

Abstract
Calcium/calmodulin-dependent protein kinase II (CaMKII) interprets information conveyed by the amplitude and frequency of calcium transients by a controlled transition from an autoinhibited basal intermediate to an autonomously active phosphorylated intermediate (De Koninck and Schulman, 1998). We used spin labelling and electron paramagnetic resonance spectroscopy to elucidate the structural and dynamic bases of autoinhibition and activation of the kinase domain of CaMKII. In contrast to existing models, we find that autoinhibition involves a conformeric equilibrium of the regulatory domain, modulating substrate and nucleotide access. Binding of calmodulin to the regulatory domain induces conformational changes that release the catalytic cleft, activating the kinase and exposing an otherwise inaccessible phosphorylation site, threonine 286. Autophosphorylation at Thr286 further disrupts the interactions between the catalytic and regulatory domains, enhancing the interaction with calmodulin, but maintains the regulatory domain in a dynamic unstructured conformation following dissociation of calmodulin, sustaining activation. These findings support a mechanistic model of the CaMKII holoenzyme grounded in a dynamic understanding of autoregulation that is consistent with a wealth of biochemical and functional data.
Keywords: calcium signalling, CaMKII, EPR, kinase, spin labelling
Introduction
Cytosolic calcium ion (Ca2+) concentrations are dynamically regulated by hormonal and electrical signals to generate spatially localized intracellular calcium transients. Signalling pathways ‘translate’ information conveyed by the frequency, amplitude, and duration of the resulting Ca2+ oscillations to yield diverse biological outputs (Berridge et al, 2003). Often Ca2+ transients are ‘sensed’ by calmodulin, a ubiquitous Ca2+-binding protein that interacts with and activates numerous enzymes (Hoeflich and Ikura, 2002). Of its targets, calmodulin notably activates the family of Ca2+–calmodulin-dependent protein kinases (Soderling and Stull, 2001; Swulius and Waxham, 2008). These include the Ser/Thr kinase Ca2+/calmodulin-dependent protein kinase II (CaMKII), which operates in various signalling pathways targeting close to 40 known substrates (Hudmon and Schulman, 2002).
Unique among protein kinases, CaMKII assembles into a dodecameric holoenzyme via a dedicated C-terminal association domain (Hoelz et al, 2003; Gaertner et al, 2004; Rosenberg et al, 2006). The oligomeric assembly shapes the readout of Ca2+ signals by CaMKII, its mechanism of activation, and its interactions with substrates. The catalytic domain is tethered to the association domain via a linker region containing the regulatory domain, which in the basal state inhibits CaMKII in part by acting as a pseudosubstrate (Kelly et al, 1988; Payne et al, 1988). Furthermore, several studies show that autoinhibition also interferes with the binding of ATP (Shields et al, 1984; King, 1988; King et al, 1988; Colbran et al, 1989; Smith et al, 1992). Calcium signals activate CaMKII in a two-step process. First, binding of Ca2+/calmodulin (Ca2+/CaM) disrupts the interaction between regulatory and kinase domains. Second, high-frequency Ca2+ pulses or longer Ca2+ transients trigger transphosphorylation at Thr286 by a neighbouring subunit's kinase domain to induce a novel conformation with enhanced affinity for Ca2+/CaM (Meyer et al, 1992; Waxham et al, 1998). Following dissociation of Ca2+/CaM, Thr286-autophosphorylated kinase retains autonomous (Ca2+ independent) activity, further sustaining the signalling response (Lai et al, 1986; Miller and Kennedy, 1986). In contrast, intrasubunit autophosphorylation at Thr305/Thr306 abrogates CaM binding and generates a Ca2+ desensitized form of the kinase (Hanson and Schulman, 1992; Colbran, 1993).
As a ubiquitously expressed protein, CaMKII has critical roles in numerous biological pathways. For example, CaMKII has several important functions in regulating normal heart function and CaMKII inhibition is a promising therapeutic strategy (Zhang et al, 2005). Most notably, in the brain CaMKII is critical for the normal regulation of synaptic transmission (reviewed by Lisman et al, 2002). Knock-in genetic mutations that remove the Thr286 or Thr305/306 autophosphorylation sites disrupt synaptic plasticity, learning and memory in mice (reviewed by Elgersma et al, 2004). CaMKII is hyperphosphorylated in a mouse model of Angelman Syndrome (Weeber et al, 2003) a complex mental disability, and several behavioural phenotypes of these mice are corrected by knock-in mutations that remove the Thr305/306 autophosphorylation sites (van Woerden et al, 2007). CaMKII is also hyperphosphorylated at Thr286 in dopamine-depleted rodent striatum, and the associated parkinsonian symptoms can be corrected by CaMKII inhibition (Picconi et al, 2004; Brown et al, 2005). Thus, developing a dynamic structural framework for CaMKII activation by CaM, for acquisition of conformational memory, and for interactions with protein partners has the potential to inform new therapeutic strategies. However, difficulties in crystallizing the intact holoenzyme assembly hindered a structural understanding of CaMKII regulation. Although the assembly has been visualized by cryoEM (Kanaseki et al, 1991; Kolodziej et al, 2000; Morris and Torok, 2001; Gaertner et al, 2004), the resolution was not conducive to a detailed analysis of subunit structures. In addition, there are no data directly describing detailed conformational rearrangements involved in the activation process.
Crystal structures of CaMKII monomers, consisting of the kinase and regulatory domains and lacking the association domain, have led to conflicting models of autoinhibition. In an inactive Caenorhabditis elegans CaMKII structure (PDB 2BDW), the regulatory domains from two monomers form an antiparallel coiled coil (Rosenberg et al, 2005). Although it partially blocks the substrate binding site, the regulatory domain does not contact the ATP-binding lobe as postulated from biochemical data (Shields et al, 1984; King, 1988; King et al, 1988; Colbran et al, 1989; Smith et al, 1992) and from structures of the homologous and monomeric CaMKI (PDB 1A06) (Goldberg et al, 1996). The regulatory domain coiled coil has been proposed to pair kinase domains in the holoenzyme stabilizing CaMKII in an inactive conformation and to explain cooperative activation by Ca2+/CaM binding (Chao et al, 2010). Previous studies support the notion that kinase domains form a dimer in the holoenzyme (Kanaseki et al, 1991; Thaler et al, 2009), but there is no direct evidence that dimerization is mediated by the regulatory domain, suggesting that the coiled-coil structure may be favoured by crystal packing. In fact, several recent crystal structures of monomeric human CaMKII, which are 77% identical to the monomeric C. elegans CaMKII, did not show the coiled-coil dimers (PDBIDs 2VZ6, 3BHH, 2V7O, 2VN9) (Rellos et al, 2010). In one structure, the regulatory domain resolved through residue 308 appears docked against the catalytic cleft (PDB 2VN9). However, this conformation may also be influenced by contacts between molecules in the crystal lattice. Furthermore, its mechanistic implications are confounded by the presence of an ATP-binding site inhibitor. In all the other structures of human CaMKII, the regulatory domain is not influenced by crystal contacts but the structures are only visible to residue 301. Thus, the conformation of the regulatory domain in the basal (apo) state is highly ambiguous.
The regulatory domain adopts a vastly different conformation in structures of C. elegans CaMKII, partially truncated in the regulatory domain (PDB 3KK8) (Chao et al, 2010), and in complexes of human CaMKII with Ca2+/CaM (PDB 2WEL) (Rellos et al, 2010). The Thr286-autophosphorylation site of one monomer is bound to the active site of another monomer presumably mimicking a transphosphorylation intermediate. Thus, the inference from functional and structural data is that regulation of CaMKII activity entails a complex series of conformational changes initiated at the regulatory domain and modulating its interactions with the kinase domain.
Existing crystal structures of monomeric kinase domains have been extrapolated to mechanistic models of cooperative Ca2+ sensing and catalytic activation of the holoenzyme (Rosenberg et al, 2005; Chao et al, 2010; Rellos et al, 2010). Experimental testing of these models in solution in the absence of constraints imposed by the crystal lattice is essential for an in-depth understanding of the complex landscape of holoenzyme activation. Motivated by these conflicting models and the dearth of available information about conformational changes associated with activation, we report the first analysis of regulatory domain dynamics in solution using systematic spin labelling and electron paramagnetic resonance (EPR) spectroscopy (Hubbell et al, 1996, 2000) of monomeric C. elegans CaMKII. We describe the secondary structure, tertiary interactions, and backbone dynamics of the regulatory domain in discrete catalytic intermediates. Our results reveal the structural and dynamic changes underlying activation by Ca2+/CaM and Thr286 autophosphorylation and suggest a model of CaMKII holoenzyme activation. Central to this model is the novel finding that the regulatory domain is in a conformeric equilibrium, which modulates the affinity for nucleotides, substrates, and CaM.
Results
General methodology
Our studies focussed on the C. elegans CaMKII kinase monomer, consisting of the catalytic and regulatory domains (residues 1–340) (Figure 1A), the structure of which was determined previously (PDB 2BDW) (Rosenberg et al, 2005). To enable site-specific incorporation of spin labels, single cysteine mutants were introduced in a cysteine-less CaMKII background hereafter referred to as WT* (see Materials and methods). Purified WT* CaMKII is partially truncated during expression (Praseeda et al, 2004) at residue 318, but retains the entire regulatory domain (Supplementary Figure S1A and B). The region beyond this residue was not resolved in the crystal structure, suggesting it is flexible and thus prone to proteolysis (Rosenberg et al, 2005; Chao et al, 2010). Using engineered constructs consisting of residues 1–318, we verified that the truncation does not affect the EPR lineshapes at representative sites along the regulatory domain (Supplementary Figure S1C). Moreover, the cysteine substitutions do not compromise the structural and functional integrity of the enzyme. The specific kinase activity of WT*, measured by phosphorylation of peptide substrates syntide-2 (Table I) or autocamtide-2 (data not shown), is similar to that of the WT C. elegans monomer, as well as the WT mouse CaMKII holoenzyme and monomer (Table I). Furthermore, structural parameters deduced from far-UV and near-UV circular dichroism (CD) analysis and analysis of the melting temperature are consistent with retention of secondary and tertiary structures (Figure 1B and C).
Figure 1.

(A) Schematic representation of the full-length CaMKII gene and the monomer construct used for these studies. The regulatory domain is highlighted in red with the CaM-binding region in orange. The R1, R2, and R3 refer to segments of the regulatory domain defined previously (Chao et al, 2010). (B) Far-UV and (C) near-UV circular dichroism analysis for the wild-type and cysteine-less CaMKII (WT*) along with their D135N inactivated counterparts. The spectra are almost superimposable confirming the lack of substantial structural changes due to the cysteine substitutions. (D) Stability curves demonstrating the lack of substantial effects on melting temperature by the cysteine substitutions.
Table 1. Activities of CaMKII variants.
| Protein | Activity (μmol/min/μg) |
|---|---|
| Mouse CaMKIIα holoenzyme WT | 5.62±0.15 |
| Mouse CaMKIIα 1–380 WT | 7.13±0.16 |
| C. elegans 1–340 WT | 6.04±0.76 |
| C. elegans 1–340 WT* | 4.59±0.05 |
| C. elegans 1–340 D135N* | 0* |
| Average and range of activities of CaMKII variants determined by incorporation of radiolabelled phosphate from [γ32P]ATP onto the peptide substrate syntide-2 (n=2). *P<0.01 versus ‘mouse CaMKIIα holoenzyme WT’ and ‘C. elegans 1–340 WT*’ by ANOVA followed by Bonferroni post-test: activities of all other kinases are not statistically different from WT mouse CaMKIIα holoenzyme. | |
To circumvent heterogeneous phosphorylation at Thr286, Thr305, Thr306, or other sites during Escherichia coli expression, we adopted the strategy used in the crystallographic studies of replacing the highly conserved D135 in the catalytic loop (Ten Eyck et al, 2008) with asparagine to inactivate WT* (Rosenberg et al, 2005) creating a protein hereafter referred to as D135N*. Analysis by mass spectrometry confirmed the absence of detectable phosphorylation in the D135N background (data not shown). The D135N substitution does not change the CD signature of the CaMKII monomer (Figure 1B and C) or the half-maximal temperature of the melting curve but increases the cooperativity of unfolding (Figure 1D). We speculate that the apparent non-cooperative WT unfolding reflects heterogeneous autophosphorylation leading to multiple protein populations, each with a different melting temperature.
Twenty-five single cysteine and three double Cys mutants were introduced into the regulatory domain of D135N*. Spin-labelled CaMKII mutants were analysed by EPR spectroscopy under conditions designed to stabilize specific intermediates on the activation pathway. Conformational changes underlying the transitions between these intermediates were deduced from analysis of spin label mobility and proximities in spin label pairs. The mobility of a spin label refers to dynamic modes on the nanosecond timescale that average the anisotropic tensors of the spin labels. These include rotation around bonds that tether the label to the backbone and large amplitude motion of the backbone (Mchaourab et al, 1996; Hubbell et al, 1998; Columbus et al, 2001; Columbus and Hubbell, 2004). The overall tumbling of the 40-kDa CaMKII monomer has a mimimal contribution to the EPR lineshape.
Dynamic equilibrium of the regulatory domain in the apo intermediate
Although the C. elegans autoinhibited non-phosphorylated kinase domain (referred to hereafter as the apo intermediate) crystallizes as a dimer, dimers could not be detected in solution using various analytical methods such as gel filtration and analytical ultracentrifugation, even at moderate concentrations (Rosenberg et al, 2005; Chao et al, 2010). However, Rellos et al (2010) reported the detection of dimeric human CaMKII isoforms in solution at 4°C, with dissociation constants in the 200–600 μM range although the interface of this dimer does not seem to involve the regulatory domain. Consistent with this observation, we found no spectroscopic evidence of regulatory domain dimerization at room temperature. Spin labels predicted to be within 5 and 13 Å (Supplementary Figure S2) at the crystallographic coiled-coil interface did not show evidence of spin–spin coupling. Short range distances between spin labels lead to extensive dipolar coupling and broadening of the EPR spectrum (Mchaourab et al, 1997; Dong et al, 2005). No such spectral broadening was observed for these sites up to concentrations of 200 μM, above which the protein aggregated non-specifically to form an insoluble precipitate. Thus, the coiled-coil regulatory domain dimer is not stable in solution at room temperature, although we cannot exclude the possibility that higher effective subunit concentrations in the holoenzyme (estimated at 3 mM) allow for this interaction (Rosenberg et al, 2005). However, consistent with our data, the coiled-coil dimer was not observed in the multiple crystals of human CaMKII isoforms (Rellos et al, 2010). Thus, multiple lines of evidence using different experimental approaches suggest that the regulatory domain dimeric interface is an artifact of the crystallization of C. elegans CaMKII.
In contrast to the rigid regulatory domain structure in the crystal, EPR lineshape analysis for a set of spin-labelled mutants in this domain reveals evidence of multiple conformational states. In the R2 and R3 segments of the regulatory domain (residues 292–297 and 298–314, respectively, defined previously (Chao et al, 2010), Figure 1A), multiple populations of spin labels with vastly distinct mobilities are detected (Figure 2A; Supplementary Figure S3A). Sharp components in these spectra (indicated with arrows in Supplementary Figure S3A) reflect spin labels undergoing fast and large amplitude motion with ∼1 ns effective correlation times (Table II). This type of motion is indicative of an unstructured backbone and is inconsistent with an ordered α-helical backbone. Another spectral component (indicated with circles in Supplementary Figure S3A) arises from spin labels undergoing slower motion indicative of a sterically restricted environment. This spin label population cannot arise from the putative coiled coil since there is no evidence of spin–spin coupling (Supplementary Figure S2).
Figure 2.

(A) Analysis of spin label dynamics at site 307. The experimental EPR spectrum was fit by two components using the MOMD analysis as described in the Materials and methods section. The fast component (green spectrum) arises from an unstructured, undocked conformation of the R3 segment while the slow (orange spectrum) component arises from a docked conformation. (B) Percentages of the fast and the slow components for representative residues in the R3 segment in the apo CaMKII state (black bars) and the ATP-bound state (purple bars). (C) Inverse correlation times (τ−1) for sites in the R1 region plotted versus residue number. The correlation time was determined from non-linear least-squares analysis of the EPR spectra (see Materials and methods). The superimposed sinusoid fit to the data has a 3.7 period. The local environment of these residues in the crystal structure (PDB 2BDW) is shown to highlight the agreement with the EPR data (image created with PyMOL, DeLano Scientific (DeLano, 2002)). Thus, residues on the surface (green) (e.g. 289) have a large τ−1 while those at the interface with the catalytic domain (orange) have small τ−1.
Table 2. Rotational correlation times.
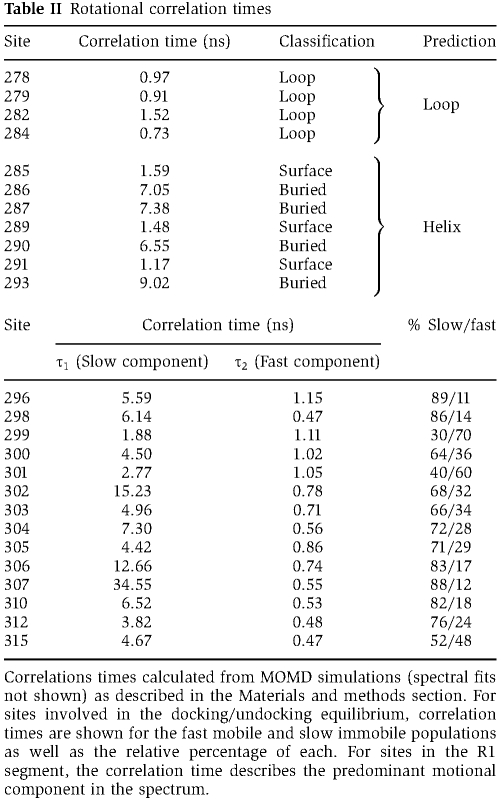
Rather, we interpret these results as evidence of an equilibrium between an undocked dynamic conformation (leading to fast spin label motion) and a structured conformation (leading to restricted motion) where the R2 and R3 segments are docked to the catalytic domain presumably near the ATP-binding site. The EPR spectra capture two extremes of this equilibrium, revealing that the underlying conformational change is slow on the EPR timescale, on the order of 100 ns. Using non-linear least-squares analysis (see Materials and methods), we determined the average rotational correlation time (Budil et al, 1996) and fraction of spin labels in each of the two populations. The component spectra representing the two conformations were summed in varying ratios to fit the EPR spectra for sites 302–307 (Figure 2B, black bars; Supplementary Figure S4).
Lineshapes for spin labels in the R1 segment (residues 278–291) consist of single predominant components (Supplementary Figure S3C and D), demonstrating structural decoupling between R1 and the rest of the regulatory domain. In particular, the spin label environment at residues 286, 287, and 293 is restricted with lineshapes similar to those observed in protein hydrophobic cores (Supplementary Figure S3C). The ordered backbone and solvent inaccessibility indicate that these residues are buried by contacts with the catalytic domain consistent with their local environments in the crystal structure (Figure 2C).
Secondary structure of the regulatory domain
In segments of secondary structure, spin label mobility varies as a function of residue number (Mchaourab et al, 1996). Quantitative analysis of spin label dynamics along the 284–293 stretch of residue (R1 helix) reveals a 3.7 periodicity strongly indicating an α-helical backbone (Figure 2C; Table II). The phase of the helix is in concordance with the crystal structure (PDB 2BDW) (Rosenberg et al, 2005), where the helix surface encompassing 286 and 287 is docked against the catalytic domain. The spin labels are uniformly mobile in the preceding 278–284 (R1 loop) segment, consistent with a local loop structure (Table II; Supplementary Figure S3D). Analysis of spin label mobility from residue 296 through residue 307 is complicated by the multicomponent nature of the EPR spectra. However, sharp and narrow spectral components for sites 299–301 (Supplementary Figure S3B) indicate a loop structure (Table II).
The presence of helical secondary structures can also be deduced from proximities between i, and i+4 residues in consecutive helical turns. The expected 5.5 Å separation between the spin labels should result in extensive broadening of the EPR lineshape due to strong dipolar coupling and direct overlap of the unpaired electron wavefunctions (Fiori et al, 1993; Mchaourab et al, 1997). In contrast, flexible unstructured segments are characterized by average distances >13 Å and broad distance distributions. Short range coupling was evident in the spectrum of spin label pairs introduced four residues apart in the 285–293 region (Supplementary Figure S5A), with a corresponding narrow distance distribution centred at 8 Å (Figure 3A, black trace). In contrast (i, i+4) spin label pairs in the calmodulin-binding segment have a multimodal distance distribution consistent with a significant fraction of unstructured backbone with some α-helical characteristics (Figure 3B; Supplementary Figure S5B). Taken together, our EPR data are consistent with a solution structure in which the R1 helix is stably docked to the C-terminal lobe of the catalytic domain and the R3 segment is in equilibrium between docked and undocked conformations, thereby inhibiting the kinase in the absence of CaM binding.
Figure 3.

Representative distance distributions between (i, i+4) spin label pairs in the R1; 286/290 (A) and R3; 300/304 (B) segments. The distributions, consisting of the probability of given a distance P(r), were obtained from fits to the EPR spectra as detailed in the Materials and methods and shown in Supplementary Figures S5 and S7A. Ca2+/CaM binding to R3 induces conformational changes along the entire regulatory segment evidenced by changes in average distance and breadth of the distribution (A, red trace) and increased dynamics of the R1 segment illustrated by sharper EPR lineshapes at sites 287 and 282 (C) and increases in the inverse correlation times (D).
The CaM-binding segment of the regulatory domain senses ATP binding
Since CaMKII activation is associated with increased affinity for ATP (see Introduction), we hypothesize that the R3 dynamic equilibrium facilitates active site exposure for binding of both nucleotide and substrate. Thus, we predict ATP binding would shift the R3 equilibrium towards the undocked population. Indeed, while the addition of ATP does not change the EPR lineshapes in the R1 segment (data not shown), it increases the fraction of spin labels that are in the undocked conformation in the R3 segment (Figure 2B; Supplementary Figure S6A and B). This allosteric modulation by ATP suggests that the R3 segment interacts with the nucleotide-binding lobe. Furthermore, it suggests that the R3 equilibrium is not an artifact arising from the truncation of the association domain. Rather it represents a mechanism to modulate ATP affinity.
To confirm that the shift in equilibrium arises from ATP binding, spin labels were introduced at representative sites along the regulatory domains in a D135N*/K42M background. The Lys to Met substitution, which is expected to abolish ATP binding, eliminates the changes in the mobile component in the R3 segment. In the apo form, the mutation shifts the equilibrium towards the docked conformation (Supplementary Figure S6C). Thus, it is likely that the shift in the R3 equilibrium results from ATP competing this segment away from the nucleotide-binding lobe.
Ca2+/CaM binding stabilizes an ordered helical conformation in the R3 segment
The CaM-binding region, which is in equilibrium between unstructured and docked conformations in the apo state, undergoes conformational transitions upon Ca2+/CaM binding. For the (i, i+4) doubly labelled 300/304 mutant, binding induces spectral broadening relative to the apo state deduced from comparison of the lineshape and intensity (Supplementary Figure S7A). The corresponding distance distribution shifts to shorter distances indicating stabilization of α-helical structure (Figure 3B). Ca2+/CaM binding also leads to a reduction in the mobile fraction of spin labels, suggesting that most R3 residues are in tertiary contacts with CaM (Supplementary Figure S7B). These changes are consistent with the crystal structure of Ca2+/CaM bound to the corresponding CaMKII peptide (residues 293–310) (PDB 1CDM) (Meador et al, 1993) and a recent structure of the CaMKII/CaM complex (PDB 2WEL) (Rellos et al, 2010).
Ca2+/CaM-mediated activation induces unfolding of the R1 helix
Concomitant with the ordering of the R3 helix, spin labels at a number of sites in the R1 segment (274–291) report substantial but reversible increases in mobility upon Ca2+/CaM binding (Supplementary Figure S7C). This is illustrated by residue 287, which in the apo state is buried at the interface with the catalytic domain (see above). Binding of Ca2+/CaM leads to a 10-fold increase in rotational correlation time (Figure 3C, red trace). This loss of tertiary contacts with the catalytic domain is accompanied by unfolding of the helix, as evidenced by increased distance between spin labels at (i, i+4) residues (Figure 3A). In addition, representative site 282, which is surface exposed in the neighbouring R1 loop, reports a three-fold increase in mobility, indicating an increase in the dynamics of the backbone upon CaM binding (Figure 3C). The increase in backbone flexibility and loss of tertiary contacts create an expanded loop spanning residues 278–291 surrounding the Thr286-phosphorylation site (Figure 3D).
Unfolding in the R1 region is consistent with a recent crystal structure of CaMKII bound to Ca2+/CaM (PDB 2WEL) (Rellos et al, 2010), in which the R1 segment is docked at the active site of an adjacent kinase monomer in the crystal seemingly positioning Thr286 for transphosphorylation. Evidence of this interaction occurring in solution was also obtained from analytical ultracentrifugation at 4°C, revealing a rather moderate KD of 50–100 μM (Rellos et al, 2010). However, the mobile EPR lineshapes in the R1 segment are inconsistent with the formation of Ca2+/CaM/CaMKII dimers in solution. It is possible that these dimers are not stable at the concentrations used in the EPR experiments (50–100 μM) at 25°C. In addition to the mobile EPR lineshapes, we failed to observe such dimers using light scattering (data not shown) in disagreement with data reported previously (Chao et al, 2010).
Autophosphorylation induces unfolding of the R1 helix
Transition of CaMKII to the autonomously active intermediate is mediated by autophosphorylation at Thr286, which in the apo state is in a solvent inaccessible environment. To investigate the conformational changes induced by Thr286 phosphorylation, we carried out mobility analysis of spin labels introduced in a phosphomimic T286E mutant on the D135N* background. This mimicry approach is necessitated by the expected heterogeneous autophosphorylation at Thr253, Thr305 and Thr306 (Miller et al, 1988; Lou and Schulman, 1989) in the WT* enzyme, which would complicate interpretation of the EPR data.
Spin labels report that the R1 helix surface buried by interactions with the catalytic domain in the autoinhibited apo D135N* monomer (i.e. residues 286 and 287) becomes exposed in the T286E/D135N* mutant, with EPR lineshapes characteristic of an unstructured backbone (Figure 4A; Supplementary Figure S8A). Local unfolding of the helix is also manifested by increased distance between spin labels introduced at (i, i+4) residues (Figure 4B). Presumably, disruption of contacts between the R1 helix and the catalytic domain accounts for the lower melting temperature of CaMKII T286E/D135N* (Figure 4C). The similarity of the EPR parameters of the T286E mutant to those observed in the Ca2+/CaM-bound conformation is consistent with the notion that Thr286 autophosphorylation prevents docking of the 287–289 segment to the catalytic domain thereby blocking reinstatement of autoinhibition after CaM dissociates. One notable difference is that the phosphorylation-mimicking mutation leaves the conformeric equilibrium of the CaM-binding R3 region intact (data not shown).
Figure 4.

(A) Comparison of the inverse rotational correlation time of spin labels in three CaMKII intermediates showing the increase in dynamics of the R1 and R2 segments in the presence of Ca2+/CaM and in the T286E mutant. (B) The phosphorylation-mimicking mutation T286E destabilizes the R1 helix leading to an increase in the width of the distance distribution between (i, i+4) spin label pairs. (C) R1 helix unfolding is manifested by a shift in the melting temperature of T286E. (D) Representative EPR lineshapes highlighting the increased dynamics of the regulatory domain in the CaM-bound T286E mutant.
Phosphorylation of CaM-bound CaMKII further increases the flexibility of the R1 region
Autophosphorylation of CaMKII at Thr286 increases Ca2+/CaM affinity by more than a 1000-fold (Meyer et al, 1992; Waxham et al, 1998). While it has been established that the origin of this effect involves the transition of CaMKII-bound CaM to a high-affinity ‘trapped’ conformation (Waxham et al, 1998), corresponding changes in CaMKII structure have yet to be defined. We observed distinct changes in backbone order upon binding of Ca2+/CaM to the T286E/D135N* mutant. EPR spectra in the R1 segment become sharper consistent with increased flexibility of the backbone (Figure 4D). While CaM binding alone induces an expanded loop conformation in the Thr286 region, addition of the phosphomimicking mutation causes further increases in mobility as illustrated by the spectral lineshape for site 289 (Figure 4D). With Ca2+/CaM bound to the T286E mutant, the spectra at sites 278–291 (Supplementary Figure S8B) are similar to those of unstructured peptides or intrinsically disordered proteins (Morin et al, 2006). Thus, we conclude the R1 helix experiences complete unfolding in the CaM-trapped intermediate (i.e. CaM bound and Thr286 phosphorylated).
Structural changes are also observed in the region consisting of residues 291–294 that has been linked to Ca2+/CaM trapping (Waxham et al, 1998). Although CaM binding has no substantial effect on EPR lineshape at residue 293 in D135N*, CaM induces a distinct increase in mobility of residues 293 in T286E/D135N* (Figure 4D), suggesting that disruption of the tertiary contacts with the catalytic domain are involved in the trapping phenomenon. Significant changes in the immobile components are also observed at sites 299 and 303 in the CaM-binding region (Supplementary Figure S8C), suggesting that the T286E mutation affects the interactions between CaM and D135N* CaMKII.
Discussion
Our results present a novel perspective on CaMKII regulatory mechanisms, revealing the link between dynamics and interactions of the regulatory domain and the activation state of the enzyme. The structural plasticity of this domain manifested by the conformeric equilibrium described here is a central element in autoregulation, serving to modulate substrate and nucleotide access and affinity for Ca2+/CaM. Our findings are summarized in the cartoon of Figure 5, illustrating how conformational changes of the regulatory domain and its multiple interactions with the kinase domain underlie transitions between CaMKII intermediates.
Figure 5.

(A) Model of the mechanism of autoinhibition and activation of CaMKII. The CaMKII monomer is represented with N- and C-lobes of the catalytic domain in blue and the regulatory domain in red. The ATP-binding and Thr286-docking sites are indicated in grey with letters ‘A’ and ‘T’, respectively. In the basal, apo intermediate, the R3 segment of the regulatory domain is in a dynamic equilibrium between docked and undocked, flexible (indicated by parentheses) conformations. ATP binding shifts the equilibrium towards the undocked conformation facilitating exposure of the catalytic cleft. CaM (yellow) binding releases autoinhibition and primes the regulatory domain for phosphorylation at residue 286. Autophosphorylation at Thr286 causes further flexibility of the regulatory domain and exposes site 293, which is implicated in CaM trapping. While CaM dissociation allows for reinstatement of the R3 equilibrium, the R1 helix remains predominantly unstructured. (B) Cartoon model of cooperative activation in the holoenzyme. CaM binding to one subunit causes undocking and unfolding of R1. The released R1 can displace the R3 segment of a neighbouring kinase subunit from the catalytic cleft. This R3 is undocked and primed for CaM association.
In the basal state, the R3 segment containing the CaM-binding region partitions between an undocked, dynamic conformation and one where it interacts with the catalytic domain reducing affinity to nucleotides and presumably substrate access to the active site. The docked conformation is consistent with the structure of the human CaMKIIδ isozyme (PDB 2VN9) placing Thr306 at the catalytic cleft (Rellos et al, 2010). We found that saturating concentrations of ATP reduce, but do not eliminate, binding of R3 at or near the active site. Thus, in the docked conformation Thr306 remains proximal to the active site when ATP is bound allowing slow basal intrasubunit autophosphorylation (Colbran, 1993). This interaction also explains in a structural context previous biochemical results, demonstrating that regulatory domain peptides compete not only with substrates but also with ATP binding (Kelly et al, 1988; Payne et al, 1988; Colbran et al, 1989; Smith et al, 1992) and that regulatory domain peptides protect reactive arginine residues in nucleotide-binding site from covalent modifications that inactivate CaMKII (King, 1988; Smith et al, 1992). Because cellular concentrations of ATP are well above the KD of binding to CaMKII, a substantial fraction of R3 is presumably in the undocked state, physiologically primed for Ca2+/CaM binding. Stable docking of the R1 helix to the catalytic domain buries Thr286, ensuring a stringent dependence on Ca2+ for activation.
Our data directly demonstrate that binding of Ca2+/CaM to the R3 segment induces significant changes in the backbone structure and interaction of the regulatory/catalytic domains that cause activation. The R3 equilibrium is shifted to the undocked conformation fully releasing the catalytic cleft for nucleotide binding. The CaM-binding region is stabilized in a helical conformation consistent with crystal structures of this peptide bound to Ca2+/CaM (PDB 1CDM) (Meador et al, 1993) as well as a recent crystal structure of monomeric CaMKII bound to Ca2+/CaM (PDB 2WEL) (Rellos et al, 2010). This conformational change propagates to the 285–293 R1 helix exposing Thr286 to the solvent. Furthermore, the unfolding of this helix effectively expands the length of the R1 region, presumably enabling the binding of Thr286 at the active site of a neighbouring subunit in the holoenzyme, a prerequisite for transphosphorylation and transition to the autonomously active intermediate. In addition, undocking of the R1 segment disrupts interactions with helix D, which was proposed to stabilize an inactive conformation of the catalytic domain (Rosenberg et al, 2005). In the CaMKIIδ/CaM structure, the orientation of the highly conserved E97 is altered to coordinate the sugar moiety of ATP, as seen in glycogen phosphorylase kinase (Huang et al, 1995). In combination, these structural changes likely mediate the 10-fold increase in nucleotide affinity following activation by Ca2+/CaM (Shields et al, 1984; King et al, 1988; Colbran, 1993).
Thr286 phosphorylation is thought to generate the autonomously active form by blocking reinstatement of autoinhibition upon dissociation of Ca2+/CaM (Lai et al, 1986; Miller and Kennedy, 1986). Indeed, we find that the phosphomimic T286E maintains the R1 helix in a predominantly unfolded and undocked conformation. However, in contrast to the CaM-bound intermediate, the R3 conformeric equilibrium is restored allowing contacts with the catalytic cleft. This may explain the lower activity of autophosphorylated CaMKII relative to the Ca2+/CaM-bound intermediate (Hanson et al, 1994). The relative autonomous activity of CaMKII holenzymes can be increased by using higher concentrations of ATP and peptide substrates (Smith et al, 1992), supporting the restoration of a competitive R3 interaction with the nucleotide-binding site. However, binding of Ca2+/CaM to the autonomously active Thr286-phosphorylated form fully activates CaMKII. In support of this conclusion, we found that the R1 segment is most strongly disordered in Ca2+/CaM-bound/T286E CaMKII, indicative of essentially complete disruption of interactions between regulatory and catalytic domains. The region between residues 291 and 294 previously implicated in the CaM-trapping phenomenon (Meyer et al, 1992; Waxham et al, 1998) also shows conformational changes unique to this intermediate. However, introduction of negative charge by the T286E mutation may not fully mimic the effects of a phosphate group, and details of the local dynamics may be somewhat different in the Thr286-phosphorylated kinase.
Assembly and interaction of the CaMKII subunits in holoenzymes allow for transphosphorylation of one subunit by a neighbouring one and mediates cooperative CaM binding and substrate phosphorylation. A model was proposed on the basis of the crystal structure of the autoinhibited kinase domain and extensive analysis of cooperative substrate phosphorylation in the holoenzyme (Chao et al, 2010). A central element in this model is dimerization of the autoregulatory domain, which locks the enzyme in an inactive conformation. Binding of Ca2+/CaM disrupts the dimer simultaneously activating the kinase domain of one subunit and releasing the CaM-binding region of the other subunit. However, we found no evidence for regulatory domain dimerization and this model cannot rationalize cooperative CaMKII activation. Explanation for the experimental Hill coefficient of 3–4 as estimated previously (Chao et al, 2010) requires the postulation of additional interactions between kinase domains. Moreover, the coiled-coil motif was not detected in a recent structure of human CaMKII kinase domains bound to ATP inhibitors (PDB 2VN9) (Rellos et al, 2010). In this structure, part of the R3 segment of the regulatory domain was folded back onto the catalytic cleft consistent with our data.
The results presented here can be extrapolated to propose an alternative model of autoinhibition and activation of the CaMKII holoenzyme. Rather than forming the coiled coil, the R3 segment is in an equilibrium that involves interactions with the catalytic domain partially blocking the nucleotide-binding region. Although the dynamics of the R3 segment are likely to be modified by its linkage to the association domain in the holoenzyme, the effects of ATP on the R3 equilibrium demonstrate its role in activation. Binding of CaM disrupts the autoregulatory/catalytic domain interactions at the R3 and R1 segments through concomitant but opposite structural changes of the backbone. Our model is also consistent with previous work showing that Ca2+/CaM binding to CaMKII holenzymes enhances affinities for nucleotides (Shields et al, 1984; King et al, 1988; Colbran, 1993).
How is cooperativity achieved in the context of this model? We propose that upon unfolding, the R1 segment becomes available for binding at the active site of a neighbouring kinase domain effectively competing with the R3 segment and displacing it. The shift in equilibrium would increase the apparent affinity to Ca2+/CaM hence accounting for the positive cooperativity with respect to Ca2+/CaM binding. To experimentally test this model, we monitored the R3 equilibrium at site 307 in the presence of a peptide corresponding to the autoregulatory domain, referred to as autocamtide-2 (AC-2). Consistent with the model's prediction, peptide binding shifts the equilibrium towards the undocked state reflected by the sharp population in the EPR spectrum (Figure 6). AC-2 mimics the unfolded and flexible R1 segment in the Ca2+/CaM-bound state, which can reach over to a neighbouring kinase domain and displace its R3 segment. The high concentrations required in the case of monomeric kinase suggest a low-binding affinity. However, this interaction will be potentiated in the holoenzyme, which concentrates subunits to an unusual extent. Moreover, the AC-2 concentration threshold for undocking R3 is reduced in the T286E mutant consistent with higher affinity for substrate in this intermediate. Similar conformation changes in the regulatory domain could be induced using a peptide based on the CaMKIIN inhibitor (Supplementary Figure S9). Thus, although phosphorylation does not perturb the R3 equilibrium, phosphorylated kinase domains are more likely to capture the unfolded R1 segment of a neighbouring subunit allowing the propagation of autophosphorylation. These results are in line with recent studies, demonstrating that addition of an inactivated kinase monomer lacking the regulatory domain (‘decoy’ kinase) leads to apparent cooperativity in the activation of an intact kinase monomer with respect to Ca2+/CaM (Chao et al, 2010). The interpretation of Figure 6 is consistent with a previous suggestion that CaM binding releases and unfolds the R1 segment to act as a ‘molecular grappling hook’ that can be captured by the decoy enzyme (Chao et al, 2010).
Figure 6.

Shifts in the R3 equilibrium induced by binding of autocamtide-2 (AC-2). EPR spectra for site 307 show that the level of the mobile spectral component arising from undocked conformation of the R3 segment increases in the presence of AC-2. The T286E mutation increases the affinity of this interaction as evidenced by the lower molar ratio of peptide to CaMKII (x axis) required to increase the mobile spin label fraction.
While grounded in analysis of solution dynamics under well-defined biochemical conditions, the ultimate test of the veracity of our model awaits structural and dynamic analyses of the holoenzyme. This will be especially pertinent because crystals of monomeric human CaMKII bound to inhibitors (PDB 2VZ6, 3BHH, 2V7O, 2VN9) (Rellos et al, 2010) suggest an interface between kinase domains different than the one originally observed (PDB 2BDW) (Rosenberg et al, 2005). Intersubunit communications predicted by cooperative CaM binding and autophosphorylation have been observed by FRET studies of catalytic domain ‘pairs’ that separate upon activation (Thaler et al, 2009). Given the strong amino-acid sequence identity between catalytic/regulatory domains of the C. elegans protein studied here and the four mammalian CaMKII isoforms (Tobimatsu and Fujisawa, 1989), it seems likely that the conformational dynamics defined here for the first time will represent core regulatory properties of all CaMKII isoforms. However, variable linker domains that couple catalytic/regulatory domains to the central ‘hub’ of the dodecameric holoenzyme likely modify these dynamics in unique ways. For example, activation properties of CaMKIIβ holoenzymes are modulated by the length and sequence of the linker (Brocke et al, 1999; Bayer et al, 2002). Elucidating the conformational dynamics associated with activation of various CaMKII holoenzymes is the next frontier in understanding CaMKII activation.
Materials and methods
CaMKII expression, purification, and labelling
C. elegans CaMKII (unc-43) cDNA derived from the splice variant K11E8.d with codons representing residues 1–340 and an N-terminal 10 histidine sequence was cloned into the pET28b vector controlled by the T7 promoter. All mutants were introduced using a modified Quikchange protocol and verified with DNA sequencing. A D135N inactivating mutant was introduced to prevent heterogeneous autophosphorylation during expression (Rosenberg et al, 2005). Native cysteines were replaced with residues found in homologs or with alanine; C30A, C64L, C115A, C126I, C199A, C272A, and C289A. Protein expression was carried out in E. coli BL21(DE3) as described previously except for a 16-h induction protocol (Rosenberg et al, 2005). Harvested cell pellets were resuspended in buffer containing 50 mM HEPES, 150 mM NaCl, 10 mM imidazole, 1 mM DTT, 0.1 mM PMSF, 10% (vol/vol) glycerol, pH 8, and lysed by sonication. CaMKII was purified from the lysate supernatant by Ni2+ affinity chromatography (Qiagen). Upon elution, 20-fold excess of (1-oxyl-2,2,5,5-tetramethylpyrrolinyl-3-methyl)-methanethiosulfonate spin label (MTSSL; Toronto Research Chemicals, Inc) was added and the mixture incubated on ice for at least 15 h. Labelled CaMKII was further purified by size exclusion chromatography on a Superdex 75 (GE Healthcare) in 50 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, 0.02% azide, pH 7.5. For these studies, the N-terminal His tag was not cleaved. Spin-labelled CaMKII mutants were washed three times immediately before experiments with the same buffer to remove residual free spin label. Protein concentration was determined using a calculated extinction coefficient of 1.065/M/cm at 280 nm.
The WT holenzyme and monomeric 1–380 truncation mutant of mouse CaMKIIα were expressed in Sf9 cells and purified as described previously (Strack et al, 2000).
SDS–PAGE analysis of purified CaMKII
SDS–PAGE and western analysis of purified proteins revealed two CaMKII bands, one at the appropriate size of 42 kDa and another slightly smaller cleavage product (Supplementary Figure S1A). The degree of cleavage varied between mutants and the smaller fragment could not be separated with standard ionic exchange or sizing chromatography.
Calmodulin expression and purification
Calmodulin was cloned into pET3a containing a T7 promoter and expressed in E. coli BL21(DE3) cells. Protein expression was induced with 0.4 mM IPTG and incubated at 30° and 250 r.p.m. for 3 h. Harvested cell pellets were resuspended in 50 mM MOPS, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, pH 7.5 and lysed by sonication. 5 mM CaCl2 was added to lysis supernatant and loaded on a High Trap Phenyl HP column (GE Healthcare) with 50 mM Tris–HCl, 1 mM CaCl2, pH 7.5. The protein was washed with 50 mM Tris–HCl, 1 mM CaCl2, 0.5 M NaCl, pH 7.5, and eluted with 10 mM Tris–HCl, 10 mM EDTA, pH 7.5. Purified protein was then desalted into 50 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, 0.02% azide, pH 7.5.
EPR spectroscopy
EPR spectra were collected at room temperature on a Bruker EMX spectrometer (X-band) at an incident power of 10 mW and 1.6 gauss modulation amplitude. Samples contained 50 μM CaMKII unless otherwise stated and all binding molecules were added in excess; 300 μM CaM/10 mM CaCl2 and/or 10 mM ATP/10 mM Mg(CH3COO)2. All proteins and reagents were prepared in the same buffer used for EPR experiments: 50 mM HEPES, 100 mM NaCl, 2.5 mM EDTA, 0.02% azide, pH 7.5.
Circular dichroism
Spectra were collected on a Jasco J-810 Spectropolarimeter in 20 mM phosphate buffer, pH 7.1. Near-UV spectra (330–250 nm) were obtained using a 0.5 mg/ml protein solution at room temperature. Far-UV spectra (260–185 nm) were collected at room temperature using a 0.1 mg/ml protein solution. Melting curves were obtained using 0.15 mg/ml protein solution by monitoring ellipticity at 222 nm in a temperature range between 5 and 95°C at a temperature slope of 60°C/h.
Activity assays
Specific kinase activities were determined as previously described (Colbran, 1993) except for the buffer conditions that matched those of the EPR analysis. Activity was determined by the protein's ability to incorporate a radioactive γ phosphate from [γ32P]ATP onto either syntide-2 (PLARTLSVAGLPGKK), a synthetic peptide based on a glycogen synthase phosphorylation site, or autocamtide-2 (KKALRRQETVDAL), a peptide based on the CaMKII regulatory domain. Assays were carried out under the following conditions: 50 mM HEPES, 100 mM NaCl, 0.4 mM [γ32P]ATP, 10 mM Mg(Ac)2, 1 μM CaM, 0.5 mM CaCl2, 20 μM syntide-2 or 10 μM autocamtide-2, 1 mM DTT, 1 mg/ml BSA. The level of radioactive γ phosphate was normalized relative to control samples where buffer was added instead of CaMKII.
Distance determinations
Dipolar coupling between spin labels was analysed by using a modification of the deconvolution method (Rabenstein and Shin, 1995). This approach requires both the dipolar broadened spectra and unbroadened reference spectra. The unbroadened spectrum is obtained by measuring the spectrum for each mutant individually followed by digital sum of the two spectra, termed the sum of singles. The sum of singles spectra is convoluted with a broadening function, which is a sum of Pake patterns weighted by a distribution of the distance between the two spin labels. The distribution is modelled as a sum of gaussians and the number of gaussians, up to 5, is chosen based on a statistical improvement in the fit of the broadened spectra with the experimental spectra of the double mutant (Sen et al, 2007).
Simulations for spectral lineshape analysis
Spectra were fit using the microscopic order/macroscopic disorder (MOMD) model (Budil et al, 1996). For the two component fits, the motion of one component was assumed to be effectively isotropic (Rx=Ry=Rz). The motion of the second component is assumed to undergo anisotropic rotation (Rx≠Ry≠Rz) within an ordering potential. The rotational correlation time, τ, for the motion is obtained from the R's: τ=1/(6 (Rx+Ry+Rz)1/3). The uncertainty in the correlation time of the isotropic component is around 15% (Columbus et al, 2001). While the uncertainty is somewhat larger for the restricted component correlation time, the differences in the spectral lineshape demonstrate that difference between the two correlation times are outside the uncertainty intervals. The restriction in motion is described by an order parameter, S, calculated from the coefficient, c20, used to describe the ordering potential. In addition to the ordering potential, the axis of the nitroxide was allowed not to be colinear with respect to the diffusion axis and this tilt is given by the angles (αD, βD).
Supplementary Material
Acknowledgments
We thank J Kuriyan for his kind gift of the C. elegans CaMKII gene, K Schey for his assistance with mass spectrometry experiments, and H Koteiche for help with the construction of the cysteine-free CaMKII. We are grateful to H Koteiche and D Claxton for critical reading of the manuscript.
Author contributions: LH and HSM designed the experiments. LH performed the experiments and analysed the data. RAS carried out the spectral simulations. LH, HSM, and RJC wrote the paper. This work was supported by an IDEA grant from Vanderbilt University to HSM, the Vanderbilt Vision Research Center (5T32 EY007135-15), and a pre-doctoral US National Research Service Award from the National Institute of Mental Health (1F31 MH86184-01) to LH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Bayer KU, De Koninck P, Schulman H (2002) Alternative splicing modulates the frequency-dependent response of CaMKII to Ca(2+) oscillations. EMBO J 21: 3590–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529 [DOI] [PubMed] [Google Scholar]
- Brocke L, Chiang LW, Wagner PD, Schulman H (1999) Functional implications of the subunit composition of neuronal CaM kinase II. J Biol Chem 274: 22713–22722 [DOI] [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ (2005) Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci 22: 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budil DE, Lee S, Saxena S, Freed JH (1996) Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg–Marquardt algorithm. J Mag Res Series A 120: 155–189 [Google Scholar]
- Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, Kuriyan J (2010) Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat Struct Mol Biol 17: 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ (1993) Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J Biol Chem 268: 7163–7170 [PubMed] [Google Scholar]
- Colbran RJ, Smith MK, Schworer CM, Fong YL, Soderling TR (1989) Regulatory domain of calcium/calmodulin-dependent protein kinase II. Mechanism of inhibition and regulation by phosphorylation. J Biol Chem 264: 4800–4804 [PubMed] [Google Scholar]
- Columbus L, Hubbell WL (2004) Mapping backbone dynamics in solution with site-directed spin labeling: GCN4-58 bZip free and bound to DNA. Biochemistry 43: 7273–7287 [DOI] [PubMed] [Google Scholar]
- Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL (2001) Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry 40: 3828–3846 [DOI] [PubMed] [Google Scholar]
- De Koninck P, Schulman H (1998) Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279: 227–230 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC. San Carlos, CA: DeLano Scientific. http://www.pymol.org [Google Scholar]
- Dong J, Yang G, Mchaourab HS (2005) Structural basis of energy transduction in the transport cycle of MsbA. Science 308: 1023–1028 [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Sweatt JD, Giese KP (2004) Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci 24: 8410–8415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori WR, Miick SM, Millhauser GL (1993) Increasing sequence length favors alpha-helix over 3(10)-helix in alanine-based peptides: evidence for a length-dependent structural transition. Biochemistry 32: 11957–11962 [DOI] [PubMed] [Google Scholar]
- Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN (2004) Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem 279: 12484–12494 [DOI] [PubMed] [Google Scholar]
- Goldberg J, Nairn AC, Kuriyan J (1996) Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell 84: 875–887 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12: 943–956 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H (1992) Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem 267: 17216–17224 [PubMed] [Google Scholar]
- Hoeflich KP, Ikura M (2002) Calmodulin in action: diversity in target recognition and activation mechanisms. Cell 108: 739–742 [DOI] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC, Kuriyan J (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell 11: 1241–1251 [DOI] [PubMed] [Google Scholar]
- Huang CY, Yuan CJ, Blumenthal DK, Graves DJ (1995) Identification of the substrate and pseudosubstrate binding sites of phosphorylase kinase gamma-subunit. J Biol Chem 270: 7183–7188 [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Cafiso DS, Altenbach C (2000) Identifying conformational changes with site-directed spin labeling. Nat Struct Biol 7: 735–739 [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Gross A, Langen R, Lietzow MA (1998) Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol 8: 649–656 [DOI] [PubMed] [Google Scholar]
- Hubbell WL, Mchaourab HS, Altenbach C, Lietzow MA (1996) Watching proteins move using site-directed spin labeling. Structure 4: 779–783 [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H (2002) Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Ann Rev Biochem 71: 473–510 [DOI] [PubMed] [Google Scholar]
- Kanaseki T, Ikeuchi Y, Sugiura H, Yamauchi T (1991) Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J Cell Biol 115: 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PT, Weinberger RP, Waxham MN (1988) Active site-directed inhibition of Ca2+/calmodulin-dependent protein kinase type II by a bifunctional calmodulin-binding peptide. Proc Natl Acad Sci USA 85: 4991–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MM (1988) Conformation-sensitive modification of the type II calmodulin-dependent protein kinase by phenylglyoxal. J Biol Chem 263: 4754–4757 [PubMed] [Google Scholar]
- King MM, Shell DJ, Kwiatkowski AP (1988) Affinity labeling of the ATP-binding site of type II calmodulin-dependent protein kinase by 5′-p-fluorosulfonylbenzoyl adenosine. Arch Biochem Biophys 267: 467–473 [DOI] [PubMed] [Google Scholar]
- Kolodziej SJ, Hudmon A, Waxham MN, Stoops JK (2000) Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J Biol Chem 275: 14354–14359 [DOI] [PubMed] [Google Scholar]
- Lai Y, Nairn AC, Greengard P (1986) Autophosphorylation reversibly regulates the Ca2+/calmodulin-dependence of Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA 83: 4253–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175–190 [DOI] [PubMed] [Google Scholar]
- Lou LL, Schulman H (1989) Distinct autophosphorylation sites sequentially produce autonomy and inhibition of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Neurosci 9: 2020–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL (1996) Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 35: 7692–7704 [DOI] [PubMed] [Google Scholar]
- Mchaourab HS, Oh KJ, Fang CJ, Hubbell WL (1997) Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry 36: 307–316 [DOI] [PubMed] [Google Scholar]
- Meador WE, Means AR, Quiocho FA (1993) Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 262: 1718–1721 [DOI] [PubMed] [Google Scholar]
- Meyer T, Hanson PI, Stryer L, Schulman H (1992) Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 256: 1199–1202 [DOI] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB (1986) Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44: 861–870 [DOI] [PubMed] [Google Scholar]
- Miller SG, Patton BL, Kennedy MB (1988) Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2(+)-independent activity. Neuron 1: 593–604 [DOI] [PubMed] [Google Scholar]
- Morin B, Bourhis JM, Belle V, Woudstra M, Carriere F, Guigliarelli B, Fournel A, Longhi S (2006) Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J Phys Chem B 110: 20596–20608 [DOI] [PubMed] [Google Scholar]
- Morris EP, Torok K (2001) Oligomeric structure of alpha-calmodulin-dependent protein kinase II. J Mol Biol 308: 1–8 [DOI] [PubMed] [Google Scholar]
- Payne ME, Fong YL, Ono T, Colbran RJ, Kemp BE, Soderling TR, Means AR (1988) Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. J Biol Chem 263: 7190–7195 [PubMed] [Google Scholar]
- Picconi B, Gardoni F, Centonze D, Mauceri D, Cenci MA, Bernardi G, Calabresi P, Di Luca M (2004) Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J Neurosci 24: 5283–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praseeda M, Beena MK, Asha SJ, Omkumar RV (2004) The C-terminus of CaMKII is truncated when expressed in E. coli. Protein Pept Lett 11: 175–179 [DOI] [PubMed] [Google Scholar]
- Rabenstein MD, Shin YK (1995) Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci USA 92: 8239–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, Knapp S (2010) Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol 8: e1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J (2006) Oligomerization states of the association domain and the holoenyzme of Ca2+/CaM kinase II. FEBS J 273: 682–694 [DOI] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung R-J, Nairn AC, Kuriyan J (2005) Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123: 849–860 [DOI] [PubMed] [Google Scholar]
- Sen KI, Logan TM, Fajer PG (2007) Protein dynamics and monomer-monomer interactions in AntR activation by electron paramagnetic resonance and double electron-electron resonance. Biochemistry 46: 11639–11649 [DOI] [PubMed] [Google Scholar]
- Shields SM, Vernon PJ, Kelly PT (1984) Autophosphorylation of calmodulin-kinase II in synaptic junctions modulates endogenous kinase activity. J Neurochem 43: 1599–1609 [DOI] [PubMed] [Google Scholar]
- Smith MK, Colbran RJ, Brickey DA, Soderling TR (1992) Functional determinants in the autoinhibitory domain of calcium/calmodulin-dependent protein kinase II. Role of His282 and multiple basic residues. J Biol Chem 267: 1761–1768 [PubMed] [Google Scholar]
- Soderling TR, Stull JT (2001) Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev 101: 2341–2352 [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 275: 23798–23806 [DOI] [PubMed] [Google Scholar]
- Swulius MT, Waxham MN (2008) Ca(2+)/calmodulin-dependent protein kinases. Cell Mol Life Sci 65: 2637–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Eyck LF, Taylor SS, Kornev AP (2008) Conserved spatial patterns across the protein kinase family. Biochim Biophys Acta 1784: 238–243 [DOI] [PubMed] [Google Scholar]
- Thaler C, Koushik SV, Puhl HL III, Blank PS, Vogel SS (2009) Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proc Natl Acad Sci USA 106: 6369–6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobimatsu T, Fujisawa H (1989) Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem 264: 17907–17912 [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10: 280–282 [DOI] [PubMed] [Google Scholar]
- Waxham MN, Tsai AL, Putkey JA (1998) A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. J Biol Chem 273: 17579–17584 [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD (2003) Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci 23: 2634–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME (2005) Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11: 409–417 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.