

Abstract
An economical and simplified procedure to derive and propagate fully functional lines of undifferentiated rat spermatogonia in vitro is presented. The procedure is based on the formulation of a new spermatogonial culture medium termed SG medium. The SG medium is composed of a 1:1 mixture of Dulbecco modified Eagle medium:Ham F12 nutrient, 20 ng/ml of GDNF, 25 ng/ml of FGF2, 100 μM 2-mercaptoethanol, 6 mM l-glutamine, and a 1× concentration of B27 Supplement Minus Vitamin A solution. Using SG medium, six individual spermatogonial lines were derived from the testes of six separate Sprague-Dawley rats. After proliferating over a 120-day period in SG medium, stem cells within the spermatogonial cultures effectively regenerated spermatogenesis in testes of busulfan-treated recipient rats, which transmitted the donor cell haplotype to more than 75% of progeny by natural breeding. Subculturing in SG medium did not require protease treatment and was achieved by passaging the loosely bound spermatogonial cultures at 1:3 dilutions onto fresh monolayers of irradiated DR4 mouse fibroblasts every 12 days. Spermatogonial lines derived and propagated using SG medium were characterized as homogeneous populations of ZBTB16+ DAZL+ cells endowed with spermatogonial stem cell potential.
Keywords: fertility, germ cell, germline, regenerative medicine, spermatogenesis, spermatogonia, spermatogonial, spermatozoa, stem cell, stem cells, transgenic rats
A newly formulated spermatogonial culture medium (i.e., SG medium) allows researchers to effectively and efficiently study the biology and applications of spermatogonial stem cells in the rat.
INTRODUCTION
The ability to conditionally induce mammalian spermatogonial stem cell lines to undergo the process of spermatogenesis in vitro for the production of male gametes would provide a long sought after technology holding potential to advance a wide scope of industries related to human health, animal husbandry, and conservation. The discovery that spermatogonial stem cells residing within fractions of dissociated mouse and rat testis cells maintain their ability to regenerate spermatogenesis in testes of recipient mice [1–3] was essential to the prospect of establishing culture systems that maintain spermatogenesis in vitro [4]. The ability to isolate and experimentally manipulate spermatogonial stem cells has opened new doors for research on spermatozoan development [5, 6], assisted reproduction [7–9], cellular therapy [8–10], and genetics [11–13]. In view of this potential, we and others have established protocols for isolating [12, 14–16], propagating [17, 18], and genetically modifying [12, 19] fully functional rat spermatogonial stem cells in culture. We initially chose the rat as a species for these studies because of its popularity as a laboratory animal model for the study of human health and disease and because of the lack of protocols for genetically modifying the rat germline using clonally expanded stem cells from culture [12]. Considering the many potential applications of the laboratory rat as a research model, a cost-effective and easy-to-prepare culture medium was sought in this study for the derivation and continuous proliferation of primary rat spermatogonial stem cell lines in vitro.
Toward this goal, media previously reported to support long-term proliferation of rodent spermatogonial stem cells in vitro represent clear methodological advances for studies on the biology and applications of spermatogonia [17, 18, 20–22]. However, such media are complex, expensive, and time-consuming to prepare and are most effective when applied in combination with feeder layers of fibroblasts [17, 18, 20–22]. For example, the medium originally described by Kanatsu-Shinohara and colleagues [20] for the successful derivation and long-term cultivation of germline stem cells from postnatal mouse testes was a pivotal breakthrough in spermatogonial research. However, that medium is based on the proprietary StemPro-34 medium (Invitrogen) plus 24 individually added components, including small molecules, fetal bovine serum, and a mixture of polypeptide growth factors. Serum-free derivatives of this StemPro-34-based spermatogonial medium have since been formulated for spermatogonial culture in which the serum has been replaced by the supplement B27 [17, 21]. On inspection of components within B27 supplement (Chart 1), we postulated that it could be used together with key growth factors in a commonly applied nutrient mixture to formulate a more efficient spermatogonial stem cell culture medium.

MATERIALS AND METHODS
Materials
Dispase, rat-tail collagen I-coated culture dishes, and gelatin-coated culture dishes were from Fisher, Inc. Invitrogen was our source for the following: PBS, nonessential amino acids, minimum essential medium vitamin solution, l-glutamine solution, trypsin-edetic acid (EDTA) solutions (0.05% w/v trypsin with 0.2 g/L of EDTA·4Na or 0.25% w/v trypsin with 0.38 g/L of EDTA·4Na), antibiotic antimycotic solution (catalog No. 15240–062), and Hoechst 33342- and Alexa Fluor 594-conjugated and goat anti-rabbit and goat anti-mouse IgGs. Bovine serum albumin (BSA) and dimethyl sulfoxide (DMSO) were from Calbiochem, Inc. Fetal bovine serum (FBS) for mouse embryonic fibroblast (MEF) medium was from Hyclone (catalog No. SH30071.03). Blocking reagent was from Roche Applied Biosciences. Sigma was our source for the following: mouse laminin, sodium bicarbonate, trypan blue, Dulbecco modified Eagle medium (DMEM [catalog No. D5648]), and DMEM:Ham F12 (1:1) nutrient mixture (DMEM:Ham F12 [catalog No. D8437]). Vendors and catalog numbers for all media components are listed in Supplemental Table S1 (available at www.biolreprod.org).
Animal Care and Use
Protocols for the use of rats in this study were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center in Dallas. Rats used for this study were housed in individually ventilated Lab Products 2100 cages in a dedicated room with atmosphere controls set to 72°F and 45%–50% humidity during a 12L:12D cycle (i.e., light cycle from 0600 to 1800 h Central Standard Time adjusted for daylight saving time). Rats were given a 5% fat diet (Teklad Irradiated 7912 LM-485 Mouse/Rat Diet; Harlan, Inc.) and a continuous supply of reverse osmosis water.
Isolating Enriched Fractions of Undifferentiated Spermatogonia
Seminiferous tubules were isolated from testes of 23- to 24-day-old wild-type Sprague-Dawley rats (Hsd:Sprague-Dawley SD; Harlan, Inc.) or homozygous SD-Tg(Gt[ROSA]26Sor-EGFP)2–4Reh transgenic rats. Rats of the Tg(Gt[ROSA]26Sor-EGFP)2–4Reh line were produced by pronuclear injection and are referred to as GCS-EGFP rats because they exhibit germ cell-specific (GCS) expression of enhanced green fluorescent protein (EGFP) [23]. The tubules were enzymatically and mechanically dissociated into a cellular suspension to generate cultures of testis cells in serum-containing medium as previously described [12, 16], except that a medium volume of 10 ml/rat was applied for all centrifugation and filtration steps. The testis cell cultures were then used to isolate enriched populations of laminin-binding spermatogonia according to previously published methods [12, 16] that describe how to first remove more than 95% of somatic testis cells from the germ cell population by negative selection on plastic and collagen, before positive selection for the spermatogonial stem cells based on their ability to bind to laminin (Fig. 1). By this procedure, the freshly isolated laminin-binding germ cell population contains more than 90% undifferentiated type A spermatogonia (ZBTB16+ DAZL+) in the single (∼88%) or paired (∼12%) cell state [15, 24]. Fractions of laminin-binding spermatogonia isolated by this procedure contain ∼4% somatic cells and ∼5% differentiating spermatogonia plus spermatocytes [15]. In this study, a single rat of this age range yielded a mean ± SD of 3.62 × 105 ± 0.93 × 105 laminin-binding spermatogonia (n = 6 rats), compared with a yield reported for this procedure when scaled for processing pools of testes from 22- to 24-day-old rats of 1.98 × 105 ± 0.56 × 105 cells/rat (n = 34 primary cultures) [16].
FIG. 1.

Methods and media for deriving and propagating RSGLs. Overview of selection steps to isolate laminin-binding undifferentiated spermatogonia in primary culture [16] for the derivation of RSGLs in SG medium. A modified version of this technique was previously described using a medium for spermatogonial culture that did not contain serum or vitamin A (SA medium) [17]. Components of SG and SA media are listed in Chart 1.
Derivation of Spermatogonial Lines
To derive rat spermatogonial stem cell lines, freshly isolated laminin-binding spermatogonia from individual rats were plated separately into gelatin-coated wells (3.5 cm) of a culture plate at ∼1.9 × 104 cells/cm2 in 0.37 ml/cm2 of spermatogonial culture medium (SG medium). Components of SG medium are given in Chart 1 and in Supplemental Table S1. As previously described using another spermatogonial medium, SA medium (which lacks serum and vitamin A) [17], spermatogonia cultured on gelatin using SG medium were observed loosely bound to the culture plate and to residual adherent somatic testis cells and in suspension; many of the spermatogonia in suspension adhered to each other as cellular “clusters” of variable size. In contrast, the small fraction of contaminating somatic testis cells attached avidly and spread out on the gelatin matrix. After an initial selection for 40–48 h on the gelatin-coated plates, spermatogonia in suspension (i.e., including loosely bound spermatogonia) were harvested free from the contaminating somatic testis cells by pipetting. Harvested spermatogonia were pelleted at 200 × g for 4 min, the supernatant was discarded, and the cellular pellet was suspended in SG medium and plated into fresh gelatin-coated wells (3.5 cm) for an additional 72–96 h. After this point (i.e., after depletion of essentially all somatic testis cells), suspensions of spermatogonia from each rat that survived through the final selection steps on gelatin were harvested into fresh SG medium and passaged into 2.2-cm culture wells (i.e., 12-well culture dish) containing feeder layers of irradiated MEFs. Methods for preparing MEF feeder layers are described herein. The initial passage of spermatogonial cultures after their plating onto MEF feeder layers required a 1:1 to 1:2 split into the same size wells at 14–21 days after their initial seeding onto the MEFs. Because irradiated MEF feeder layers are not as effective after 14 days in culture, fresh MEFs (2 × 104/cm2) were “spiked” into the ongoing spermatogonial cultures on Days 12–14 to bypass the need to passage the spermatogonia before expanding to larger numbers. Once established by the second or third passage on MEFs, cultures of spermatogonia were passaged at ∼1:3 dilutions onto a fresh monolayer of MEFs every 10–14 days at ∼3 × 104 cells/cm2 for longer than 5 mo (i.e., ∼12 passages). For passaging, cultures were first harvested by gently pipetting them free from the MEFs. After harvesting, the clusters of spermatogonia were dissociated by gentle trituration with 20–30 strokes through a p1000 pipette in their SG culture medium. The dissociated cells were pelleted at 200 × g for 4 min, and the number of cells recovered during each passage was determined by counting them on a hemocytometer (spermatogonial clusters were not disrupted for counting until the second passage on MEFs). As verified by expression of the GCS-EGFP marker transgene, spermatogonia were easily distinguished during counting as the predominant population of smaller round cells with smooth surfaces, compared with occasionally observed larger and often irregular-shaped irradiated MEFs. All culture steps for deriving and propagating spermatogonial lines when in SG medium were performed at 37°C and 5% CO2. The doubling time for the number of GCS-EGFP+ cells that could be harvested from cultures of each spermatogonial line after subsequent passages between Days 30 and 150 in culture on MEFs was calculated by nonlinear regression analysis using the least squares fit model set for automatic outlier exclusion provided as the exponential growth equation in the GraphPad Prism program (version 5.01; GraphPad Software, Inc.).
Preparation of Fibroblast Feeder Layers
Primary stocks of DR4 MEFs were purchased from ATCC, Inc., and were expanded in DMEM supplemented with 1.5 g/L of sodium bicarbonate and 15% heat-inactivated FBS (MEF medium) at 37°C and 5% CO2 for up to 4 passages following their thawing and initial plating (i.e., passage 0) from the vial received from the manufacturer. Following expansion to passages 3 and 4, secondary stocks of MEFs were irradiated (120 Gy) and then cryopreserved in liquid nitrogen for future use according to the manufacturer's protocol. Before use for culture with spermatogonia, the MEFs were thawed and plated into gelatin-coated dishes (4.5 × 105 cells/cm2) in MEF medium for 16–48 h, rinsed one time with PBS, and then preincubated in SG medium for an additional 16–48 h. The SG medium used for preincubation was then discarded, and spermatogonia were passaged onto the MEFs in fresh SG medium.
Germ Cell Transplantation and Progeny Genotyping
Wild-type Sprague-Dawley rats at age 12 days were injected (i.p.) with 12.5 mg/kg of busulfan (4 mg/ml in 50% DMSO) and then used as recipient males at age 24 days. Busulfan is a spermatogonial toxin commonly used to kill spermatogonia in recipient rat testes before transplantation because it increases the colonization efficiency by the donor stem cells [12, 25, 26]. Donor cells from rat spermatogonial lines GCS9 (recipients R880–R884) and GCS10 (recipients R898–R901) were loaded into injection needles fashioned from 1000-μl glass capillary tubes at a concentration of 3 × 105 cells/65 μl of SG medium containing 0.04% (w/v) trypan blue, and then the entire volume was transplanted into the seminiferous tubules of anesthetized rats by retrograde injection through the rete testes [25, 27]. RSGL-GCS9 was harvested from passage 10, which corresponded to 120 days in culture before their transplantation. RSGL-GCS10 was harvested from passage 9, which corresponded to 111 days in culture before their transplantation. At the indicated date of analysis, recipient males were killed, and their testes were dissected out to analyze relative levels of donor cell-derived spermiogenesis based on the percentage of tubular cross-sections colonized by green fluorescent elongating spermatids. Only the right testes of R880 and R898 were transplanted. Both testes from R880 and R898 were isolated for analysis at Days 64 and 75 after transplantation, respectively, for analytical purposes. Recipient males transplanted with GCS-EGFP spermatogonia were paired with wild-type female Sprague-Dawley rats of similar age at 75 days after transplantation. Transgenic rat progeny from wild-type recipients and wild-type females were determined by quantitative PCR analysis of genomic DNA using primers specific to the GCS-EGFP transgene and the 18S ribosomal subunit; relative transgene copy numbers in F2 progeny from hemizygous crosses were verified by Southern dot blot hybridization analysis of the genomic DNA using a probe specific for the GCS-EGFP transgene. Genotyping results were also confirmed in representative progeny by direct visualization of transgene expression in testes and ovaries using a Nikon SMZ1500 fluorescence stereomicroscope.
Immunocytochemistry
Cultures of germ cells (2 cm2) were washed twice with DMEM:Ham F12 medium (0.6 ml/well) and then fixed for 7.5 min with 4% paraformaldehyde and 1 M sodium phosphate (pH 7.2) (0.4 ml/well). After fixation, the cells were washed three times with PBS (0.6 ml/well) and then incubated for 15 min in PBS containing 0.1% (v/v) Triton X-100 (0.4 ml/well). The cells were then washed three times in PBS (0.6 ml/well), and nonspecific protein-binding sites were blocked by incubating the cells in 0.1% w/v blocking reagent (0.4 ml/well; Roche Applied Biosciences]) for 1.5 h at 22–24°C. The blocking reagent was then removed, and the cells were incubated for 16 h at 22–24°C in primary antibodies (0.4 ml/well). The mouse anti-human ZBTB16 IgG (Calbiochem, Inc.) and the purified nonimmune mouse IGHG1 (Santa Cruz, Inc.) fractions were each diluted to 1 μg/ml in blocking reagent. The anti-DAZL-3 IgG and the preimmune-3 IgG fractions [12] were diluted to 250 ng/ml in blocking reagent. Following incubation in primary antibodies, the cells were washed three times for 5 min with 0.6 ml/well of 10 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween-20 (pH 7.5) (TBST) to remove unbound IgG. The cells were then incubated for 40 min at 22–24°C in conjugated secondary antibody (0.4 ml/well) diluted to 4 μg/ml in PBS containing 5 μg/ml of Hoechst 33342 dye. Following incubation in secondary antibodies, the cells were washed three times for 5 min with TBST (0.6 ml/well) to remove unbound IgG and dye before viewing in fresh PBS (0.4 ml/well) using an inverted Olympus IX70 microscope (Olympus, Inc.).
RESULTS
Effects of Key Components in Rat Spermatogonial Culture Media
To formulate a more efficient and economical medium for propagating rat spermatogonia in culture, we proceeded to evaluate distinct groups of components in one such culture medium for rat spermatogonia that lacks serum and vitamin A (i.e., SA medium [Chart 1]) [17]. Although SA medium was effective for culturing primary germlines derived from enriched fractions of laminin-binding undifferentiated spermatogonia (Fig. 2A) [17], several added components are redundant in SA medium because of their presence in the B27 supplement (i.e., insulin, transferrin, selenium, putrescine, progesterone, biotin, and BSA) (Chart 1). Therefore, these redundant components were subtracted from SA medium to yield SR medium, which was then tested for the ability to support the proliferation of a previously established spermatogonial line derived from GCS-EGFP transgenic rats using SA medium [17]. The transgenic rat line GCS-EGFP was named based on its germline-specific expression of EGFP during all stages of male and female gametogenesis [23] and has provided a unique reagent for studying spermatogonia in culture [15–17, 24]. Based on growth rates of GCS-EGFP spermatogonia in vitro on feeder layers of MEFs (Fig. 2A), as well as in vivo spermatogenesis colony-forming assays (Fig. 2C, left), no significant differences were observed between SA and SR media.
TABLE 1.
Germline transmission from rat spermatogonial lines GCS9 and GCS10 by natural breeding of recipient founders.

FIG. 2.

Growth factor requirements for SA medium. A) Growth curves of spermatogonia maintained in SA and SR media. Growth curves were obtained based on the yield of GCS-EGFP+ germ cells harvested from cultures at times of passaging. B) Growth curves of spermatogonia maintained in SR medium, SR medium without LIF and EGF (SR-LE), SR-LE without GDNF (SR-LEG), and SR-LE without FGF2 (SR-LEF). C) In vivo spermatogenesis colony-forming potential of a spermatogonial line derived from GCS-EGFP rats using SA medium. The line was maintained in SA medium for ∼6 mo and then switched to fresh SA, SR, and SR-LE media for an additional 2 mo (left) or switched to fresh SR-LE, SR-LEG, and SR-LEF media for an additional week (right) before transplantation. Spermatogonia from each culture condition were transplanted into rat testes, and the resulting numbers of EGFP+ spermatogenic colonies per testis were scored 1–2 mo later. Numbers of colonies formed per testis per 104 germ cells transplanted are plotted as the mean ± SEM (n). SR-LE medium without GDNF (SR-LEG) significantly reduced colony formation, compared with SR-LE at the 1-wk time point; *P < 0.02 by two-tailed Student t-test.
Media used for propagating rat spermatogonial stem cells contain different combinations of glial cell line-derived neurotrophic factor (GDNF), fibroblast growth factor 2 (FGF2 [also known as basic FGF]), epidermal growth factor (EGF), leukemia inhibitory factor (LIF), and soluble GDNF receptor alpha 1 (GFRA1) [17, 18]. Therefore, we next assessed the necessity of these factors in SR medium. While the removal of EGF and LIF from SR medium (i.e., SR-LE medium [Chart 1]) did not seem to affect spermatogonial proliferation in vitro (Fig. 2B), nor the spermatogenesis colony-forming potential of the cultures when transplanted into testes (Fig. 2C, right), the removal of these two factors together with the removal of FGF2 or GDNF from SR medium had clear detrimental effects on spermatogonial proliferation in vitro (Fig. 2B) and on the colony-forming potential of the spermatogonia in vivo (Fig. 2C, right). Therefore, added EGF and LIF did not seem beneficial for propagating the rat spermatogonial line (RSGL) in SR medium, which (like SA medium) did not contain GFRA1. Accordingly, the combination of GDNF and FGF2 was most effective at stimulating proliferation of rat spermatogonia in SR medium.
Formula and Effects of the New SG Medium
In agreement with these findings, a DMEM:Ham F12 nutrient mixture supplemented with GDNF, FGF2, B27 Supplement Minus Vitamin A, l-glutamine, and 2-mercaptoethanol was found to support the continued propagation (>2 million-fold expansion in cell number) of a previously established RSGL on MEFs following its initial derivation, propagation, and cryopreservation in SA medium (Fig. 3A). The newly formulated spermatogonial medium was termed SG medium and further eliminated the need to add minimum essential medium vitamins, estradiol, pyruvate, lactate, ascorbate, nonessential amino acids, glucose, and StemPro supplement (Chart 1). The SG medium was also used at a 100% success rate to derive new proliferating spermatogonial lines from individual wild-type rats (n = 3) and homozygous GCS-EGFP transgenic rats (n = 3) on a Sprague-Dawley background by our previously established methods (Fig. 3B) [17] but without the need to further enrich the starting spermatogonial population by flow cytometry or magnetic cell sorting techniques. The proliferating germlines derived in SG medium were characterized as undifferentiated spermatogonia based on coexpression of the marker proteins ZBTB16 (previously know as PLZF) and DAZL (Fig. 3C) [28, 29] and on their ability to effectively colonize the seminiferous tubules of busulfan-treated rats (Fig. 3D). The lines were subcultured in SG medium by pipetting and did not require protease treatment. The newly derived lines showed mean ± SD doubling times of 8.4 ± 0.2 days (n = 4) for the total GCS-EGFP+ germ cell population when exponential growth curves were fit between Culture Days 30 and 150 after their initial seeding onto MEFs in SG medium as freshly isolated laminin-binding spermatogonia. For comparison, spermatogonial lines derived and propagated in SA medium showed mean ± SD doubling times of 6.5 ± 1.8 days (n = 4) for the total GCS-EGFP+ population when analyzed between Culture Days 30 and 150 after their initial seeding onto MEFs.
FIG. 3.

A new medium for deriving and propagating RSGLs. A) Growth rate of an established RSGL during culture in SA and SG media. Spermatogonial line RSGL-GCS3 was initially derived from GCS-EGFP rats and was propagated for 100 days in SA medium before being cryopreserved in SA freezing medium [17]. The line was thawed following a 5–6 mo (i.e., 172 days) storage period at −196°C, plated directly into SG medium on MEFs, and then expanded for an additional 100 days in SG medium. Plots start during the culture phase on MEFs at passage 2. B) Growth rates of RSGLs derived in SG medium. Left: Spermatogonial lines (RSGL-WT1 and RSGL-WT2) derived from wild-type Sprague-Dawley rats. Right: Spermatogonial lines (RSGL-GCS9 and RSGL-GCS10) derived from transgenic GCS-EGFP Sprague-Dawley rats. Plots start during the culture phase on MEFs at passages 3–5. C) Expression of ZBTB16 and DAZL by spermatogonial line GCS9 after proliferating in SG medium for 111 days (passage 9) on feeder layers of MEFs. Coexpression of ZBTB16, DAZL, and the GCS-EGFP transgene was determined by immunocytochemistry, which characterized the spermatogonial lines as undifferentiated spermatogonia. Sg, spermatogonia; M, MEFs. D) Colonization of rat seminiferous tubules by RSGL-GCS9 after proliferating in culture in SG medium for 120 days (i.e., passage 10). Shown are green fluorescent (top) and brightfield (bottom) images of testes from R880 at 64 days following transplantation with (R indicates right testis) or without (L indicates left testis) cells from line GCS9.
To determine the spermatogenic potential of the new RSGLs that were derived using SG medium, RSGL-GCS9 and RSGL-GCS10 from GCS-EGFP rats were propagated for 111 and 120 days in culture, respectively, for a total of 9–10 passages before being transplanted into testes of busulfan-treated wild-type rats at ∼3 × 105 cells/testis (i.e., after expanding in cell number by >20 000-fold after their initial seeding onto MEFs) (Table 1). At 75 days after transplantation, recipients of RSGL-GCS9 and RSGL-GCS10 were paired with wild-type females, with whom they sired a total of 28 litters between 104 and 263 days after transplantation (Table 1). On average, recipients of RSGL-GCS9 and RSGL-GCS10 yielded a mean ± SD of 78.9% ± 10.4% and 67.2% ± 16.4% germline transmissions, respectively, of the GCS-EGFP transgene to F1 progeny from spermatozoa produced by the donor stem cells (19 litters and 193 total pups for GCS9 recipients vs. 9 litters and 107 total pups for GCS10 recipients) (Fig. 4A and Table 1). Resulting nonmendelian ratios (i.e., <100% transgenic F1 progeny) were due to competition from residual wild-type spermatozoa produced by the recipients (Fig. 4A and Table 1). However, transmission of the GCS-EGFP transgene to F2 progeny from crosses between hemizygous F1 cousins yielded mendelian ratios (21% wild-type, 51% hemizygous, and 28% homozygous [3 litters and 47 total pups]) (Fig. 4, B and C).
FIG. 4.

Functional analysis of RSGLs derived in SG medium. A) Germline transmission from RSGL-GCS9 following its development into functional spermatozoa in recipient rat testes. Recipient/founder rats were transplanted with cells from line GCS9 after proliferating for 120 days in SG medium. Recipients were then paired with wild-type female rats 75 days after transplantation. Transmission of the donor cell haplotype to progeny was based on inheritance of the GCS-EGFP transgene (Table 1). B) Transmission of donor cell haplotypes from wild-type recipient/founders (F0) R883 and R901, which were transplanted with lines GCS9 and GCS10, respectively. Spermatogonial lines GCS9 and GCS10 were derived from siblings that were each homozygous for the GCS-EGFP transgene. Thus, F1 progeny represent cousins, some of which were crossed to rederive transgenic F2 progeny homozygous for the TgGCS-EGFP allele. C) Stable transmission of donor stem cell transgene to F2 progeny. Expression of the GCS-EGFP transgene (green fluorescence) in testes (top) and ovaries (bottom) of wild-type (−/−) and hemizygous (−/+) F2 progeny derived from a cross between hemizygous F1 progeny from recipients R883 and R901. For testes, the image shown in the left panel represents brightfield microscopy of the same specimen shown in the right panel by green fluorescent microscopy. Bars = 1 cm in images of testes and 100 μm in images of ovaries. D) Spermatogonial lines derived in SG medium show long-term spermatogenesis colony-forming potential. Left: Expression of the GCS-EGFP transgene in the testes of wild-type recipient R881 at 263 days following transplantation with RSGL-GCS9. Bar = 1 cm. Right: Histological testis sections from recipient R881 at 263 days after transplantation (TP) with line GCS9. Representative seminiferous tubules show normal spermatogenesis regenerated from the donor cells (green [EGFP]), which express markers for different generations of spermatogenic cells (red [immunolabeling for SYCP3, DAZL, and CREM tau]). Bar = 50 μm.
Testes from recipients transplanted with RSGL-GCS9 and RSGL-GCS10 were next analyzed histologically for long-term spermatogenesis colony-forming potential (Fig. 4D and Table 1). When evaluated at 206–263 days following transplantation, a mean ± SEM of 92.5% ± 2.4% (n = 4) and 81.5% ± 9.5% (n = 3) of seminiferous tubule cross-sections that were colonized by RSGL-GCS9 and RSGL-GCS10, respectively, showed development of EGFP+ spermatogonia to the elongating spermatid stage (Fig. 4D and Table 1). Therefore, spermatogonial lines derived in SG medium were classified as essentially pure cultures of undifferentiated spermatogonia containing fully functional spermatogonial stem cells. Moreover, when the product of the rate of change in total germ cell number for RSGL-GCS9 (Fig. 5A) and the mean rate of change in spermatogenesis colony-forming potential for RSGL-GCS9 (Fig. 5A) was plotted as a function of culture time in SG medium (Fig. 5B), it was evident that rat spermatogonial stem cells were also proliferating within the cultures of undifferentiated spermatogonia (Fig. 5C). Most important to future applications of the spermatogonial lines derived in this study, the resulting >4000-fold increase in spermatogonial stem cell number yielded by their proliferation in SG medium was achieved using a thawed stock of RSGL-GCS9 that had previously been preserved at −196°C for 384 days in SG medium containing 10% DMSO (i.e., SG freezing medium) (Fig. 5).
FIG. 5.
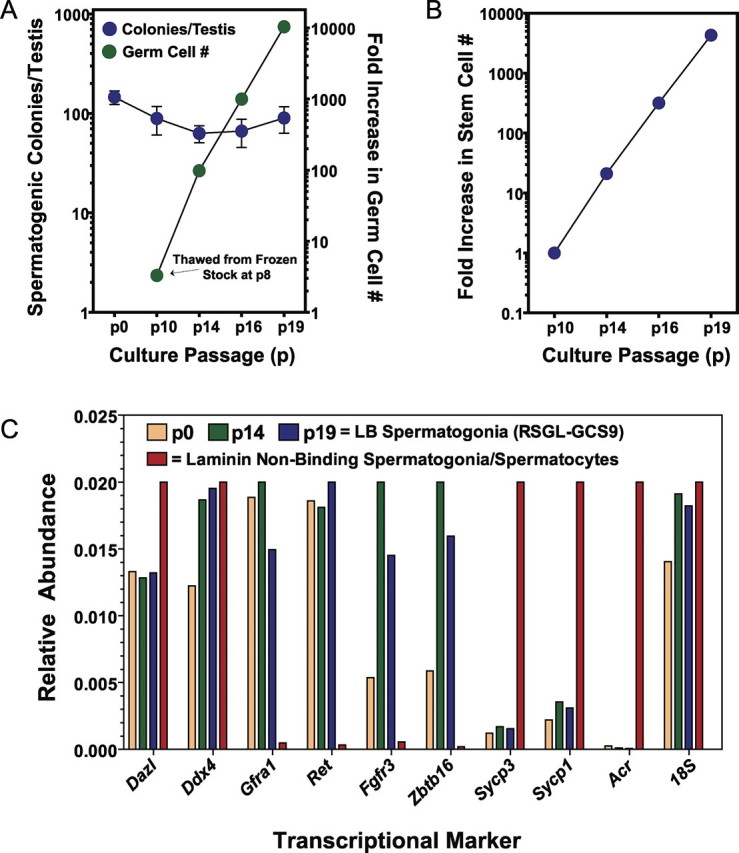
The SG medium supports proliferation of rat spermatogonial stem cells. A) Growth rate and in vivo spermatogenesis colony-forming activity of RSGL-GCS9 after thawing from cryopreserved stocks at passage 8 and continued subculturing over subsequent passages in SG medium. Aliquots of GCS-EGFP+ cells harvested at passages (p) 0, 10, 14, 16, and 19 were transplanted into recipient rats to determine the number of GCS-EGFP+ colonies formed per testis per 10 000 donor cells at 30 days after transplantation [16], which were a mean ± SD of 146 ± 56 at p0, 89 ± 69 at p10, 63 ± 30 at p14, 66 ± 51 at p16, and 90 ± 65 at p19 (n = 6–7 transplanted testes scored per point). Cells from p0 represent freshly isolated laminin-binding spermatogonia isolated from 23- to 24-day-old rats. Time lapsed between p8, p10, p14, p16, and p19 was 25, 40, 24, and 33 days, respectively (i.e., 122 total days after thaw at p8). B) Fold increase in spermatogonial stem cell number as a function of culture time for RSGL-GCS9 in SG medium. The change in stem cell number was calculated by multiplying the change in total germ cell number by the change in number of spermatogenic colonies formed per testis per 104 donor cells transplanted from p10, p14, p16, and p19 cultures that were propagated in SG medium. Values plotted were from A and were normalized to 1 at p10. C) Transcriptional profile of RSGL-GCS9 at p10 and p19 compared with freshly isolated p0 laminin-binding (LB) spermatogonia and laminin-nonbinding spermatogonia/spermatocytes using molecular markers for spermatogenic cells as determined by real-time PCR [17]. Rat Ddx4 (also known as rat Vasa-like gene) and Dazl mRNAs are specifically expressed by germ cells; rat Gfra1, Ret, Fgfr3, and Zbtb16 mRNAs are enriched in undifferentiated spermatogonia; rat Sycp1, Sycp3, and Acr mRNAs are markers for differentiating spermatogonia and spermatocytes.
DISCUSSION
The present study describes the use of SG medium in technically straightforward, highly reproducible, and cost-effective methods to derive and cultivate primary RSGLs that maintain their ability to effectively develop into functional spermatozoa when transplanted back into rat testes. The process of derivation and cultivation of primary RSGLs in SG medium does not require fluorescence or magnetic cell sorting and can be achieved with the testes of a single animal using basic cell culture techniques and reagents (Fig. 1). Subculturing in SG medium does not require protease treatment, and SG medium supplemented with 10% DMSO can be used to cryopreserve functional rat spermatogonial stem cell lines for periods longer than 1 yr (Figs. 3–5). Formulation of SG medium can be made with a routinely applied base medium plus six added commercially available components. This saves valuable time and money relative to other media used to derive and propagate RSGLs [17, 18]. For example, the defined rat spermatogonial medium rat serum free-culture medium (RSFM) [18] was formulated based on modifications to a defined medium initially reported for the propagation of mouse spermatogonia [22]. The RSFM is composed of 20 individually added components and, based on transplantation assays, supported the propagation of functional rat spermatogonial stem cells for up to 7 mo in culture on feeder layers of the STO SNL76/6 fibroblast line (doubling time for stem cells, ∼10.8 days) [18]. Such fully defined serum-free media should prove beneficial for the controlled study and optimization of culture conditions favorable for spermatogonial renewal and differentiation in vitro. Therefore, it will be important to try to increase the proliferation rate of spermatogonia in new formulas of defined media by determining if distinct components in RSFM and SG medium are cooperative.
A second rat spermatogonial medium (i.e., SA medium) supported increased rates in the expansion of germ cell numbers in culture [17]. A doubling time of ∼3.5 days was estimated for this spermatogonial line at time points after 150 days in culture [17]. Based on this doubling time, the total spermatogonial population expanded in number ∼2.4 times faster in SA medium than in SG medium (i.e., total spermatogonial doubling time, ∼8.4 days in SG medium). However, when analyzed between Culture Days 30 and 150 after plating onto MEFs, spermatogonial lines derived in SA medium showed on average 6.5-day doubling times for the total spermatogonial population. Although SA medium is complex and time-consuming to prepare and costs about three times more per liter than SG medium, undifferentiated spermatogonia can be expanded in number ∼1.3 times faster in SA medium than in SG medium during the first 5 mo of culture. These data indicate that the rate at which rat spermatogonial numbers expand over multiple passages in culture could potentially be accelerated by making simple modifications to SG medium. If this small change in the rate of cell proliferation was achieved using SG medium, it would translate into substantial conservation of resources when applied to an exponential growth scale (i.e., ∼16 000-fold vs. ∼250 000-fold expansion in spermatogonial number over a 120-day period). As shown in Figure 5 and as reported for RSFM [18], media modifications that speed up rat spermatogonial proliferation in vitro can be tested for their effectiveness at accelerating the proliferation of functional spermatogonial stem cells within culture. Such modifications may include the addition of components that 1) increase the rate of renewing stem cell divisions, 2) block the loss of stem cell numbers due to differentiation, 3) enhance the survival of the stem cells as they proliferate in culture, and 4) increase the plating efficiency of stem cells that survive each passage.
Various combinations of growth factors are utilized for propagating mouse and RSGLs in serum-free culture media, including LIF, EGF, FGF2, GDNF, and soluble GFRA1 [17, 18, 21, 22]. Instrumental to our knowledge on how to propagate spermatogonia in culture was a study [30] showing the progressive loss of undifferentiated spermatogonia in mice made haplodeficient in the expression of GDNF. Indeed, GDNF has since proven to be an essential growth factor for propagating rodent spermatogonial stem cells in vitro, and depending on the genetic background of the test species, GDNF is maximally effective when used in combination with FGF2 [18, 22]. Herein, we show the combination of GDNF and FGF2 to be most effective at supporting the proliferation of RSGLs in a simplified formula of SA medium lacking EGF and LIF (Chart 1 and Fig. 2). Furthermore, GDNF and FGF2 were added as components of SG medium to effectively propagate fully functional RSGLs in culture (Figs. 3–5 and Table 1). As shown herein with RSGLs derived in SG medium (Fig. 5), the maintenance of rat spermatogonial stem cells in culture is tightly associated with the expression of spermatogonial transcripts encoding known receptor subunits for GDNF and FGF2 [15, 17]. This is evidenced by the rapid down-regulation of Gfra1, Ret, and Fgfr3 transcripts in cultures of undifferentiated rat spermatogonia after being stimulated to initiate spermatogenesis in vitro [15]. Based on transplantation assays, these effects on gene expression were linked to factors stimulating a loss in total numbers of spermatogonial stem cells in the face of increasing spermatogonial numbers within the cultures [15, 24]. Thus, the combined effects of GDNF and FGF2 regulate pathways required to expand the numbers of rodent spermatogonial stem cells during culture in vitro [31–34] and to maintain the process of spermatogenesis in vivo [30, 35].
Given the importance of the laboratory rat as a model for the study of spermatogenesis and fertility, as well as its roles as a physiological model in essentially every other field of research on human health and disease, a concerted effort should be made to further optimize culture conditions for propagating and genetically modifying rat spermatogonial stem cells in vitro. Most important, because of apparent conservation in the functional roles of key growth factors in mammalian spermatogonial stem cell biology [3, 36], SG medium could serve as an additional guide for culturing undifferentiated spermatogonia from a variety of species. When transplanted into the environment of mouse seminiferous tubules, donor spermatogonia from rats [3] and hamsters [36] are able to proliferate and undergo spermatogenesis to produce spermatozoa. In addition, donor spermatogonia from rabbits, dogs, bulls, horses, and pigs are able to proliferate or develop into colonies of aligned spermatogonia within seminiferous tubules of mice, although they do not complete spermatogenesis [37, 38]. In fact, GDNF has already been reported as a component in media used to stimulate the proliferation of hamster and bovine spermatogonial stem cells during culture in vitro [39, 40].
Establishing culture conditions for propagating functional spermatogonial stem cells from these species will provide the foundation for conditionally stimulating their subsequent development through the process of spermatogenesis in vitro. In addition to providing much needed in vitro models for studying the molecular basis of spermatogenesis (and thus fertility), such culture systems would clearly have the potential to advance genetic research by spurring the invention of new protocols in transgenesis that do not rely on embryonic stem cells and the micromanipulation of embryos [11]. As described herein with the use of SG medium, establishment of such technically amenable protocols based on the nature of spermatogonia would theoretically broaden the means of the biomedical research community to study gene function in their species of choice, including the laboratory rat.
Supplementary Material
Acknowledgments
We thank Bray Denard, Tuyetanh Nguyen, and James Shirley for their help with these experiments. We also thank Dr. Robert E. Hammer for his help with these experiments and for his analysis of the manuscript before submission.
Footnotes
1Supported by a grant from the National Center for Research Resources to F.K.H. (NIH: 1R21RR023958) and by the Cecil H. & Ida Green Center for Reproductive Biology Sciences. A portion of these studies was also funded by the Howard Hughes Medical Institute to D.L. Garbers before his death.
REFERENCES
- Brinster RL, Avarbock MR. Germline transmission of donor haplotype following spermatogonial transplantation. Proc Natl Acad Sci U S A 1994; 91: 11303 11307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Zimmermann JW. Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A 1994; 91: 11298 11302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouthier DE, Avarbock MR, Maika SD, Hammer RE, Brinster RL. Rat spermatogenesis in mouse testis. Nature 1996; 381: 418 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano M, Avarbock MR, Leonida EB, Brinster CJ, Brinster RL. Culture of mouse spermatogonial stem cells. Tissue Cell 1998; 30: 389 397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahato D, Goulding EH, Korach KS, Eddy EM. Spermatogenic cells do not require estrogen receptor-alpha for development or function. Endocrinology 2000; 141: 1273 1276 [DOI] [PubMed] [Google Scholar]
- Mahato D, Goulding EH, Korach KS, Eddy EM. Estrogen receptor-alpha is required by the supporting somatic cells for spermatogenesis. Mol Cell Endocrinol 2001; 178: 57 63 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Dobrinski I, Avarbock MR, Brinster RL. Transplantation of male germ line stem cells restores fertility in infertile mice. Nat Med 2000; 6: 29 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann BP, Sukhwani M, Lin CC, Sheng Y, Tomko J, Rodriguez M, Shuttleworth JJ, McFarland D, Hobbs RM, Pandolfi PP, Schatten GP, Orwig KE. Characterization, cryopreservation, and ablation of spermatogonial stem cells in adult rhesus macaques. Stem Cells 2007; 25: 2330 2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shao S, Meistrich ML. The radiation-induced block in spermatogonial differentiation is due to damage to the somatic environment, not the germ cells. J Cell Physiol 2007; 211: 149 158 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, Baba S, Kato T, Kazuki Y, Toyokuni S, Toyoshima M, Ogura A, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell 2004; 119: 1001 1012 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ikawa M, Takehashi M, Ogonuki N, Miki H, Inoue K, Kazuki Y, Lee J, Toyokuni S, Oshimura M, Ogura A, Shinohara T. Production of knockout mice by random or targeted mutagenesis in spermatogonial stem cells. Proc Natl Acad Sci U S A 2006; 103: 8018 8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamra FK, Gatlin J, Chapman KM, Grellhesl DM, Garcia JV, Hammer RE, Garbers DL. Production of transgenic rats by lentiviral transduction of male germ-line stem cells. Proc Natl Acad Sci U S A 2002; 99: 14931 14936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano M, Brinster CJ, Orwig KE, Ryu BY, Avarbock MR, Brinster RL. Transgenic mice produced by retroviral transduction of male germ-line stem cells. Proc Natl Acad Sci U S A 2001; 98: 13090 13095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu BY, Orwig KE, Kubota H, Avarbock MR, Brinster RL. Phenotypic and functional characteristics of spermatogonial stem cells in rats. Dev Biol 2004; 274: 158 170 [DOI] [PubMed] [Google Scholar]
- Hamra FK, Schultz N, Chapman KM, Grellhesl DM, Cronkhite JT, Hammer RE, Garbers DL. Defining the spermatogonial stem cell. Dev Biol 2004; 269: 393 410 [DOI] [PubMed] [Google Scholar]
- Hamra FK, Chapman KM, Wu Z, Garbers DL. Isolating highly pure rat spermatogonial stem cells in culture. Methods Mol Biol 2008; 450: 163 179 [DOI] [PubMed] [Google Scholar]
- Hamra FK, Chapman KM, Nguyen DM, Williams-Stephens AA, Hammer RE, Garbers DL. Self renewal, expansion, and transfection of rat spermatogonial stem cells in culture. Proc Natl Acad Sci U S A 2005; 102: 17430 17435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu BY, Kubota H, Avarbock MR, Brinster RL. Conservation of spermatogonial stem cell self-renewal signaling between mouse and rat. Proc Natl Acad Sci U S A 2005; 102: 14302 14307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwig KE, Avarbock MR, Brinster RL. Retrovirus-mediated modification of male germline stem cells in rats. Biol Reprod 2002; 67: 874 879 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 2003; 69: 612 616 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Miki H, Inoue K, Ogonuki N, Toyokuni S, Ogura A, Shinohara T. Long-term culture of mouse male germline stem cells under serum- or feeder-free conditions. Biol Reprod 2005; 72: 985 991 [DOI] [PubMed] [Google Scholar]
- Kubota H, Avarbock MR, Brinster RL. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc Natl Acad Sci U S A 2004; 101: 16489 16494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronkhite JT, Norlander C, Furth JK, Levan G, Garbers DL, Hammer RE. Male and female germline specific expression of an EGFP reporter gene in a unique strain of transgenic rats. Dev Biol 2005; 284: 171 183 [DOI] [PubMed] [Google Scholar]
- Hamra FK, Chapman KM, Nguyen D, Garbers DL. Identification of neuregulin as a factor required for formation of aligned spermatogonia. J Biol Chem 2007; 282: 721 730 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Dobrinski I, Brinster RL. Recipient preparation is critical for spermatogonial transplantation in the rat. Tissue Cell 1999; 31: 461 472 [DOI] [PubMed] [Google Scholar]
- Ryu BY, Orwig KE, Avarbock MR, Brinster RL. Stem cell and niche development in the postnatal rat testis. Dev Biol 2003; 263: 253 263 [DOI] [PubMed] [Google Scholar]
- Dym M. The mammalian rete testis: a morphological examination. Anat Rec 1976; 186: 493 523 [DOI] [PubMed] [Google Scholar]
- Buaas FW, Kirsh AL, Sharma M, McLean DJ, Morris JL, Griswold MD, de Rooij DG, Braun RE. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet 2004; 36: 647 652 [DOI] [PubMed] [Google Scholar]
- Reijo RA, Dorfman DM, Slee R, Renshaw AA, Loughlin KR, Cooke H, Page DC. DAZ family proteins exist throughout male germ cell development and transit from nucleus to cytoplasm at meiosis in humans and mice. Biol Reprod 2000; 63: 1490 1496 [DOI] [PubMed] [Google Scholar]
- Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, Hess MW, Raatikainen-Ahokas A, Sainio K, Rauvala H, Lakso M, Pichel JG, Westphal H, Saarma M, Sariola H. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 2000; 287: 1489 1493 [DOI] [PubMed] [Google Scholar]
- Lee J, Kanatsu-Shinohara M, Inoue K, Ogonuki N, Miki H, Toyokuni S, Kimura T, Nakano T, Ogura A, Shinohara T. Akt mediates self-renewal division of mouse spermatogonial stem cells. Development 2007; 134: 1853 1859 [DOI] [PubMed] [Google Scholar]
- Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc Natl Acad Sci U S A 2006; 103: 9524 9529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley JM, Brinster RL. Regulation of spermatogonial stem cell self-renewal in mammals. Annu Rev Cell Dev Biol 2008; 24: 263 286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braydich-Stolle L, Kostereva N, Dym M, Hofmann MC. Role of Src family kinases and N-Myc in spermatogonial stem cell proliferation. Dev Biol 2007; 304: 34 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jijiwa M, Kawai K, Fukihara J, Nakamura A, Hasegawa M, Suzuki C, Sato T, Enomoto A, Asai N, Murakumo Y, Takahashi M. GDNF-mediated signaling via RET tyrosine 1062 is essential for maintenance of spermatogonial stem cells. Genes Cells 2008; 13: 365 374 [DOI] [PubMed] [Google Scholar]
- Ogawa T, Dobrinski I, Avarbock MR, Brinster RL. Xenogeneic spermatogenesis following transplantation of hamster germ cells to mouse testes. Biol Reprod 1999; 60: 515 521 [DOI] [PubMed] [Google Scholar]
- Dobrinski I, Avarbock MR, Brinster RL. Transplantation of germ cells from rabbits and dogs into mouse testes. Biol Reprod 1999; 61: 1331 1339 [DOI] [PubMed] [Google Scholar]
- Dobrinski I, Avarbock MR, Brinster RL. Germ cell transplantation from large domestic animals into mouse testes. Mol Reprod Dev 2000; 57: 270 279 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Muneto T, Lee J, Takenaka M, Chuma S, Nakatsuji N, Horiuchi T, Shinohara T. Long-term culture of male germline stem cells from hamster testes. Biol Reprod 2008; 78: 611 617 [DOI] [PubMed] [Google Scholar]
- Aponte P, Soda T, Teerds K, Mizrak S, van de Kant H, de Rooij D. Propagation of bovine spermatogonial stem cells in vitro. Reproduction 2008; 136: 543 547 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.