

Abstract
Genetic deficiency in granulocyte-macrophage colony-stimulating factor (CSF2, GM-CSF) results in altered placental structure in mice. To investigate the mechanism of action of CSF2 in placental morphogenesis, the placental gene expression and cell composition were examined in Csf2 null mutant and wild-type mice. Microarray and quantitative RT-PCR analyses on Embryonic Day (E) 13 placentae revealed that the Csf2 null mutation caused altered expression of 17 genes not previously known to be associated with placental development, including Mid1, Cd24a, Tnfrsf11b, and Wdfy1. Genes controlling trophoblast differentiation (Ascl2, Tcfeb, Itgav, and Socs3) were also differentially expressed. The CSF2 ligand and the CSF2 receptor alpha subunit were predominantly synthesized in the placental junctional zone. Altered placental structure in Csf2 null mice at E15 was characterized by an expanded junctional zone and by increased Cx31+ glycogen cells and cyclin-dependent kinase inhibitor 1C (CDKN1C+, P57Kip2+) giant cells, accompanied by elevated junctional zone transcription of genes controlling spongiotrophoblast and giant cell differentiation and secretory function (Ascl2, Hand1, Prl3d1, and Prl2c2). Granzyme genes implicated in tissue remodeling and potentially in trophoblast invasion (Gzmc, Gzme, and Gzmf) were downregulated in the junctional zone of Csf2 null mutant placentae. These data demonstrate aberrant placental gene expression in Csf2 null mutant mice that is associated with altered differentiation and/or functional maturation of junctional zone trophoblast lineages, glycogen cells, and giant cells. We conclude that CSF2 is a regulator of trophoblast differentiation and placental development, which potentially influences the functional capacity of the placenta to support optimal fetal growth in pregnancy.
Keywords: cytokines, gene regulation, placenta, pregnancy, trophoblast
Mice with a null mutation in the Csf2 gene have altered placental gene expression as well as dysregulated differentiation and/or functional maturation of trophoblast glycogen cells and giant cells in the placental junctional zone.
INTRODUCTION
The placenta is essential for the growth and development of the fetus, and its function is a key determinant of postnatal health [1]. It acts to transfer nutrients, oxygen, and wastes between the mother and fetus; to modulate maternal physiology; to prevent immunological rejection of the conceptus; and to sequester a uterine blood supply. This functional diversity depends on multiple placental trophoblast cell lineages that perform specialized roles [2–4]. These arise from the proliferation and differentiation of trophoblast stem cell precursors that organize into specific layered structures as gestation proceeds. Precise spatial and temporal regulation of the morphogenic process is required to control trophoblast differentiation along distinct lineage pathways. Alterations in lineage allocation lead to insufficient or overabundant populations of terminally differentiated cells, resulting in compromised function. Impaired trophoblast differentiation is implicated in common disorders of pregnancy such as unexplained miscarriage, fetal growth restriction, preeclampsia, and preterm birth [5]. Even moderately impaired fetal growth increases the risk of cardiovascular disease, obesity, and diabetes mellitus in adult life [1]. Therefore, understanding the regulation and molecular basis of placental differentiation has major implications for postnatal health and the etiology of adult disease.
The trophoblast differentiation pathways underpinning placental morphogenesis have been well characterized in the mouse [4, 6]. Trophoblast stem cells reside in the extraembryonic ectoderm of the early conceptus. These cells have the capacity to differentiate into chorionic ectoderm and ectoplacental cone trophoblast cell lineages [7], which further differentiate into the trophoblast subtypes of the labyrinthine zone and the junctional zone, respectively [8, 9]. These two distinct zones comprise the major structural elements of the mature chorioallantoic mouse placenta and are discernible from Embryonic Day (E) 10.5. The labyrinth, which is proximal to the fetus, consists of maternal blood spaces juxtaposed with fetal capillaries and separated by three layers of trophoblast cells. These form the interface for maternal-fetal nutrient and gas exchange in the placenta. The junctional zone is proximal to the uterine tissue and is made up of three differentiated trophoblast cell populations, namely, spongiotrophoblast cells, glycogen cells, and giant cells. Spongiotrophoblast cells comprise the most abundant population, and these cells overlie the placental labyrinth to provide structural support and to secrete peptide hormones [10, 11]. Spongiotrophoblast cells also act as precursors for the terminally differentiated trophoblast glycogen cells (characterized by their accumulation of glycogen) that reside at the placental-decidual interface [4, 12]. Once differentiated, they infiltrate the decidual stroma, localizing near spiral arteries that supply the placental labyrinth [13, 14]. These cells lyse just before term to provide a substantial glycogen energy source for the final phase of fetal growth [15]. Secondary trophoblast giant cells are large polyploid cells arising directly from ectoplacental cone trophoblasts [16] that invade developing decidual vessels and maternal spiral arteries [14]. They secrete a range of cytokines and hormones that promote systemic and local adaptations in the mother and are vital for fetal growth and survival [17–19].
The correct spatiotemporal balance between progenitor cell proliferation and differentiation into the specialized trophoblast cell lineages seems to be controlled by the actions of growth factors and other signals in the microenvironment that influence target cell gene expression profiles, including fate-determining genes. The hemopoietic cytokine granulocyte-macrophage colony-stimulating factor (CSF2, GM-CSF) has been implicated in trophoblast cell signaling [20]. It was originally identified as a regulator of myeloid leukocyte survival, proliferation, differentiation, and function [21], and it operates through a heterodimeric receptor complex composed of a CSF2-specific α subunit (CSF2RA, GM-CSFRα) and a βc subunit [22]. CSF2 synthesis is induced in uterine luminal epithelial cells during early pregnancy [23], when it acts to promote preimplantation embryonic development [24] and to initiate adaptation for pregnancy in the maternal immune response [25].
In midgestation, CSF2 is synthesized by stromal cells, uterine natural killer cells, and endothelial cells in the maternal decidua, as well as by trophoblast cells within the junctional zone but not the labyrinth tissue of the placenta [26–28]. Immunohistochemical investigations in first-trimester human placental tissue show that the receptor CSF2RA is present in cytotrophoblasts and all invading extravillous trophoblasts but is only weakly expressed in syncytial trophoblasts [29]. In culture, CSF2 has trophic effects on ectoplacental trophoblast outgrowth and increases DNA synthesis and giant cell formation in primary mouse placental cells [30, 31]. In human trophoblasts, CSF2 promotes syncytialization and placental lactogen secretion [32]. Administration of exogenous CSF2 to mice protects against embryonic and fetal loss [33–35] that is associated with changes in placental development [33].
Studies in mice genetically deficient in CSF2 support a potential role for this cytokine in regulating placental morphogenesis. Mice with a null mutation in Csf2 (Csf2−/− mice) exhibit fetal growth restriction in utero and have elevated rates of late gestation fetal loss and death in the first week after birth [36, 37]. We previously reported that the structure of the mature placenta is altered in Csf2−/− mice, with a decreased labyrinthine zone:junctional zone ratio [37], and postulated that this causes placental insufficiency and underpins the accompanying fetal growth impairment [37]. The aim of this study was to define the role of CSF2 in the molecular regulation of trophoblast cell lineage allocation during placental development. We utilized Csf2−/− mice and quantified the transcription of genes identified as known markers of trophoblast cell subtypes or key regulators of specific differentiation pathways, with particular emphasis on those implicated in junctional zone development.
MATERIALS AND METHODS
Animals, Collection of Tissues, and Reproductive Outcome Parameters
Mice homozygous for a null mutation in the Csf2 gene (Csf2−/− mice) [37, 38] in a C57BL/6 background were bred from homozygous breeding pairs and were housed together with a wild-type C57BL/6 (WT) breeding colony in the specific pathogen-free facility at the University of Adelaide Medical School Animal House. The absence of full-length Csf2 transcripts in Csf2−/− mice was confirmed by quantitative real-time RT-PCR (qRT-PCR) (described herein). Mice were provided with food and water ad libitum and were utilized according to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes with approval from the University of Adelaide Animal Ethics Committee.
For timed matings, adult virgin Csf2−/− and WT females (age 7–10 wk) were housed with adult stud males of the same genotype (2:1) and were allowed to mate naturally. The day on which a copulation plug was evident was designated E1 of pregnancy. On E13, E15, or E18, mice were killed by cervical dislocation, uteri were recovered, and viable and resorbing implantation sites were counted. For E15 and E18 implantation sites, fetal membranes and decidual tissue were removed, and fetal and placental weights were recorded. Whole placentae collected on E13 were snap frozen in liquid nitrogen for gene expression analysis by microarray and qRT-PCR. Placentae obtained on E15 were cut midsagittally and fixed in 4% paraformaldehyde in 70 mM phosphate buffer for structural analyses or were dissected under a light microscope (SZ-PT; Olympus, Tokyo, Japan) to separate labyrinthine and junctional zone (spongiotrophoblast plus giant cell layer) tissue before snap freezing in liquid nitrogen and storage at −80°C for later RNA extraction. Placentae recovered on E18 were fixed in 4% paraformaldehyde in 70 mM phosphate buffer for structural analysis.
RNA Extraction and Generation of cDNA
Total RNA was isolated from whole placentae collected on E13 after homogenization using a Qiagen (Clifton Hill, Australia) RNeasy midi kit according to the manufacturer's instructions with on-column DNase I (Qiagen) treatment. Quantity and quality of the resulting RNA were assessed by A260:A280 ratio with a UV spectrophotometer (Beckman Coulter Australia, Gladesville, Australia). Total RNA (2.5 μg) was reverse transcribed with Superscript III (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer's instructions with 200-ng random sequence oligohexamers (Geneworks, Adelaide, Australia) and 500-ng oligo dT18 (Proligo, Lismore, Australia) at 52°C for 1 h using a Geneamp PCR System 2700 thermocycler (Applied Biosystems, Inc., Foster City, CA). Total RNA was also isolated from labyrinthine and junctional zone tissues recovered on E15 using Trizol reagent (Invitrogen Life Technologies), and contaminating DNA was removed by DNase I treatment (DNA-free kit; Ambion, Austin, TX), both according to the manufacturer's instructions. The quantity of RNA extracted was determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, PA). Samples were considered sufficiently pure if the A260:A280 ratio was >1.6. Total RNA (2 μg) was reverse transcribed to cDNA as already described.
Microarray Analysis
Four biological replicates of 50 μg of RNA from Csf2−/− and WT E13 placental tissue, each pooled from four or five whole placentae from two mothers, were processed at the Australian Genome Research Facility in Melbourne, Australia, for single-cycle labeling and hybridization to GeneChip mouse genome 430_2.0 chips (Affymetrix, Santa Clara, CA). This 3′ expression microarray chip is composed of >45 000 probe sets covering >39 000 transcripts and variants (a complete list is available at www.affymetrix.com). RNA integrity analysis, hybridization, and washing were performed according to the manufacturer's instructions. Data were analyzed using the algorithm MAS5 in GeneChip operating software (version 1.4; Affymetrix). CEL files for each chip were scaled globally to a target intensity of 150 for generation of CHP files to assess quality control parameters. Differential expression between groups was determined using a fold change analysis with t-tests and the IlluminaGUI package [39] as implemented by Bioconductor in the program R (open source software available at www.bioconductor.org). This package is designed to analyze Sentrix Illumina beadchip data, so Affymetrix identifications were substituted with Illumina mouse6 version 1.1 identifications. The fold changes for all probes were calculated from the ratio of the mean intensities of the two groups (i.e., from MAS5 CHP file values) (n = 4/group). In a first-tier analysis, program default cutoffs were used to identify differentially expressed genes meeting high-stringency criteria (fold change >2; P < 0.05, difference between mean intensity >100). Low-stringency criteria (fold change >1.2; P < 0.05, difference between mean intensity >10) were then applied in a second-tier analysis to assess the effect of the Csf null mutation on the transcription of genes known to have essential roles in placental development [6, 18, 40].
Quantitative Real-Time RT-PCR
Real-time RT-PCR was performed using a Rotor-Gene 6000 thermal cycler (Corbett Life Sciences, Sydney, Australia) and SYBR Green I chemistry (Applied Biosystems, Inc.) to detect synthesized products. All primer sets were used with reaction cycle conditions of 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec and 60°C for 60 sec, and products were melted over 60–90°C to generate dissociation curves. Primer pairs specific for placental genes (Table 1) were designed to bracket exon-exon boundaries where possible using published murine sequences and Primer Express software (Applied Biosystems, Inc.) and were synthesized by Sigma Genosys (Castle Hill, Australia). Equal volumes of each placental cDNA preparation were pooled and used to generate standard cDNA for validation and optimization of primer pairs. Amplicons were checked for the absence of primer dimers and nonspecific amplification by dissociation curves and were validated by gel electrophoresis and DNA sequencing (Southpath and Flinders Sequencing Facility, Adelaide, Australia). The PCR reaction conditions were optimized for each primer pair, such that amplification efficiency >85% and a linear standard curve were achieved to a dilution of 1:4092 of standard cDNA. Nontemplate control samples containing water substituted in place of cDNA were included in all assays to confirm the absence of nonspecific amplification products. A cycle threshold was calculated for each sample using Rotor-Gene 6000 software. For each gene of interest, the relative mRNA abundance was calculated by comparison with standard curves generated from serial dilutions of pooled placental cDNA and were then normalized to the housekeeping gene Ywhaz (Table 1). Preliminary experiments revealed that Ywhaz expression was unaltered by the Csf2 null mutation.
TABLE 1.
Sequences and Ensembl accession numbers for qRT-PCR primers.
Placental Histology
Midsagittal cross-sections of placental tissue were stained with Masson trichrome using standard protocols. From each of six WT and six Csf2−/− mice, two to three placentae were randomly selected for histological assessment. The cross-sectional areas of the placental junctional zone and labyrinth were measured on digital images of complete midsagittal sections using an Olympus BH-2 microscope with 2× objective and 3.3× ocular lenses and videoimage analysis software (Video Pro; Leading Edge, Adelaide, Australia). The proportion (%) of each region in the placenta was estimated by dividing the cross-sectional area of that region by the total midsagittal cross-sectional area of the placenta. An estimate of the volume of these regions was calculated by multiplying their proportion by the total placental weight.
Junctional Zone Morphology
Immunohistochemical staining of midsagittal cross-sections of placental tissue was used to detect cyclin-dependent kinase inhibitor 1C (CDKN1C, P57Kip2) and gap junction beta-3 protein (connexin-31, Cx31) in the placental junctional zone. For Cx31 immunolabeling, sections were treated for antigen retrieval by microwaving for 10 min at 150 W in citrate buffer (pH 6.0), and endogenous peroxidase was then quenched by incubation with 3% H2O2 in water for 30 min. Sections were permeabilized in PBS containing 0.2% Triton X-100 for 10 min and were blocked with 10% normal swine serum and 1% bovine serum albumin (BSA) in PBS before addition of rabbit anti-Cx31 antibody (catalog No. CX31-A; Alpha Diagnostics, San Antonio, TX) and then diluted 1:25 for 16 h at room temperature. Bound antibody was detected using biotinylated goat anti-rabbit IgG (catalog No. E0432; DAKO, Carpinteria, CA) diluted 1:200 for 2 h, followed by streptavidin-conjugated horseradish peroxidase (catalog No. S000–03; Rockland Immunochemicals, Gilbertsville, PA) diluted 1:500 for 1 h, both at room temperature. For CDKN1C, sections were microwaved for 5 min and 15 sec at 650 W in Tris-edetic acid (EDTA) (pH 9.0) (10 mM Tris base, 1 mM EDTA, 0.05% Tween 20) for antigen retrieval, and endogenous peroxidase activity was quenched as already described. Sections were blocked for nonspecific binding with 10% rabbit serum and 1% BSA in PBS before addition of goat anti-P57Kip2 (CDKN1C) antibody (catalog No. M-20; Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:50 for 16 h at room temperature. Bound antibody was detected with horseradish peroxidase-conjugated rabbit anti-goat IgG (catalog No. P0160; DAKO) diluted 1:200 for 2 h at room temperature. Immunolabeling was visualized by incubating sections with diaminobenzidine (Sigma, St. Louis, MO) and by counterstaining with hematoxylin before mounting in DPX (Sigma). Negative controls used irrelevant rabbit and goat affinity-purified IgG or the primary antibody diluent alone.
Analyses of Cx31 and CDKN1C staining were performed on digital images of sections captured using a NanoZoom digital scanner (Hamamatsu, Herrsching am Ammersec, Germany). The total cross-sectional area of Cx31 staining was measured in complete midsagittal sections as already described using 20× objective and 3.3× ocular lenses. The proportion (%) of Cx31 staining was estimated by dividing the area of Cx31 positivity by the total area of the placenta or junctional zone. In addition, the areas of Cx31+ cells in sections of junctional zone tissue were quantified using a 40× objective. This was calculated as the average area of all Cx31+ cells within eight fields per placental section, where two fields were randomly selected in each of four quadrants of junctional zone tissue. The total number of Cx31+ cells was estimated by dividing the total area of Cx31 staining by the mean Cx31+ cell area. In addition, the spatial distribution of glycogen cells in the junctional zone was quantified using an arbitrary scale 1) as located within the junctional zone in close proximity to the labyrinth; 2) as distributed across the junctional zone, including at the giant cell border; or 3) as localized solely at the spongiotrophoblast-giant cell layer border. The numbers of CDKN1C+ glycogen cells and giant cells per midsagittal section were counted manually in digital images with the tissue identity masked. Giant cells were identified by their very large obviously polyploid nucleus and location at the junctional zone-decidual border.
Statistical Analysis
All data were analyzed using SPSS version 13 (SPSS Inc., Chicago, IL). The effects of genotype on fetal and placental weight data were analyzed by one-way ANOVA. The effects of genotype on placental gene expression and morphology were analyzed by linear mixed model repeated-measures ANOVA with post hoc Bonferroni t-test using the mother as the subject and the fetus or placenta as the repeated measure. The number of viable implantation sites per litter was included as a covariate when required. Associations between parameters were assessed using Pearson two-tailed bivariate correlation analyses. Data are expressed as mean ± SEM or as estimated marginal mean ± SEM as appropriate. Data were considered statistically significant at P < 0.05.
RESULTS
Csf2 Null Mutation Alters Fetal and Placental Growth Trajectory
Csf2−/− mice in a mixed SV129/C57BL/6 background have fewer viable implantations and reduced fetal growth compared with WT controls [37]. The genetic background of Csf2−/− mice used in the present study was pure C57BL/6, so initially the effect of CSF2 deficiency was evaluated in the C57BL/6 strain. Csf2−/− or WT mice were mated with males of the same genotype and were killed on E15 or E18. There was no significant effect of the Csf2 null mutation on total number of implantations, fetal viability, or resorption rate at either time point (Table 2). Effects on the growth of gestational tissues were evident at E15, with the fetal weight:placental weight ratio (an indicator of placental efficiency) reduced in Csf2−/− implantation sites by 17% (P < 0.001) and associated with a 9% reduction in fetal weight (P = 0.001), but there was no significant difference in placental weight. At E18, there was no significant difference between genotypes in fetal weight; however, placental weight was increased by 13% (P < 0.001), and the fetal weight:placental weight ratio remained 9% lower in Csf2−/− compared with WT implantation sites (P = 0.002) (Table 2).
TABLE 2.
The effect of Csf2 null mutation on litter composition and fetal and placental weights on E15 and E18.

Csf2 Null Mutation Alters Development of the Placental Junctional Zone
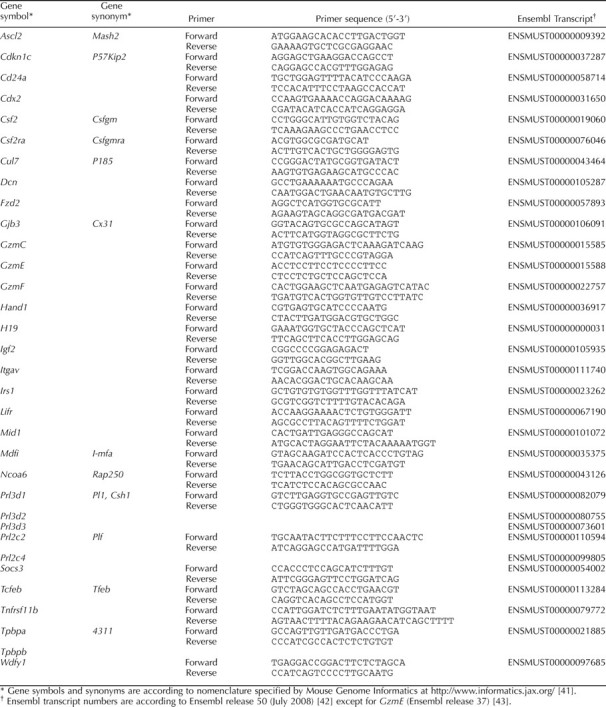
To investigate whether the increase in placental weight relative to fetal weight in Csf2−/− implantation sites was associated with altered placental structure, the area and estimated volume of the junctional and labyrinthine zones were evaluated histologically in midsagittal sections of placentae recovered on E15 and on E18 from WT and Csf2−/− female mice mated with males of the same genotype (Fig. 1, A and B). The Csf2 null mutation increased the cross-sectional area of the junctional zone by 18% on E15 (P = 0.023 [Fig. 1C]) and by 26% on E18 (P = 0.009 [Fig. 1D]), but the labyrinth was not affected. This corresponded to increases in the volume of junctional zone tissue of 24% at E15 (P = 0.005 [Fig. 1E]) and 20% at E18 (P = 0.046 [Fig. 1F]) and to reductions in the labyrinthine zone:junctional zone volume ratio of 21% at E15 (P = 0.001) and 20% at E18 (P = 0.05) in Csf2−/− mice (Fig. 1G). The relative change from E15 to E18 in the proportion of the tissue comprising labyrinthine and junctional zone regions was not affected by genotype, with the labyrinthine zone:junctional zone volume ratio increasing by 49% (P < 0.001) and 60% (P < 0.001), respectively, in WT and Csf2−/− placentae (Fig. 1G). Together, these data indicate that overgrowth of the junctional zone occurs in Csf2−/− mice and suggest that altered junctional zone structure may contribute to the adverse effects of CSF2 deficiency on fetal development.
FIG. 1.
The Csf2 null mutation causes enlargement of the placental junctional zone. A and B) Representative midsagittal sections of E15 placentae stained with Masson trichrome to distinguish the labyrinthine zone (LZ) and junctional zone (JZ) in WT mice (A) and Csf2−/− mice (B); the JZ-LZ boundary is indicated by a dotted line; bar = 450 μm. C and D) The midsagittal cross-sectional area of LZ and JZ at E15 (C) and at E18 (D) in WT and Csf2−/− mice. E and F) The volume of LZ and JZ at E15 (E) and at E18 (F) in WT and Csf2−/− mice. G) The LZ volume:JZ volume ratio at E15 and at E18 in WT and Csf2−/− mice. Data are expressed as estimated marginal mean (adjusted for the number of viable pups) ± SEM, with two to three placentae per mother randomly selected for histological analysis from six WT and six Csf2−/− mice. The effect of genotype was analyzed by linear mixed model repeated measures with post hoc Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.05).
Csf2 Null Mutation Alters Placental Gene Expression on E13
To investigate the molecular basis of the effect of the Csf2 null mutation on placental development, we began by performing a microarray experiment on whole placenta recovered on E13 from WT and Csf2−/− female mice mated with males of the same genotype. This developmental stage was selected as the earliest time at which the full range of differentiated trophoblast cell lineages is present in the junctional zone [10]. Arrays were hybridized with cDNA from each of four biological replicates of Csf2−/− and WT placentae. The full microarray data set is provided as Supplemental Table S1 (available at www.biolreprod.org). Seventeen genes were identified as differentially expressed and meeting stringent criteria (fold change >2.0; P < 0.05, difference between mean intensity >100) (Table 3). The gene showing the greatest induction by the Csf2 null mutation was Mid1 (midline 1 [up 17.0-fold]), while the gene most suppressed was Wdfy1 (WD repeat and FYVE domain containing 1 [down 5.6-fold]).
TABLE 3.
Genes identified as differentially expressed using high stringency criteria in microarray analysis of mRNA expression in E13 placental tissue of Csf2-/- and WT mice.*

Microarray data were further interrogated for the effect of the Csf2 null mutation on expression of a panel of genes identified as essential regulators of placental morphogenesis [6, 18, 40]. Several genes known to be markers of differentiation of placental trophoblast cells, including genes implicated in junctional zone morphogenesis, showed altered expression in Csf2 null mutant placentae according to low-stringency criteria (fold change >1.2; P < 0.05, difference between mean intensity >10). These included genes involved in spongiotrophoblast differentiation (Arnt [down 1.3-fold] and Tpbpa [up 1.3-fold]), as well as genes associated with giant cell differentiation or function (Ccne1 [up 1.2-fold], Prl2c2 [up 1.4-fold], and Socs3 [down 1.6-fold]) and glycogen cell differentiation (Igf2r [up 1.3-fold]) (Table 4). Several other genes expressed in trophoblast stem cells or the labyrinthine zone showed small but statistically significant changes in expression (Table 4).
TABLE 4.
Genes known to be markers of trophoblast cell lineages or regulators of placental morphogenesis meeting low stringency criteria in microarray analysis of mRNA expression in E13 placental tissue of Csf2-/- and WT mice.*

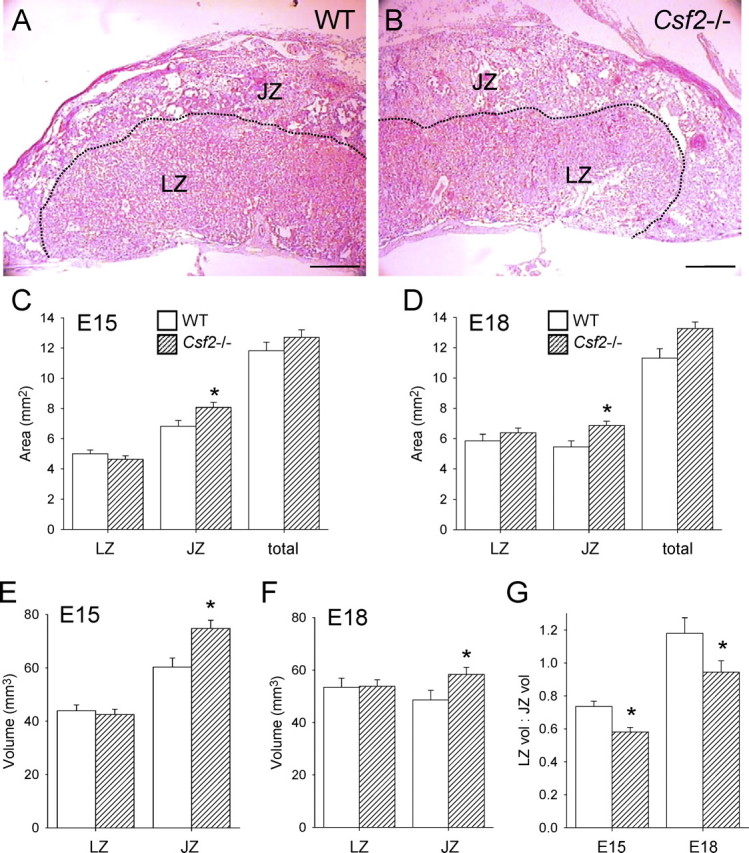
In an effort to validate the microarray data, qRT-PCR was performed on mRNA extracted from whole placental tissue of WT and Csf2−/− placentae at E13. Among the genes identified by microarray analysis using stringent criteria, four of six genes evaluated were confirmed as differentially expressed by qRT-PCR. These were Mid1 (up 19.8-fold, P < 0.001), Cd24a (up 2.0-fold, P = 0.019), Tnfrsf11b (down 31.0-fold, P = 0.029), and Wdfy1 (down 3.6-fold, P < 0.001) (Fig. 2, A–D). In contrast, significant differences in expression were not confirmed for Fzd2 or Irs1 (Fig. 2, E and F). Among six genes identified using low-stringency criteria as potentially influenced at E13 by the Csf2 genotype, two were confirmed as differentially expressed by qRT-PCR, namely, Socs3 (down 1.2-fold, P = 0.001) (Fig. 2G) and Itgav (down 1.2-fold, P = 0.015) (Fig. 2H). Cebpb, Fgfr2, Prl2c2, and Tpbpa were not differentially expressed when evaluated by qRT-PCR (data not shown).
FIG. 2.
The Csf2 null mutation alters placental gene expression at E13. Messenger RNA transcripts identified as differentially expressed by microarray analysis (A–F) or selected on the basis of known roles in placental trophoblast cell differentiation (G–N) were quantified by qRT-PCR in placental tissue collected from WT and Csf2−/− mice at E13 and were normalized to the internal control, Ywhaz mRNA. Transcripts analyzed were Mid1 (A), Cd24a (B), Tnfrsf11b (C), Wdfy1 (D), Fzd2 (E), Irs1 (F), Socs3 (G), Itgav (H), Cdkn1c (I), Ascl2 (J), Tcfeb (K), Gzmc (L), Gzme (M), and Gzmf (N). Data are expressed as mean ± SEM. Values are from two to three placentae from each of five WT and six Csf2−/− mice, and data are expressed as mean ± SEM. The effect of genotype was analyzed by linear mixed model repeated measures with post hoc Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.05).
Genes known to have key roles in trophoblast differentiation but not differentially expressed in the microarray experiment were also evaluated by qRT-PCR in E13 placentae. Genes with elevated expression in response to the Csf2 null mutation were Ascl2 (up 1.4-fold, P = 0.001) (Fig. 2J), which is associated with spongiotrophoblast differentiation, and Tcfeb (up 1.1-fold, P = 0.046) (Fig. 2K), which is associated with labyrinthine zone development. No change was evident in Cdkn1c (Fig. 2I) or in Cul7, Cdx2, Lifr, H19, or Ncoa6 (data not shown).
Genes encoding members of the granzyme family of enzymes that are implicated in extracellular matrix remodeling were also evaluated by qRT-PCR after their notable appearance in a preliminary microarray experiment (data not shown). Granzyme genes showed reduced expression in response to the Csf2 null mutation, including Gzmc (down 3.1-fold, P = 0.044) (Fig. 2L), Gzme (down 2.0-fold, P = 0.040) (Fig. 2M), and Gzmf (down 2.0-fold, P = 0.037) (Fig. 2N). No change was evident in Gzmb, Gzmd, or Gzmg (data not shown).
Csf2 Null Mutation Alters Junctional Zone Gene Expression on E15
To gain further insight into the effect of the Csf2 null mutation on junctional zone gene expression, placentae were recovered on E15 from WT and Csf2−/− mice and were dissected into junctional zone and labyrinthine tissue before gene expression analysis by qRT-PCR. The dissected junctional zone tissue was enriched for spongiotrophoblast and giant cells, as confirmed by diminished expression of labyrinthine-specific genes Cdx2 and Tcfeb (P < 0.001 and P = 0.034, respectively (Fig. 3A), and by increased transcription of genes characteristically expressed by spongiotrophoblast cells (Ascl2, Tpbpa, and Hand1) and by giant cells (Prl3d1 and Hand1) (P < 0.001 for all) (Fig. 3B).
FIG. 3.

The labyrinthine and junctional zones express different markers of differentiation. Messenger RNA transcripts known to be characteristically expressed in the labyrinthine zone (LZ) (A) or by spongiotrophoblast cells and/or giant cells in the junctional zone (JZ) (B) were quantified by qRT-PCR in labyrinthine and junctional zone tissue dissected from WT placentae at E15 and were normalized to the internal control, Ywhaz mRNA. Transcripts analyzed were Cdx2 and Tfeb, associated with labyrinthine development (A), and Ascl2, Tbpba, Hand1, and Prl3d1, associated with junctional zone development (B). Data are expressed as mean ± SEM and are shown relative to junctional zone content (assigned an arbitrary value of 100). Values are from two placentae randomly selected from each of 15 WT mice. The effect of tissue compartment was analyzed by linear mixed model repeated measures with post hoc Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.05).
Within E15 junctional zone tissue, the Csf2 null mutation led to increased transcription of genes associated with spongiotrophoblast cell and giant cell differentiation, including Ascl2 (up 1.6-fold, P = 0.01), Hand1 (up 1.4-fold, P = 0.038), Prl3d1 (up 1.9-fold, P = 0.032), and Prl2c2 (up 1.6-fold, P = 0.027) (Fig. 4, A, B, D, and E) [6, 18, 40]. Ncoa6, a gene associated with spongiotrophoblast and glycogen cell development [44], was also increased in junctional zone tissue of Csf2−/− mice (up 1.8-fold, P = 0.033) (Fig. 4C). In contrast, the Csf2 null mutation reduced junctional zone expression of granzyme genes, including Gzmc (down 1.4-fold, P = 0.001), Gzme (down 1.3-fold, P = 0.023), and Gzmf (down 1.4-fold, P < 0.001) (Fig. 4, F–H). There was a trend toward increased transcription of the glycogen cell-expressed gene Igf2 (up 1.3-fold, P = 0.09), as well as Cul7 (up 1.7-fold, P = 0.07), in response to the Csf2 null mutation, while Mdfi, Tpbpa, Cdkn1c, H19, Socs3, Itgav, Lifr, Vegf, Dcn, and Gjb3 mRNA abundance was unaltered (data not shown). In addition, Csf2 deficiency increased expression of Ascl2 (up 1.6-fold, P = 0.001) and Tcfeb (up 1.9-fold, P = 0.014) in placental labyrinthine tissue (data not shown).
FIG. 4.

The Csf2 null mutation alters placental junctional zone gene expression at E15. Messenger RNA transcripts implicated in junctional zone development were quantified by qRT-PCR in dissected junctional zone tissue from WT and Csf2−/− placentae at E15 and were normalized to the internal control, Ywhaz mRNA. Transcripts analyzed were Ascl2 (A), Hand1 (B), Ncoa6 (C), Prl3d1 (D), Prl2c2 (E), Gzmc (F), Gzme (G), and Gzmf (H). Data are expressed as mean ± SEM. Values are from two placentae from each of 15 WT and 13 Csf2−/− mice. The effect of genotype was analyzed by linear mixed model repeated measures with Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.05).
Csf2 and Csf2ra Are Predominantly Expressed in the Placental Junctional Zone
To investigate the potential for CSF2 signaling within the labyrinthine and junctional zones, qRT-PCR for mRNA encoding CSF2 (Csf2) and the ligand-specific CSF2 receptor α subunit (CSF2RA, Csf2ra) was performed in dissected E15 placenta from WT and Csf2−/− mice. In WT placenta, expression of Csf2 mRNA was 87-fold greater in the junctional zone than in the labyrinthine zone (P = 0.002) (Fig. 5A). The transcription of Csf2ra was 6.5-fold and 11.4-fold greater in the junctional zone compared with the labyrinthine zone in WT and Csf2−/− placental tissue, respectively (P < 0.01 for both) (Fig. 5B). The Csf2 null mutation was associated with 1.8-fold higher expression of Csf2ra in junctional zone tissue compared with WT tissue (P = 0.048) (Fig. 5B).
FIG. 5.

Csf2 and Csf2ra are predominantly expressed in the junctional zone of the placenta. Messenger RNA transcripts encoding CSF2 (Csf2) (A) and GM-Rα (Csf2ra) (B) were quantified by qRT-PCR in dissected labyrinthine zone (LZ) and junctional zone (JZ) tissues dissected from WT and Csf2−/− placentae at E15 and were normalized to the internal control, Ywhaz mRNA. Data are expressed as mean ± SEM. Values are from two placentae from each of nine WT and seven Csf2−/− mice. The effect of genotype and tissue compartment was analyzed by linear mixed model repeated measures with Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.05 compared with JZ; #P < 0.01 compared with WT).
Csf2 Null Mutation Alters Glycogen Cell and Giant Cell Abundance
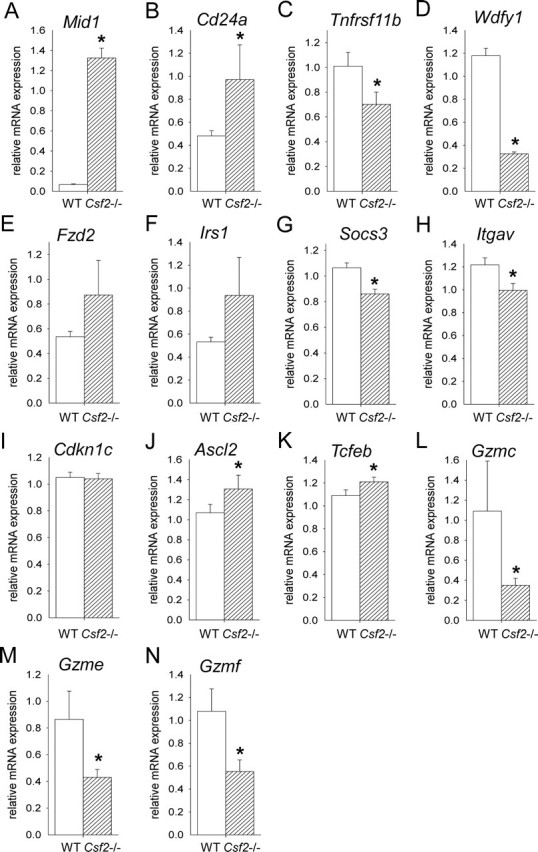
To evaluate whether the changes in junctional zone gene expression were associated with altered trophoblast cell lineage allocation, junctional zone tissue from WT and Csf2−/− females mated with males of the same genotype was assessed on E15 for known markers of terminally differentiated trophoblast glycogen cells and giant cells, namely, Cx31 and CDKN1C. The gap junction protein Cx31 was exclusively and intensely expressed on the cytoplasmic membrane of large clusters of glycogen cells located in the junctional zone predominantly at the spongiotrophoblast-giant cell border (Fig. 6, A and B), with occasional weak expression in small nests of cells within the labyrinth. The identity of Cx31+ cells as glycogen cells was confirmed on the basis of their vacuolated appearance and positive staining with periodic acid-Schiff stain (data not shown). The absolute area of placental tissue occupied by Cx31+ glycogen cells was elevated by 73.3% in Csf2−/− mice (P < 0.001) (Fig. 6E), such that they occupied 46.1% of the junctional zone area (compared with 33.5% in WT mice, P = 0.01) and 29.0% of the whole placenta area (compared with 19.3% in WT mice, P = 0.01). Cx31+ glycogen cells were more intensely stained and larger in size in placentae from Csf2−/− mice (Fig. 6, C and D), with a 13.5% increase in cell area (P = 0.018) (Fig. 6F). The estimated number of Cx31+ glycogen cells per midsagittal cross-sectional section of the placenta was increased by 52% in Csf2−/− compared with WT placentae (P = 0.005) (Fig. 6G). When a semiquantitative scoring system was used to evaluate their spatial location, glycogen cells in Csf2−/− placentae were less evidently accumulated at the spongiotrophoblast cell-giant cell border than in WT placentae and instead were often distributed across the full depth of the junctional zone compartment (P = 0.063) (Fig. 6H).
FIG. 6.
The Csf2 null mutation increases the number, size, and location of Cx31+ glycogen cells in the placental junctional zone. Representative midsagittal cross-sections of E15 placentae stained immunohistochemically to detect Cx31 in junctional zone tissue of WT mice (A and C) and Csf2−/− mice (B and D). LZ, labyrinthine zone; JZ, junctional zone; Gc, glycogen cells; the JZ-LZ boundary is indicated by a dotted line; bars = 200 μm (A and B) and 60 μm (C and D). Tissue stained with irrelevant rabbit IgG was comparable to the photomicrograph shown in Figure 7E. E) The area of Cx31+ staining in midsagittal cross-sections of placentae from WT and Csf2−/− mice. F) The size of Cx31+ glycogen cells in midsagittal cross-sections of placentae from WT and Csf2−/− mice. G) The number of Cx31+ glycogen cells per midsagittal cross-section of the placental junctional zone, estimated by dividing the area of the junctional zone by the average area of Cx31+ glycogen cells. H) The spatial distribution of glycogen cells in the junctional zone, scored as relative proximity to giant cell (Gi) border as detailed in Materials and Methods. Values are from two to three placentae from each of six WT and six Csf2−/− mothers, and data are expressed as mean ± SEM. The effect of genotype was analyzed by linear mixed model repeated measures with post hoc Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (*P < 0.018).
Intense nuclear staining for CDKN1C was localized to a subset of glycogen cells, as well as most giant cells, which could be distinguished on the basis of their size and location at the placental-decidual border (Fig. 7, A–D). Nuclei of some spongiotrophoblast cells were also labeled. The number of CDKN1C+ glycogen cells was increased by 28% in Csf2−/− placentae (P = 0.060) (Fig. 7F), and the number of CDKN1C+ giant cells was increased by 64% (P = 0.027) (Fig. 7G). Together, these data show that CSF2 deficiency is associated with increased numbers of glycogen cells and giant cells in the mature placenta and indicate that expanded populations of these cells largely account for the increase in junctional zone area observed at this time point.
FIG. 7.
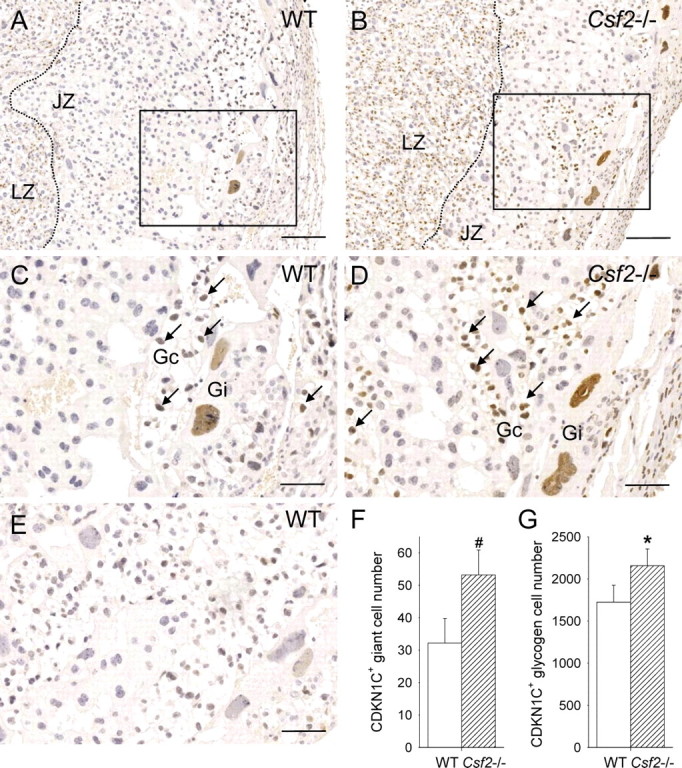
The Csf2 null mutation increases the number of CDKN1C+ glycogen cells and giant cells in the placental junctional zone. Representative midsagittal cross-sections of E15 placentae stained immunohistochemically to detect CDKN1C in junctional zone tissue of WT mice (A and C) and Csf2−/− mice (B and D) compared with tissue stained with irrelevant goat IgG (E). LZ, labyrinthine zone; JZ, junctional zone; Gc, glycogen cells; Gi, giant cells; arrows indicate CDKN1C+ glycogen cells; the JZ-LZ boundary is indicated by a dotted line; bars = 200 μm (A and B) and 95 μm (C and D). F) The number of CDKN1C+-stained glycogen cells per midsagittal cross-sections of placentae from WT and Csf2−/− mice. G) The number of CDKN1C+-stained giant cells per midsagittal cross-sections of placentae from WT and Csf2−/− mice. Values are from two to three placentae from each of six WT and six Csf2−/− mice, and data are expressed as mean ± SEM. The effect of genotype was analyzed by linear mixed model repeated measures with post hoc Bonferroni t-test using the mother as the subject and the placenta as a repeated measure (#P = 0.060; *P = 0.027).
DISCUSSION
Mice genetically deficient in CSF2 have compromised reproductive capacity that is characterized by fetal and postnatal growth impairment and by increased perinatal mortality, accompanied by altered placental structure in late gestation [37]. The present study shows that CSF2 deficiency causes altered expression of key genes associated with trophoblast differentiation in the junctional zone of the placenta, resulting in an increase in the numbers of terminally differentiated glycogen cells and giant cells. The adverse effects of CSF2 deficiency on reproductive outcome are thus likely mediated at least in part via the actions of this cytokine in controlling trophoblast differentiation and in contributing to placental composition and function.
The Csf2 null mutation in the C57BL/6 background altered the fetal and placental growth trajectory in late gestation, but this was less striking than that in the mixed SV129/C57BL/6 genetic background. Fetal growth restriction was present at E15 but not at E18, although a reduced fetal weight:placental weight ratio (a hallmark of reduced placental efficiency) was evident at both times. In addition, a high rate of perinatal mortality (with death of >50% of pups by weaning) occurs in the C57BL/6 Csf2−/− colony (data not shown). These effects are qualitatively similar to those that we and others have reported in SV129/C57BL/6 Csf2 null mutant mice, although fetal loss in the mixed background occurred in utero and after birth [36, 37]. The findings in both lines suggest that poor perinatal viability might result from placental insufficiency in utero, as no developmental anomalies other than late-onset pulmonary pathology have been described [37, 38] and the fostering of Csf2−/− pups to WT mothers did not reduce postnatal loss (data not shown); however, a direct link is not confirmed because undefined fetal abnormalities cannot be excluded.
In both genetic backgrounds, the most overt effect of CSF2 deficiency on placental structure was an increase in the relative proportion of tissue occupied by the junctional zone relative to the labyrinthine layer. The present analysis showed that the Csf2 null mutation increased the cross-sectional area and volume of the junctional zone at E15 and at E18, resulting in a heavier placenta at E18. Although the proportion of placenta comprising the labyrinthine zone was reduced, there was no substantive effect of genotype on labyrinthine area or volume.
Restriction of CSF2 actions predominantly to the junctional zone fits with the compartmentalized synthesis of CSF2 ligand and CSF2RA receptor. Our qRT-PCR data on Csf2 expression confirm an earlier in situ hybridization study [27] showing that Csf2 is expressed by trophoblast cells in the junctional zone, as well as by decidual leukocytes and endothelial cells. It remains to be determined whether spongiotrophoblasts and/or glycogen cells are the source of Csf2 transcripts. Increased expression of Csf2ra in junctional zone tissue of placentae from Csf2−/− mice suggests that CSF2 may downregulate its signaling pathway in placental cells, as occurs in myeloid cells [22].
Terminally differentiated trophoblast cell populations in the junctional zone include spongiotrophoblasts, glycogen cells, and giant cells. Glycogen cells were most profoundly affected by CSF2 deficiency. The proportion of the placenta occupied by Cx31+ glycogen cells was substantially increased in response to the Csf2 null mutation, with accumulation of cells throughout the junctional zone. Even accounting for their enlarged morphology, the absolute number of glycogen cells in Csf2 null placentae was estimated to be increased by 50%. Cx31 expression also appeared more intensely in glycogen cells of Csf2 null placenta. Cx31 is a gap junction protein that mediates intercellular communication by directing the exchange of ions, secondary messengers, and metabolites between cells [45, 46]. It is thought to aid in the early differentiation of glycogen cells by operating downstream of ASCL2 (Mash2) [46].
The increased number of Cx31+ glycogen cells was not accompanied by any effect of the Csf2 null mutation on the transcription of Gjb3 mRNA. However, there was a trend toward increased Igf2 expression in Csf2−/− placentae, and Igf2r was identified in the microarray experiment as moderately upregulated in Csf2 null mice. Igf2 is most abundantly synthesized by glycogen cells [12] and is essential for their differentiation and glycogen synthesis [47].
CDKN1C protein was utilized as another means of localizing glycogen cells in the placenta on E15. CDKN1C is a cyclin-dependent kinase inhibitor that negatively regulates cell proliferation expressed by trophoblast glycogen cells [13] and by giant cells [10]. Despite no change in placental expression of Cdkn1c mRNA, a trend to increased numbers of glycogen cells synthesizing CDKN1C protein was seen in E15 Csf2−/− placentae. The difference between mRNA and protein abundance for Gjb3 and Cdkn1c might be accounted for by the relative insensitivity of qRT-PCR for genes expressed by subpopulations of cells or by the possibility of posttranscriptional regulation of these molecules.
These observations do not concur with our previous findings of fewer L185+ glycogen cells in the junctional zone of E15 placentae from Csf2−/− mice [37]. Cx31 is expressed exclusively by glycogen cells in the mouse placenta [48] and identifies precursor and mature glycogen cells [10]. L185 labels only a subset of glycogen cells [37], suggesting that it may be associated with a glycogen cell function induced by CSF2, although the identity of the L185 target epitope is unknown.
In addition to glycogen cells, the number of placental giant cells expressing CDKN1C on E15 was increased in the junctional zone of Csf2−/− placentae, associated with increased expression of the giant cell-specific genes Prl3d1 and Prl2c2. These genes encode the placental hormones prolactin 1 and proliferin, which exert endocrine effects on several maternal organs, modulating maternal metabolism and adaptation to pregnancy [17, 49]. Proliferin also promotes giant cell invasion and angiogenesis, facilitating placental access to maternal blood vessels in early gestation [17].
CSF2 deficiency caused increased expression of a range of genes implicated at different stages in the control of trophoblast differentiation from ectoplacental cone precursors into mature junctional zone lineages. Most strikingly, the Csf2 null mutation increased labyrinthine and junctional zone transcription of Ascl2 encoding the basic helix-loop-helix transcription factor ASCL2. ASCL2 is localized to the ectoplacental cone trophoblast and chorion in early gestation and later is expressed predominantly by patches of spongiotrophoblast cells in the junctional zone and in the labyrinth [50, 51]. ASCL2 maintains progenitor trophoblasts in a proliferative state and suppresses their terminal differentiation into giant cells, with placentae in Ascl2−/− mice showing deficiency in spongiotrophoblast cells and excessive giant cell differentiation [50, 52]. Expression of the nuclear transcriptional coactivator Ncoa6, which is involved in directing differentiation of progenitor trophoblast into spongiotrophoblast and glycogen cells but not giant cells [44], was also increased in the junctional zone in the absence of CSF2.
Increased transcription in Csf2−/− junctional zone tissue of Hand1, a basic helix-loop-helix transcription factor controlling giant cell differentiation [53, 54], may contribute to the expanded giant cell population seen in the absence of CSF2. In contrast, the absence of CSF2 reduced placental transcription of Socs3 encoding the suppressor of cytokine signaling 3. Socs3 participates in a negative feedback loop with CSF2 and suppresses giant cell differentiation in early gestation [55, 56]. Socs3−/− placentae show impaired labyrinthine and junctional zone development, suggesting a role for Socs3 in maintaining extraembryonic ectodermal progenitor cells at the expense of giant cells [56]. Reduced Socs3 expression might thus contribute to the placental phenotype of Csf2−/− mice by permitting overcommitment of trophoblasts to the giant cell lineage. Additional genes upregulated in the microarray experiment were Ccne1 (encoding cyclin E1, an essential determinant of giant cell reduplication [57]) and Tpbpa (encoding 4311, which is expressed exclusively in spongiotrophoblast cells and not in giant cells [58]).
In addition, 17 genes not known to be linked with placental morphogenesis were identified in the microarray experiment as differentially expressed in Csf2−/− placentae. Of these, Mid1, Erdr1, Tubb3, Kif5c, Prdx4, Crim1, Matn2, Gp49a, Dd3y, Timd2, and Wdfy1 are not previously reported as being expressed in placental tissue. The most upregulated gene, Mid1, encodes MID1, a 72-kDa ring finger protein [59] that has ubiquitin ligase activity and regulates ventral midline formation in embryogenesis [60]. The most suppressed gene, Wdfy1, encodes WD repeat and FYVE domain containing 1 protein and may act as a signaling factor in the PI3 kinase signaling pathway [61, 62]. Two additional genes confirmed by qRT-PCR as differentially expressed were Cd24a and Tnfrsf11b. The Csf2 null mutation increased expression of Cd24a, which encodes a mucin-like cell adhesion molecule, CD24, that shows reduced placental expression in preeclampsia [63]. Tnfrsf11b, which was downregulated in Csf2 null mutant placentae, encodes osteoprotegerin, a cytokine that may protect gestational tissues from apoptosis [64]. Another gene strongly downregulated by the Csf2 null mutation was Lilrb4, which encodes a leukocyte immunoglobulin-like receptor, CD85K, that is implicated in the signaling pathway through which placental HLA-G exerts immunosuppressive effects in T lymphocytes [65]. The full significance of these genes in placental morphogenesis and their specific roles in mediating the effects of CSF2 deficiency on placental structure and function warrant further investigation.
Aberrant expression of Ascl2, Hand1, Ncoa6, Socs3, and other genes in junctional zone tissue of Csf2−/− placentae could be due to direct transcriptional induction or repression of these genes by CSF2 or, perhaps more likely, due to the downstream consequence of an altered developmental program diverted earlier in gestation by the absence of cytokine. In support of this, altered placental morphogenesis can be programmed at the blastocyst stage by depriving embryos of CSF2 [66]. A possible explanation is that CSF2 normally represses the progression of progenitor trophoblast along the ectoplacental cone cell lineage pathway into cells that later constitute the junctional zone, so that precocious or increased junctional zone development ensues in the absence of CSF2. Alternatively, trophoblast terminal differentiation and placental functional maturation could be delayed in the absence of CSF2, which would explain the altered pattern of Ascl2 transcription that we observed. Placental Ascl2 expression normally declines with the advancement of gestation, initially and predominantly occurring in the labyrinth, followed by the junctional zone [51]. Increased Ascl2 in both compartments could reflect the absence of signals inducing trophoblast terminal differentiation in Csf2−/− placentae. This notion is consistent with in vitro evidence that CSF2 induces trophoblast differentiation into giant cells in mice [30, 31] and promotes term human cytotrophoblast fusion and syncytialization in vitro [32], but it seems inconsistent with the finding of elevated numbers of apparently fully differentiated glycogen cells and giant cells in the Csf2−/− placenta. Alternatively, increased Ascl2 transcription may reflect a larger proliferating pool of undifferentiated trophoblast stem cell precursors in either or both compartments due to reduced fate commitment in the absence of CSF2. A larger pool of precursor cells would be expected to feed a higher rate of differentiation into giant cells and glycogen cells.
Another explanation for the accumulation of Cx31+ and CDKN1C+ glycogen cells could be impaired departure of glycogen cells destined to invade the maternal decidua. Normally, glycogen cells infiltrate decidual tissue as early as E15 and localize around maternal spiral arterioles supplying the labyrinth [12–14, 67]. Altered expression of several genes associated with placental trophoblast cell migration and invasion supports the validity of this postulate. In the absence of CSF2, expression of the genes Gzmc, Gzme, and Gzmf encoding the serine proteases granzymes C, E, and F, respectively, were diminished in the placental junctional zone on E15. Granzymes are localized to glycogen cells in the mouse placenta [68], where they act to restructure the uterine-placental interface through hydrolysis of the extracellular matrix [69] and apoptosis of trophoblasts [70]. Granzymes may also contribute to terminal differentiation of giant cells [71]. Placental transcription of the gene Itgav encoding the integrin subunit αv was reduced on E13 in Csf2−/− placentae. Integrin-αv forms five different adhesion receptors with various β subunits and interacts with a range of extracellular matrix ligands [72] that have fundamental roles in the formation and structural organization of the junctional zone [73–75]. Reduced expression of Itgav and the granzyme genes in the absence of CSF2 would be expected to inhibit glycogen cells from exiting the junctional zone, causing their accumulation in and enlargement of junctional zone tissue. Improper function of glycogen cells can cause fetal growth restriction [76] and thus might contribute to the perinatal morbidity that is characteristic of Csf2−/− mice.
Expression of the transcription factor Tcfeb, which is essential for labyrinthine vasculogenesis [77], was also increased in the Csf2−/− mutant placenta, suggesting that placental endothelial cells might also be affected by the Csf2 null mutation. Because expansion of the labyrinth is driven by fetal capillary invasion of the junctional zone [78, 79], an increase in Tcfeb in the labyrinth on E15 might compensate for delayed development of this compartment. This would be consistent with the relative acceleration in labyrinthine tissue growth seen between E15 and E18 in Csf2−/− compared with WT placentae, as expansion of the labyrinth normally occurs at its fastest rate before E16.5 [79]. Indeed, while the most obvious effects of CSF2 deficiency were evident in the junctional zone, related or independent effects in the labyrinth cannot be discounted, and further studies are needed to investigate this.
In conclusion, this study demonstrates that the absence of CSF2 in mice results in a dysmorphic placenta with altered placental trophoblast cell composition. Most notably, the Csf2 null mutation affects the placental junctional zone and causes accumulation of glycogen cells and giant cells, resulting from increased generation of these cell lineages or, possibly, from their reduced demise in mid to late gestation. While we cannot exclude the possibility of indirect effects of maternal CSF2 deficiency acting via the immune compartment [20, 25], the expression of CSF2 and its receptor in the placenta (as well as in vitro evidence of CSF2 signaling in trophoblasts) suggests that the cellular and gene expression changes reported herein occur due to direct targeting of trophoblasts by CSF2. Anomalous placental development is likely a key contributing factor in altered fetal growth course and in increased postnatal mortality seen in Csf2−/− progeny. These data indicate an important role of CSF2 in regulating the cellular composition of the placenta in a manner likely to influence fetal growth and postnatal health.
Supplementary Material
Acknowledgments
We thank Rebecca Skinner, Alison Care, and Camilla Dorian for technical assistance and Ashley Dunn (Ludwig Institute for Cancer Research, Melbourne, Australia) for provision of Csf2 null mutant mice.
Footnotes
1Supported by NHMRC Australia Project and Fellowship Grants.
REFERENCES
- Barker DJ, Clark PM. Fetal undernutrition and disease in later life. Rev Reprod 1997; 2: 105 112 [DOI] [PubMed] [Google Scholar]
- Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet 2001; 2: 538 548 [DOI] [PubMed] [Google Scholar]
- Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, Fisher SJ. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest 2004; 114: 744 754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DG, Cross JC. Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev Biol 2005; 284: 12 24 [DOI] [PubMed] [Google Scholar]
- Khong TY. Placental vascular development and neonatal outcome. Semin Neonatol 2004; 9: 255 263 [DOI] [PubMed] [Google Scholar]
- Cross JC. How to make a placenta: mechanisms of trophoblast cell differentiation in mice: a review. Placenta 2005; 26 (suppl A): S3 S9 [DOI] [PubMed] [Google Scholar]
- Gardner RL, Papaioannou VE, Barton SC. Origin of the ectoplacental cone and secondary giant cells in mouse blastocysts reconstituted from isolated trophoblast and inner cell mass. J Embryol Exp Morphol 1973; 30: 561 572 [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science 1998; 282: 2072 2075 [DOI] [PubMed] [Google Scholar]
- Uy GD, Downs KM, Gardner RL. Inhibition of trophoblast stem cell potential in chorionic ectoderm coincides with occlusion of the ectoplacental cavity in the mouse. Development 2002; 129: 3913 3924 [DOI] [PubMed] [Google Scholar]
- Coan P, Conroy N, Burton G, Ferguson-Smith A. Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dyn 2006; 235: 3280 3294 [DOI] [PubMed] [Google Scholar]
- Malassine A, Frendo JL, Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update 2003; 9: 531 539 [DOI] [PubMed] [Google Scholar]
- Redline RW, Chernicky CL, Tan HQ, Ilan J. Differential expression of insulin-like growth factor-II in specific regions of the late (post day 9.5) murine placenta. Mol Reprod Dev 1993; 36: 121 129 [DOI] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith A, Burton G. Comparative developmental anatomy of the murine and human definitive placentae. Placenta 2002; 23: 3 19 [DOI] [PubMed] [Google Scholar]
- Adamson SL, Lu Y, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C, Cross JC. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol 2002; 250: 358 373 [DOI] [PubMed] [Google Scholar]
- Bouillot S, Rampon C, Tillet E, Huber P. Tracing the glycogen cells with protocadherin 12 during mouse placenta development. Placenta 2006; 27: 882 888 [DOI] [PubMed] [Google Scholar]
- Gardner RL, Davies TJ. Lack of coupling between onset of giant transformation and genome endoreduplication in the mural trophectoderm of the mouse blastocyst. J Exp Zool 1993; 265: 54 60 [DOI] [PubMed] [Google Scholar]
- Linzer DIH, Fisher SJ. The placenta and the prolactin family of hormones: regulation of the physiology of pregnancy. Mol Endocrinol 1999; 13: 837 840 [DOI] [PubMed] [Google Scholar]
- Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol 2007; 304: 567 578 [DOI] [PubMed] [Google Scholar]
- Soares MJ, Konno T, Alam SMK. The prolactin family: effectors of pregnancy-dependent adaptations. Trends Endocrinol Metab 2007; 18: 114 121 [DOI] [PubMed] [Google Scholar]
- Robertson SA. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor Rev 2007; 18: 287 298 [DOI] [PubMed] [Google Scholar]
- Gasson J. Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood 1991; 77: 1131 1145 [PubMed] [Google Scholar]
- Guthridge MA, Stomski FC, Thomas D, Woodcock JM, Bagley CJ, Berndt MC, Lopez AF. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells 1998; 16: 301 313 [DOI] [PubMed] [Google Scholar]
- Robertson SA, Mayrhofer G, Seamark RF. Uterine epithelial cells synthesize granulocyte-macrophage colony-stimulating factor and interleukin-6 in pregnant and nonpregnant mice. Biol Reprod 1992; 46: 1069 1079 [DOI] [PubMed] [Google Scholar]
- Robertson SA, Sjoblom C, Jasper MJ, Norman RJ, Seamark RF. Granulocyte-macrophage colony-stimulating factor promotes glucose transport and blastomere viability in murine preimplantation embryos. Biol Reprod 2001; 64: 1206 1215 [DOI] [PubMed] [Google Scholar]
- Robertson SA, O'Connell AC, Hudson SN, Seamark RF. Granulocyte-macrophage colony-stimulating factor (GM-CSF) targets myeloid leukocytes in the uterus during the post-mating inflammatory response in mice. J Reprod Immunol 2000; 46: 131 154 [DOI] [PubMed] [Google Scholar]
- Crainie M, Guilbert L, Wegmann T. Expression of novel cytokine transcripts in the murine placenta. Biol Reprod 1990; 43: 999 1005 [DOI] [PubMed] [Google Scholar]
- Kanzaki H, Crainie M, Lin H, Yui J, Guilbert LJ, Mori T, Wegmann TG. The in situ expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) mRNA at the maternal-fetal interface. Growth Factors 1991; 5: 69 74 [DOI] [PubMed] [Google Scholar]
- Jokhi PP, King A, Loke YW. Production of granulocyte-macrophage colony-stimulating factor by human trophoblast cells and by decidual large granular lymphocytes. Hum Reprod 1994; 9: 1660 1669 [DOI] [PubMed] [Google Scholar]
- Jokhi PP, King A, Jubinsky PT, Loke YW. Demonstration of the low affinity alpha subunit of the granulocyte-macrophage colony-stimulating factor receptor (GM-CSF-R alpha) on human trophoblast and uterine cells. J Reprod Immunol 1994; 26: 147 164 [DOI] [PubMed] [Google Scholar]
- Athanassakis I, Bleackley R, Paetkau V, Guilbert L, Barr P, Wegmann T. The immunostimulatory effect of T cells and T cell lymphokines on murine fetally derived placental cells. J Immunol 1987; 138: 37 44 [PubMed] [Google Scholar]
- Armstrong D, Chaouat G. Effects of lymphokines and immune complexes on murine placental cell growth in vitro. Biol Reprod 1989; 40: 466 474 [DOI] [PubMed] [Google Scholar]
- Garcia-Lloret MI, Morrish DW, Wegmann TG, Honore L, Turner AR, Guilbert LJ. Demonstration of functional cytokine-placental interactions: CSF-1 and GM-CSF stimulate human cytotrophoblast differentiation and peptide hormone secretion. Exp Cell Res 1994; 214: 46 54 [DOI] [PubMed] [Google Scholar]
- Chaouat G, Menu E, Clark DA, Dy M, Minkowski M, Wegmann TG. Control of fetal survival in CBA × DBA/2 mice by lymphokine therapy. J Reprod Fertil 1990; 89: 447 458 [DOI] [PubMed] [Google Scholar]
- Tartakovsky B, Ben-Yair E. Cytokines modulate preimplantation development and pregnancy. Dev Biol 1991; 146: 345 352 [DOI] [PubMed] [Google Scholar]
- Savion S, Zeldich E, Orenstein H, Shepshelovich J, Torchinsky A, Carp H, Toder V, Fein A. Cytokine expression in the uterus of mice with pregnancy loss: effect of maternal immunopotentiation with GM-CSF. Reproduction 2002; 123: 399 409 [DOI] [PubMed] [Google Scholar]
- Seymour JF, Lieschke GJ, Grail D, Quilici C, Hodgson G, Dunn AR. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood 1997; 90: 3037 3049 [PubMed] [Google Scholar]
- Robertson SA, Roberts CT, Farr KL, Dunn AR, Seamark RF. Fertility impairment in granulocyte-macrophage colony-stimulating factor-deficient mice. Biol Reprod 1999; 60: 251 261 [DOI] [PubMed] [Google Scholar]
- Stanley E, Lieschke G, Grail D, Metcalf D, Hodgson G, Gall J, Maher D, Cebon J, Sinickas V, Dunn A. Granulocyte-macrophage colony-stimulation factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A 1994; 91: 5592 5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze JL, Eggle D. IlluminaGUI: graphical user interface for analyzing gene expression data generated on the Illumina platform. Bioinformatics 2007; 23: 1431 1433 [DOI] [PubMed] [Google Scholar]
- Simmons DG, Natale DR, Begay V, Hughes M, Leutz A, Cross JC. Early patterning of the chorion leads to the trilaminar trophoblast cell structure in the placental labyrinth. Development 2008; 135: 2083 2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene symbols and synonyms were retrieved from the Mouse Genome Database (MGD), Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, Maine. 2004. World Wide Web (URL: http://www.informatics.jax.org/) (January 2009). [Google Scholar]
- Transcript numbers were retrieved from the Ensembl release 50 Mus musculus database July 2008), European Bioinformatics Institute, Hinxton, Cambridge, England. World Wide Web (URL: http://jul2008.archive.ensembl.org/Mus_musculus/index.html/) (October 2008). [Google Scholar]
- Transcript numbers were retrieved from the Ensembl release 37 Mus musculus database February 2006), European Bioinformatics Institute, Hinxton, Cambridge, England. World Wide Web (URL: http://feb2006.archive.ensembl.org/Mus_musculus/index.html/) (October 2008). [Google Scholar]
- Antonson P, Schuster GU, Wang L, Rozell B, Holter E, Flodby P, Treuter E, Holmgren L, Gustafsson JA. Inactivation of the nuclear receptor coactivator RAP250 in mice results in placental vascular dysfunction. Mol Cell Biol 2003; 23: 1260 1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Winterhager E, Reuss B, Traub O, Butterweck A, Willecke K. Expression of the gap junction proteins connexin31 and connexin43 correlates with communication compartments in extraembryonic tissues and in the gastrulating mouse embryo, respectively. J Cell Sci 1996; 109: 191 197 [DOI] [PubMed] [Google Scholar]
- Kibschull M, Nassiry M, Dunk C, Gellhaus A, Quinn JA, Rossant J, Lye SJ, Winterhager E. Connexin31-deficient trophoblast stem cells: a model to analyze the role of gap junction communication in mouse placental development. Dev Biol 2004; 273: 63 75 [DOI] [PubMed] [Google Scholar]
- Lopez MF, Dikkes P, Zurakowski D, Villa-Komaroff L. Insulin-like growth factor II affects the appearance and glycogen content of glycogen cells in the murine placenta. Endocrinology 1996; 137: 2100 2108 [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q, Kibschull M, Schnichels M, Kretz M, Petrasch-Parwez E, Strotmann J, Reucher H, Lynn BD, Nagy JI, Lye SJ, Winterhager E, Willecke K. Characterization of Connexin31.1-deficient mice reveals impaired placental development. Dev Biol 2007; 312: 258 271 [DOI] [PubMed] [Google Scholar]
- Carney EW, Prideaux V, Lye SJ, Rossant J. Progressive expression of trophoblast-specific genes during formation of mouse trophoblast giant cells in vitro. Mol Reprod Dev 1993; 34: 357 368 [DOI] [PubMed] [Google Scholar]
- Guillemot F, Nagy A, Auerbach A, Rossant J, Joyner AL. Essential role of Mash-2 in extraembryonic development. Nature 1994; 371: 333 336 [DOI] [PubMed] [Google Scholar]
- Rossant J, Guillemot F, Tanaka M, Latham K, Gertenstein M, Nagy A. Mash2 is expressed in oogenesis and preimplantation development but is not required for blastocyst formation. Mech Dev 1998; 73: 183 191 [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gertsenstein M, Rossant J, Nagy A. Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev Biol 1997; 190: 55 65 [DOI] [PubMed] [Google Scholar]
- Riley P, Anson-Cartwright L, Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet 1998; 18: 271 275 [DOI] [PubMed] [Google Scholar]
- Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nat Genet 1998; 18: 266 270 [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Carpino N, Cross JC, Torres M, Parganas E, Ihle JN. SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. EMBO J 2003; 22: 372 384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci U S A 2001; 98: 9324 9329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, Amati B. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J 2003; 22: 4794 4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescisin KR, Varmuza S, Rossant J. Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev 1988; 2: 1639 1646 [DOI] [PubMed] [Google Scholar]
- Dal Zotto L, Quaderi NA, Elliott R, Lingerfelter PA, Carrel L, Valsecchi V, Montini E, Yen CH, Chapman V, Kalcheva I, Arrigo G, Zuffardi O, et al. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Mol Genet 1998; 7: 489 499 [DOI] [PubMed] [Google Scholar]
- Schweiger S, Schneider R. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays 2003; 25: 356 366 [DOI] [PubMed] [Google Scholar]
- Ridley SH, Ktistakis N, Davidson K, Anderson KE, Manifava M, Ellson CD, Lipp P, Bootman M, Coadwell J, Nazarian A, Erdjument-Bromage H, Tempst P, et al. FENS-1 and DFCP1 are FYVE domain-containing proteins with distinct functions in the endosomal and Golgi compartments. J Cell Sci 2001; 114: 3991 4000 [DOI] [PubMed] [Google Scholar]
- Fritzius T, Burkard G, Haas E, Heinrich J, Schweneker M, Bosse M, Zimmermann S, Frey AD, Caelers A, Bachmann AS, Moelling K. A WD-FYVE protein binds to the kinases Akt and PKCzeta/lambda. Biochem J 2006; 399: 9 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy B, Berkes E, Rigo B, Ban Z, Papp Z, Hupuczi P. Under-expression of CD24 in pre-eclamptic placental tissues determined by quantitative real-time RT-PCR. Fetal Diagn Ther 2008; 23: 263 266 [DOI] [PubMed] [Google Scholar]
- Lonergan M, Aponso D, Marvin KW, Helliwell RJ, Sato TA, Mitchell MD, Chaiwaropongsa T, Romero R, Keelan JA. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), TRAIL receptors, and the soluble receptor osteoprotegerin in human gestational membranes and amniotic fluid during pregnancy and labor at term and preterm. J Clin Endocrinol Metab 2003; 88: 3835 3844 [DOI] [PubMed] [Google Scholar]
- Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H, Tsumoto K, Kumagai I, Kohda D, Maenaka K. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem 2006; 281: 10439 10447 [DOI] [PubMed] [Google Scholar]
- Sjoblom C, Roberts CT, Wikland M, Robertson SA. Granulocyte-macrophage colony-stimulating factor alleviates adverse consequences of embryo culture on fetal growth trajectory and placental morphogenesis. Endocrinology 2005; 146: 2142 2153 [DOI] [PubMed] [Google Scholar]
- Teesalu T, Blasi F, Talarico D. Expression and function of the urokinase type plasminogen activator during mouse hemochorial placental development. Dev Dyn 1998; 213: 27 38 [DOI] [PubMed] [Google Scholar]
- Allen MP, Nilsen-Hamilton M. Granzymes D, E, F, and G are regulated through pregnancy and by IL-2 and IL-15 in granulated metrial gland cells. J Immunol 1998; 161: 2772 2779 [PubMed] [Google Scholar]
- Hirst CE, Buzza MS, Sutton VR, Trapani JA, Loveland KL, Bird PI. Perforin-independent expression of granzyme B and proteinase inhibitor 9 in human testis and placenta suggests a role for granzyme B-mediated proteolysis in reproduction. Mol Hum Reprod 2001; 7: 1133 1142 [DOI] [PubMed] [Google Scholar]
- Kusakabe K, Li ZL, Kiso Y, Otsuki Y. Perforin improves the morphogenesis of mouse placenta disturbed by IL-2 treatment. Immunobiology 2005; 209: 719 728 [DOI] [PubMed] [Google Scholar]
- Maltepe E, Krampitz GW, Okazaki KM, Red-Horse K, Mak W, Simon MC, Fisher SJ. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development 2005; 132: 3393 3403 [DOI] [PubMed] [Google Scholar]
- Hynes RO. Targeted mutations in cell adhesion genes: what have we learned from them? Dev Biol 1996; 180: 402 412 [DOI] [PubMed] [Google Scholar]
- Bader BL, Rayburn H, Crowley D, Hynes RO. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all [alpha]v integrins. Cell 1998; 95: 507 519 [DOI] [PubMed] [Google Scholar]
- Rout UK, Wang J, Paria BC, Armant DR. Alpha5beta1, alphaVbeta3 and the platelet-associated integrin alphaIIbbeta3 coordinately regulate adhesion and migration of differentiating mouse trophoblast cells. Dev Biol 2004; 268: 135 151 [DOI] [PubMed] [Google Scholar]
- Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science 1994; 266: 1508 1518 [DOI] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA. Protein kinase B{alpha}/Akt1 regulates placental development and fetal growth. J Biol Chem 2003; 278: 32124 32131 [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Tessarollo L, Reid S, Jenkins N, Copeland N. The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development 1998; 125: 4607 4616 [DOI] [PubMed] [Google Scholar]
- Cross JC. Genetic insights into trophoblast differentiation and placental morphogenesis. Semin Cell Dev Biol 2000; 11: 105 113 [DOI] [PubMed] [Google Scholar]
- Coan PM, Ferguson-Smith AC, Burton GJ. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol Reprod 2004; 70: 1806 1813 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.