 2007; 7: 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol 2001; 234: 339 351 [DOI] [PubMed] [Google Scholar]
- Pesce M, De Felici M. Apoptosis in mouse primordial germ cells: a study by transmission and scanning electron microscope. Anat Embryol (Berl) 1994; 189: 435 440 [DOI] [PubMed] [Google Scholar]
- Pesce M, Farrace MG, Piacentini M, Dolci S, De Felici M. Stem cell factor and leukemia inhibitory factor promote primordial germ cell survival by suppressing programmed cell death (apoptosis). Development 1993; 118: 1089 1094 [DOI] [PubMed] [Google Scholar]
- Ratts VS, Flaws JA, Kolp R, Sorenson CM, Tilly JL. Ablation of bcl-2 gene expression decreases the numbers of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology 1995; 136: 3665 3668 [DOI] [PubMed] [Google Scholar]
- Vaskivuo TE, Anttonen M, Herva R, Billig H, Dorland M, te Velde ER, Stenback F, Heikinheimo M, Tapanainen JS. Survival of human ovarian follicles from fetal to adult life: apoptosis, apoptosis-related proteins, and transcription factor GATA-4. J Clin Endocrinol Metab 2001; 86: 3421 3429 [DOI] [PubMed] [Google Scholar]
- Hussein MR. Apoptosis in the ovary: molecular mechanisms. Hum Reprod Update 2005; 11: 162 177 [DOI] [PubMed] [Google Scholar]
- Tilly JL. Apoptosis and ovarian function. Rev Reprod 1996; 1: 162 172 [DOI] [PubMed] [Google Scholar]
- Vaskivuo TE, Tapanainen JS. Apoptosis in the human ovary. Reprod Biomed Online 2003; 6: 24 35 [DOI] [PubMed] [Google Scholar]
- Jefferson W, Newbold R, Padilla-Banks E, Pepling M. Neonatal genistein treatment alters ovarian differentiation in the mouse: inhibition of oocyte nest breakdown and increased oocyte survival. Biol Reprod 2006; 74: 161 168 [DOI] [PubMed] [Google Scholar]
- Albamonte MS, Willis MA, Albamonte MI, Jensen F, Espinosa MB, Vitullo AD. The developing human ovary: immunohistochemical analysis of germ-cell-specific VASA protein, BCL-2/BAX expression balance and apoptosis. Hum Reprod 2008; 23: 1895 1901 [DOI] [PubMed] [Google Scholar]
- Hiura M, Fujita H. Electron microscopic observations on the elimination of the oocyte through the peritoneal epithelium in the neonatal mouse ovary. Cell Tissue Res 1977; 182: 73 79 [DOI] [PubMed] [Google Scholar]
- De Felici M, Lobascio AM, Klinger FG. Cell death in fetal oocytes: many players for multiple pathways. Autophagy 2008; 4: 240 242 [DOI] [PubMed] [Google Scholar]
- Wartenberg H, Ihmer A, Schwarz S, Miething A, Viebahn C. Mitotic arrest of female germ cells during prenatal oogenesis. A colcemid-like, non-apoptotic cell death. Anat Embryol (Berl) 2001; 204: 421 435 [DOI] [PubMed] [Google Scholar]
- Bristol-Gould SK, Kreeger PK, Selkirk CG, Kilen SM, Mayo KE, Shea LD, Woodruff TK. Fate of the initial follicle pool: empirical and mathematical evidence supporting its sufficiency for adult fertility. Dev Biol 2006; 298: 149 154 [DOI] [PubMed] [Google Scholar]
- Faddy MJ, Jones EC, Edwards RG. An analytical model for ovarian follicle dynamics. J Exp Zool 1976; 197: 173 185 [DOI] [PubMed] [Google Scholar]
- Faddy MJ, Telfer E, Gosden RG. The kinetics of pre-antral follicle development in ovaries of CBA/Ca mice during the first 14 weeks of life. Cell Tissue Kinet 1987; 20: 551 560 [DOI] [PubMed] [Google Scholar]
- Hirshfield AN, Midgley AR., Jr Morphometric analysis of follicular development in the rat. Biol Reprod 1978; 19: 597 605 [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hamalainen T, Peng SL, Lan ZJ, Cooney AJ, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 2008; 319: 611 613 [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol 1991; 124: 43 101 [DOI] [PubMed] [Google Scholar]
- Pedersen T, Peters H. Proposal for a classification of oocytes and follicles in the mouse ovary. J Reprod Fertil 1968; 17: 555 557 [DOI] [PubMed] [Google Scholar]
- Peters H. The development of the mouse ovary from birth to maturity. Acta Endocrinol (Copenh) 1969; 62: 98 116 [DOI] [PubMed] [Google Scholar]
- Devine PJ, Payne CM, McCuskey MK, Hoyer PB. Ultrastructural evaluation of oocytes during atresia in rat ovarian follicles. Biol Reprod 2000; 63: 1245 1252 [DOI] [PubMed] [Google Scholar]
- Fenwick MA, Hurst PR. Immunohistochemical localization of active caspase-3 in the mouse ovary: growth and atresia of small follicles. Reproduction 2002; 124: 659 665 [DOI] [PubMed] [Google Scholar]
- Glamoclija V, Vilovic K, Saraga-Babic M, Baranovic A, Sapunar D. Apoptosis and active caspase-3 expression in human granulosa cells. Fertil Steril 2005; 83: 426 431 [DOI] [PubMed] [Google Scholar]
- Kugu K, Ratts VS, Piquette GN, Tilly KI, Tao XJ, Martimbeau S, Aberdeen GW, Krajewski S, Reed JC, Pepe GJ, Albrecht ED, Tilly JL. Analysis of apoptosis and expression of bcl-2 gene family members in the human and baboon ovary. Cell Death Differ 1998; 5: 67 76 [DOI] [PubMed] [Google Scholar]
- Yuan W, Giudice LC. Programmed cell death in human ovary is a function of follicle and corpus luteum status. J Clin Endocrinol Metab 1997; 82: 3148 3155 [DOI] [PubMed] [Google Scholar]
- D'Herde K, De Prest B, Roels F. Subtypes of active cell death in the granulosa of ovarian atretic follicles in the quail (Coturnix coturnix japonica). Reprod Nutr Dev 1996; 36: 175 189 [DOI] [PubMed] [Google Scholar]
- Guthrie HD, Welch GR, Cooper BS, Zakaria AD, Johnson LA. Flow cytometric determination of degraded deoxyribonucleic acid in granulosa cells to identify atretic follicles during preovulatory maturation in the pig. Biol Reprod 1994; 50: 1303 1311 [DOI] [PubMed] [Google Scholar]
- Kovacs J, Forgo V, Peczely P. The fine structure of the follicular cells in growing and atretic ovarian follicles of the domestic goose. Cell Tissue Res 1992; 267: 561 569 [DOI] [PubMed] [Google Scholar]
- Nishihara S, Manabe N, Nakayama M, Wada S, Inoue N, Miyamoto H. Changes in cell adhesion molecules during follicular atresia in porcine ovaries. J Reprod Dev 2000; 46: 325 334 [Google Scholar]
- Piquette GN, Tilly JL, Prichard LE, Simon C, Polan ML. Detection of apoptosis in human and rat ovarian follicles. J Soc Gynecol Investig 1994; 1: 297 301 [DOI] [PubMed] [Google Scholar]
- Van Wezel IL, Dharmarajan AM, Lavranos TC, Rodgers RJ. Evidence for alternative pathways of granulosa cell death in healthy and slightly atretic bovine antral follicles. Endocrinology 1999; 140: 2602 2612 [DOI] [PubMed] [Google Scholar]
- Hurst PR, Mora JM, Fenwick MA. Caspase-3, TUNEL and ultrastructural studies of small follicles in adult human ovarian biopsies. Hum Reprod 2006; 21: 1974 1980 [DOI] [PubMed] [Google Scholar]
- Baker TG. A quantitative and cytological study of germ cells in human ovaries. Proc R Soc Lond B Biol Sci 1963; 158: 417 433 [DOI] [PubMed] [Google Scholar]
- Bristol-Gould SK, Kreeger PK, Selkirk CG, Kilen SM, Cook RW, Kipp JL, Shea LD, Mayo KE, Woodruff TK. Postnatal regulation of germ cells by activin: the establishment of the initial follicle pool. Dev Biol 2006; 298: 132 148 [DOI] [PubMed] [Google Scholar]
- Alvero AB, Montagna MK, Mor G. Correlation of caspase activity and in vitro chemo-response in epithelial ovarian cancer cell lines. Methods Mol Biol 2008; 414: 79 82 [DOI] [PubMed] [Google Scholar]
- Kenny HA, Bernard DJ, Horton TH, Woodruff TK. Photoperiod-dependent regulation of inhibin in Siberian hamsters: I. Ovarian inhibin production and secretion. J Endocrinol 2002; 174: 71 83 [DOI] [PubMed] [Google Scholar]
- McClellan KA, Gosden R, Taketo T. Continuous loss of oocytes throughout meiotic prophase in the normal mouse ovary. Dev Biol 2003; 258: 334 348 [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J 1997; 326 (pt 1): 1 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson SA, Sherman Y, Gavrieli Y. Identification of dying cells–in situ staining. Methods Cell Biol 1995; 46: 29 39 [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992; 119: 493 501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 1995; 21: 1465 1468 [DOI] [PubMed] [Google Scholar]
- Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet 1999; 64: 1282 1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26: 239 257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka Y, Tsunekawa N, Takahashi Y, Matsui Y, Satoh M, Noce T. Expression and intracellular localization of mouse Vasa-homologue protein during germ cell development. Mech Dev 2000; 93: 139 149 [DOI] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998; 12: 1304 1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JB, Duckett R, Myers M, Britt KL, Mladenovska T, Findlay JK. Quantification of healthy follicles in the neonatal and adult mouse ovary: evidence for maintenance of primordial follicle supply. Reproduction 2006; 132: 95 109 [DOI] [PubMed] [Google Scholar]
- de Bruin JP, Dorland M, Spek ER, Posthuma G, van Haaften M, Looman CW, te Velde ER. Ultrastructure of the resting ovarian follicle pool in healthy young women. Biol Reprod 2002; 66: 1151 1160 [DOI] [PubMed] [Google Scholar]
- Hirshfield AN, DeSanti AM. Patterns of ovarian cell proliferation in rats during the embryonic period and the first three weeks postpartum. Biol Reprod 1995; 53: 1208 1221 [DOI] [PubMed] [Google Scholar]
- Escobar ML, Echeverria OM, Ortiz R, Vazquez-Nin GH. Combined apoptosis and autophagy, the process that eliminates the oocytes of atretic follicles in immature rats. Apoptosis 2008; 13: 1253 1266 [DOI] [PubMed] [Google Scholar]
- Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev 1996; 17: 121 155 [DOI] [PubMed] [Google Scholar]
- Flaws JA, Hirshfield AN, Hewitt JA, Babus JK, Furth PA. Effect of bcl-2 on the primordial follicle endowment in the mouse ovary. Biol Reprod 2001; 64: 1153 1159 [DOI] [PubMed] [Google Scholar]
- Greenfeld CR, Pepling ME, Babus JK, Furth PA, Flaws JA. BAX regulates follicular endowment in mice. Reproduction 2007; 133: 865 876 [DOI] [PubMed] [Google Scholar]
- Perez GI, Robles R, Knudson CM, Flaws JA, Korsmeyer SJ, Tilly JL. Prolongation of ovarian lifespan into advanced chronological age by Bax-deficiency. Nat Genet 1999; 21: 200 203 [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6: 1221 1228 [DOI] [PubMed] [Google Scholar]
- Rucker EB, 3rd, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, Hennighausen L. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol 2000; 14: 1038 1052 [DOI] [PubMed] [Google Scholar]
- Perez GI, Tao XJ, Tilly JL. Fragmentation and death (a.k.a. apoptosis) of ovulated oocytes. Mol Hum Reprod 1999; 5: 414 420 [DOI] [PubMed] [Google Scholar]
- Mitchell PA, Burghardt RC. The ontogeny of nexuses (gap junctions) in the ovary of the fetal mouse. Anat Rec 1986; 214: 283 288 [DOI] [PubMed] [Google Scholar]
- Duerrschmidt N, Zabirnyk O, Nowicki M, Ricken A, Hmeidan FA, Blumenauer V, Borlak J, Spanel-Borowski K. Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology 2006; 147: 3851 3860 [DOI] [PubMed] [Google Scholar]
- Lobascio AM, Klinger FG, Scaldaferri ML, Farini D, De Felici M. Analysis of programmed cell death in mouse fetal oocytes. Reproduction 2007; 134: 241 252 [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol 2004; 14: 70 77 [DOI] [PubMed] [Google Scholar]