

Abstract
Membrane proteins and membrane lipids are frequently organized in submicron-sized domains within cellular membranes. Factors thought to be responsible for domain formation include lipid–lipid interactions, lipid–protein interactions and protein–protein interactions. However, it is unclear whether the domain structure is regulated by other factors such as divalent cations. Here, we have examined in native plasma membranes and intact cells the role of the second messenger Ca2+ in membrane protein organization. We find that Ca2+ at low micromolar concentrations directly redistributes a structurally diverse array of membrane proteins via electrostatic effects. Redistribution results in a more clustered pattern, can be rapid and triggered by Ca2+ influx through voltage-gated calcium channels and is reversible. In summary, the data demonstrate that the second messenger Ca2+ strongly influences the organization of membrane proteins, thus adding a novel and unexpected factor that may control the domain structure of biological membranes.
Keywords: Ca2+ , electrostatics, microdomains, plasma membrane, signalling
Introduction
Biological membranes contain a remarkably high diversity of lipids and proteins. The fluid-mosaic model (Singer and Nicolson, 1972), introduced by Singer and Nicholson >35 years ago, is still the most prevalent model describing membrane structure and dynamics. The model proposes that membrane proteins are solubilized in a sea of lipids in which individual proteins are disorganized and freely diffusing over large distances. However, in recent years it has become clear that biological membranes are dominated by proteins much more than previously envisioned (Takamori et al, 2006; Dupuy and Engelman, 2008). Moreover, most, if not all, membrane proteins are organized in submicron-sized clusters, referred to as microdomains or rafts, rather than being uniformly distributed as freely diffusing entities (Jacobson et al, 2007; Pike, 2009; Lingwood and Simons, 2010).
Although at present it is generally accepted that biological membranes are laterally compartmentalized, the underlying mechanisms are still controversial. One of the most popular hypotheses suggests that lipid–lipid interactions constitute the primary force for driving membrane compartmentalization. According to the original version of the lipid-raft hypothesis (Simons and Ikonen, 1997), clustering of sphingolipid-head groups in the outer leaflet of the plasma membrane creates gaps between the fatty acyl chains in the hydrophobic interior that are filled with cholesterol molecules. The resulting arrangement stiffens the aliphatic side chains and thus resembles a liquid-ordered phase. Preferential partitioning of certain proteins into this phase would then lead to local high concentrations, resulting in the formation of a microdomain (in this case called lipid-raft). Other models invoke alternative mechanisms for explaining membrane compartmentalization. For instance, the picket-fence hypothesis suggests that the subplasmalemmal cytoskeleton acts as a diffusion barrier (Ritchie and Kusumi, 2004), capable of forming confined zones on the membrane surface that trap membrane proteins. However, these physical models are insufficient to explain the high degree of specificity and diversity in micropatterning. Most importantly, there are many examples of protein isoforms or proteins of similar structure and size that segregate into separate domains (Uhles et al, 2003; Kai et al, 2006; Low et al, 2006; Sieber et al, 2006), suggesting that microdomains are stabilized by specific protein–protein interactions. Thus, a picture is emerging according to which a combination of factors including specific protein–protein and protein–lipid interactions, together with a propensity of certain membrane lipids to undergo local phase transitions, defines the structure and dimensions of membrane microdomains.
In the present study, we have investigated whether microdomain organization is influenced by divalent cations, particularly Ca2+. The motivation for this study was two-fold. First, the cytoplasmic face of the plasma membrane and that of other intracellular membranes contain negatively charged membrane lipids at relatively high concentrations (van Meer et al, 2008), resulting in a fixed negative surface potential. A number of mechanisms have been suggested how Ca2+-mediated electrostatics drive the association of proteins with the negatively charged lipids, including electrostatic switches involving Ca2+ binding to C2-domains, Ca2+-induced conformational changes or Ca2+-phospolipid bridging (Hurley and Misra, 2000; McLaughlin and Murray, 2005; Mulgrew-Nesbitt et al, 2006). Second, if Ca2+ indeed has an impact on membrane domains, this could be of physiological relevance because the intracellular Ca2+ concentration is normally low but is transiently upregulated during a multitude of signalling events. In particular, Ca2+ influx mediated by calcium channels results in locally very high ion concentrations with increases up to 1000-fold over basal levels (Rizzuto and Pozzan, 2006). At such influx sites, strong electrostatic effects are expected to be exerted on the surrounding proteins.
Here, we have systematically explored the effect of Ca2+ on membrane protein organization by elevating Ca2+ uniformly at the inner plasmalemmal leaflet or locally through calcium channels. The data indicate that Ca2+ within a physiological concentration range suffices to strongly redistribute membrane proteins, which may have a profound impact on the activity of membrane proteins and hence a variety of biological processes.
Results
Ca2+ directly modulates membrane protein organization
Under resting conditions, the intracellular Ca2+ concentration is maintained at a concentration of about 100 nM. During signalling, Ca2+ increases up to several micromolar or even up to several hundred μM near to the mouth of Ca2+ channels (Heidelberger et al, 1994; Bollmann and Sakmann, 2005). To test whether increased Ca2+ levels have any influence on membrane protein organization, intracellular Ca2+ was increased in neuroendocrine PC12 cells for 5 min using the Ca2+ carrier ionomycin. For microscopic analysis of possible changes in the distribution of membrane proteins, cells were subjected to a brief ultrasound pulse that removes their upper parts and leaves behind the basal plasma membranes adhered to a glass coverslip. The so-called plasma membrane sheets were fixed and immunostained, selecting antibodies for a variety of structurally different proteins (Figure 1). Those included a protein with multiple transmembrane domains (synaptophysin), two topologically different proteins each containing a single span transmembrane domain (transferrin receptor and syntaxin 1), two structurally very similar proteins that are membrane associated via palmitoyl–anchors (SNAP23 and SNAP25) and a peripherally associated membrane protein (Munc18). As shown in Figure 1, in all cases Ca2+-dependent alterations in the immunostaining patterns were observed. In general, signals appeared more clustered and immunostaining intensities decreased to 10–60%. For the two most affected membrane-anchored proteins, we also analysed whether the effect is reversible and observed a recovery of immunostaining intensity within less than an hour (Supplementary Figure S1). In order to study these effects in more detail, and to provide more defined Ca2+ concentrations in the experiment, we incubated plasma membranes directly with buffered Ca2+ solutions (for the effect of the incubation process in the absence of any divalent ions see Supplementary Figure S2). As plasma membrane sheets are ‘unroofed cells’, they provide biochemical access to membrane proteins at the inner plasmalemmal leaflet in the absence of cytosol, membrane trafficking and actin-cortex remodelling, whereas native membrane protein complexes remain functionally intact in the presence of MgATP (Lang, 2003; Barszczewski et al, 2008). As shown in Figure 2A and B, addition of 54 μM Ca2+ in the presence of MgATP resulted in changes in membrane protein staining patterns that were similar to those observed upon ionomycin treatment of intact cells. The strong decrease was caused primarily by Ca2+, as 2 mM MgATP in the absence of Ca2+ had no clear effect (Supplementary Figure S2). We next tested buffers containing Ca2+ as the only divalent cation. As shown in Figure 2C, already submicromolar Ca2+ (850 nM) sufficed to trigger redistribution, whereas 225 nM Ca2+ had no effect (Figure 2D). The observation that both Ca2+-dependent clustering of membrane proteins and reduction of immunoreactivity is reproduced on freshly prepared membrane sheets excludes several of the possible causes for these effects such as membrane trafficking, actin remodelling, translocation of cytoplasmic factors or proteolysis, since proteases are expected to be washed away and protease inhibitors do not abolish the Ca2+ effect (Supplementary Figure S3). Hence, Ca2+-mediated remodelling of membrane proteins is not a secondary effect but due to a direct interaction of Ca2+ with the plasma membrane and occurs well within the physiological range of Ca2+ concentrations.
Figure 1.

Rising intracellular Ca2+ dramatically diminishes membrane protein immunostaining intensities. (A, B) PC12 cells were treated for 5 min with 20 μM ionomycin in the absence or presence of extracellular Ca2+, followed by the generation of membrane sheets and immunostaining for a variety of structurally diverse membrane proteins. (A) (Upper panels) TMA-DPH staining for the visualization of phospholipid membranes, indicating location and integrity of the basal plasma membranes; (lower panels) immunostaining for transferrin receptor (left) or SNAP25 (right). (B) Quantification of Ca2+-dependent decreases in immunostaining intensity. Remaining fluorescence after Ca2+ treatment was normalized to the values obtained in the absence of Ca2+ and expressed as percentage. Values are means±s.e.m. (n=3 independent experiments; 31–78 membrane sheets were analysed for each condition in one experiment).
Figure 2.

Ca2+ acts directly at the plasma membrane via electrostatic mechanisms. (A–D) Freshly prepared membrane sheets were treated in the presence (A, B) or absence (C, D) of magnesium for 10 min at 37°C with a defined Ca2+ concentration, fixed, immunostained for membrane proteins as indicated (Syp, TfR and Syx1 refer to synaptophysin, transferrin receptor and syntaxin 1 respectively) and analysed as in Figure 1. (A) Magnified views from immunostained membrane sheets after treatments with no (upper panels) or 54 μM (lower panels) Ca2+. (B–D) Change in immunostaining intensity induced by 54 μM (B), 850 nM (C) or 225 nM (D) Ca2+. Residual fluorescence intensities were normalized to the intensities obtained in the absence of Ca2+ and expressed as percentage. Values are means±s.e.m. (n=3–6 independent experiments; 18–106 membrane sheets were analysed for each condition in one experiment). (E) At pH 7.4, the amino acids Asp (D) and Glu (E) are negatively charged whereas Arg (R) and Lys (K) carry positive charges. For each membrane protein, the number of the negatively and positively charged amino acids within the cytoplasmic regions was determined and %DE–%RK plotted against fluorescence remaining after Ca2+ treatment with 850 nM (black) or 54 μM Ca2+ (grey).
We did not observe any correlations between the magnitude of the Ca2+ effect and the type of membrane protein (e.g. see the related SNAREs SNAP23 and SNAP25 that respond differently to Ca2+), the nature of membrane protein anchoring or the molecular mass of the membrane protein. However, sequence analysis of the proteins investigated revealed that the sensitivity towards Ca2+-dependent redistribution correlates positively with the abundance of negatively charged side chains (Figure 2E). Hence, electrostatic mechanisms control membrane protein organization, most likely involving a role for Ca2+ ions in compensating negative repulsive charges between the negatively charged phospholipid bilayer and the negatively charged membrane protein or between the negatively charged proteins themselves.
Relationship between redistribution and decrease in immunostaining intensities
The data presented above show that Ca2+ leads to a loss of immunostaining intensity. As degradation and trafficking can be ruled out, Ca2+ converts membrane proteins into a state in which they are less accessible to antibody staining. What may be the reason for this effect? Two scenarios are possible: First, negatively charged proteins may cluster to a higher degree when Ca2+ compensates the charges, and thus become more crowded or more tightly packed, increasing steric hindrance for antibody binding. Second, negatively charged proteins may not change their lateral position but, mediated by compensating Ca2+ charges, undergo a conformational change or move closer to the negatively charged plasma membrane, in turn leading to masking of the antibody binding site.
To find evidence for these possibilities, we selected the two most negatively charged proteins studied, syntaxin and SNAP25, and expressed them with a fluorescent protein tag. In addition, the proteins were visualized by antibody staining after incubation in the absence or presence of Ca2+. Hence, comparison of fluorescent protein fluorescence and immunostaining allows clarification whether immunostaining visualizes membrane protein distribution representatively or if masked pools form.
As shown for syntaxin (Supplementary Figure S4), both in the absence and presence of Ca2+, the images in the fluorescent protein channel and the immunostaining channel showed the same pattern within the technical limit (correlation coefficients of 0.67–0.73, see also Supplementary Figure S4), indicating that the antibody, though binding is expected to be substochiometric, is capable to recognize properly the distribution of syntaxin. Hence, the changes of the immunostaining pattern after Ca2+ increase, noticed by a higher degree of clustering (see larger dark areas forming between syntaxin clusters in Supplementary Figure S4 and quantification of clustering in Figure 3C), reports the true change in lateral distribution. In this case, decrease in immunostaining intensity is probably caused by a tighter protein packing leading to a decrease in steric accessibility.
Figure 3.

Decrease in immunostaining intensity correlates with redistribution of membrane proteins into more clustered patterns. (A, D) Syntaxin (A) and SNAP25 (D) stainings on membrane sheets incubated with 54 μM Ca2+ for times as indicated. (Upper panels) Images at the same scaling, illustrating that staining becomes dimmer over time. (Lower panels) In order to illustrate that dimming correlates with a higher degree of protein clustering images were background subtracted, and mean intensity normalized to the mean intensity of the image from the directly fixed membrane sheet. Presenting images at the same scaling illustrates that the available signal becomes more punctate upon Ca2+ treatment. For syntaxin (B, C) and SNAP25 (E, F) mean staining intensity (B, E) and pixel s.d. of the mean (normalized to mean background subtracted intensity, related to the 0 s value) (C, F) were plotted against incubation time without Ca2+ (green traces) or with 54 μM Ca2+ (red traces). Values are means±s.e.m. (n=3 independent experiments; 26–74 membrane sheets were analysed for each condition in one experiment).
For SNAP25 (Supplementary Figure S5) we observe that the similarity of the two channels is lower but still high (correlation coefficients of 0.52–0.56, see also Supplementary Figure S5). However, Ca2+ leads to occasionally occurring larger SNAP25 patches and the masking of a more uniformly distributed SNAP25 pool (Supplementary Figure S5).
Hence, in the case of SNAP25 the dramatic change of the immunostaining pattern is not solely reflected by a tighter packing, but also by a conformational change leading to masking of the epitope recognized by the antibody.
We next asked whether increase in clustering and decrease in immunostaining are coupled processes. As shown in Figure 3, the time course of intensity diminishment and increase in s.d. of pixel intensity, used here for quantifying the clustered nature of the signal distribution, were similar. This suggests that tighter clustering is the mechanism causing the diminishment of immunostaining intensity.
In summary, the data suggest that decrease in immunostaining is caused by a higher degree of clustering. In addition, formation of a masked pool can also contribute to the observed decrease in immunostaining.
Protein mobility measurements for probing the clustered state in live cells
We next turned to membrane mobility measurements by fluorescence recovery after photobleaching (FRAP) as alternative approach for probing if clustering depends on Ca2+. In these experiments, in the basal plasma membrane fluorescently labelled proteins are bleached in a region of interest (ROI) and the kinetics of fluorescence recovery in the ROI, depending on the exchange between bleached and unbleached proteins, are monitored. If the studied protein diffuses freely in an unrestricted manner (e.g. is not trapped in clusters), then fluorescence in the ROI is expected to reach essentially its initial value and maximal recovery is 100%. However, if proteins are clustered, then the shape of the recovery curve is determined by the speed of the freely diffusing proteins, the size of the clusters and the exchange rate between the free and clustered molecules (Sieber et al, 2007). Though the sizes of the incompletely recovering fraction and the clustered pool are not necessarily the same or linearly related to each other, incomplete recovery indicates protein clustering.
Figure 4 shows recovery curves from cells prior to stimulation, demonstrating for syntaxin and SNAP25 incompletely recovering fractions of 15 and 23%, respectively. When intracellular Ca2+ was raised, incomplete recovery increased by about 2–3-fold (for syntaxin and SNAP25 to 43 and 49%, respectively), whereas the mobile fraction diffused more rapidly. The effect was stronger for syntaxin, what is expected as GFP attachment to SNAP25 reduces the intracellular negative charge to a value lower than for syntaxin (Supplementary Table S1).
Figure 4.

Membrane protein mobility measured by FRAP reveals that Ca2+ increases the fraction of immobile membrane proteins and the diffusion rate of the mobile fraction. (A, B) Fluorescence recovery after photobleaching (FRAP) experiments on PC12 cells expressing syntaxin 1A-GFP (A) or GFP-SNAP25 (B). Fluorescence in the basal plasma membrane was bleached in a squared region of interest (ROI; indicated by the white boxes) and fluorescence recovery by membrane protein diffusion was monitored (upper panels in (A, B)). Then cells were treated for 5 min with the Ca2+ carrier ionomycin in the presence of extracellular Ca2+ and a second recovery curve was monitored (lower panels in (A, B)), showing that Ca2+ caused a diminishment in maximal recovery (for averaged data see (C, D); for quantification see Figure S6) and an acceleration of the diffusion rate (for quantification see Supplementary Figure S6 and (E)). In contrast, recovery traces were essentially unchanged when in control experiments in the second FRAP measurement extracellular Ca2+ was chelated by EGTA (data not shown). (C, D) Averaged recovery traces from three independent experiments for syntaxin (C) and SNAP25 (D). Green and red traces refer to the first FRAP experiment in low calcium Ringer solution and the second FRAP experiment in Ringer containing the Ca2+ carrier ionomycin, respectively. Values are given as mean±s.d. (n=3 individual traces; for one individual trace the recovery curves from 3 to 6 cells were averaged). For quantification of the effects, the hyperbola function 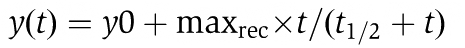 , which yields the maximal recovery (maxrec) and the half-time of recovery (t1/2), was fitted to the averaged traces obtained from one individual experiment. Before fitting, the traces were rescaled setting the pre-bleach and the post-bleach values to 100 and 0%, respectively. Averaged fits are shown in Supplementary Figure S6 and as black lines with adjusted scaling in (C, D) for comparison to averaged raw data. From the individual fits maximal recovery and half-time of recovery were further used for calculation of the immobile fraction (immobile fraction=100% − maximal recovery) and the apparent lateral diffusion coefficient. The size of the immobile pools increased after rising Ca2+ from 15 to 43% for syntaxin and from 23 to 49% for SNAP25. Values are given as mean±s.e.m. (n=3). (E) Ca2+-induced change of the apparent lateral diffusion coefficient. Values are given as mean±s.e.m. (n=3).
, which yields the maximal recovery (maxrec) and the half-time of recovery (t1/2), was fitted to the averaged traces obtained from one individual experiment. Before fitting, the traces were rescaled setting the pre-bleach and the post-bleach values to 100 and 0%, respectively. Averaged fits are shown in Supplementary Figure S6 and as black lines with adjusted scaling in (C, D) for comparison to averaged raw data. From the individual fits maximal recovery and half-time of recovery were further used for calculation of the immobile fraction (immobile fraction=100% − maximal recovery) and the apparent lateral diffusion coefficient. The size of the immobile pools increased after rising Ca2+ from 15 to 43% for syntaxin and from 23 to 49% for SNAP25. Values are given as mean±s.e.m. (n=3). (E) Ca2+-induced change of the apparent lateral diffusion coefficient. Values are given as mean±s.e.m. (n=3).
In the FRAP experiments, we also noticed that Ca2+ induced an overall dimming of the GFP fluorescence. This may be explained by GFP self-quenching, caused by the tighter packing of the proteins in the clusters and the associated decrease in the mean distance between the GFP molecules. To study this effect in more detail, we applied total internal reflection fluorescence (TIRF) microscopy, which allows for selective imaging of the plasma membrane, that is without blurring by out-of-focus fluorescence. As shown in Supplementary Figure S7, Ca2+ treatment diminished fluorescence of syntaxin 1A-GFP and GFP-SNAP25 to 59 and 43%, respectively. While endocytotic removal of the proteins may be responsible for some of the reduction, the magnitude of reduction far exceeds the turnover rates of plasma membrane proteins (e.g. 8 h for SNAP25 in PC12 cells (Lane and Liu, 1997)). Thus, we conclude that Ca2+-induced clustering is indeed responsible for GFP self-quenching, which needs to be taken into account in the interpretation of the FRAP experiments. Since freely diffusing proteins are not expected to undergo self-quenching, free and clustered proteins may contribute differentially to fluorescence recovery. In fact, the pre-bleaching signal could be the lower the more clustered the proteins, leading to an underestimate of the Ca2+-mediated size increase of the immobile fraction.
Taken together, our data show that Ca2+ mediates an increase in the clustering of membrane proteins in the plasma membrane, whereas the mobile fraction can diffuse faster. This is in line with a recent study showing that divalent ions lead to the formation of membrane protein aggregates, increasing diffusion rates between the aggregates (Frick et al, 2007). Interestingly, Ca2+ had a more than two-fold stronger effect on the apparent lateral diffusion coefficient of syntaxin when compared with SNAP25. As both constructs have approximately the same negative charge (Supplementary Table S1), this could be due to the different underlying clustering mechanisms, as syntaxin has been reported to cluster, supported by cholesterol, via protein–protein interactions (Sieber et al, 2006) and SNAP25 probably by the involvement of palmitoyl–anchor interactions with rafts (Salaün et al, 2005), independent from protein–protein interactions (Halemani et al, 2010).
Redistribution triggered by local Ca2+ transients
The data document that Ca2+ mediates membrane protein redistribution. However, our in vitro conditions, in which Ca2+ is uniformly distributed and applied for prolonged times, are not directly comparable to physiological situations, as in vivo Ca2+ increases are often transient and locally restricted to for example an open calcium channel from which Ca2+ diffuses into the intracellular space where it is quickly bound to Ca2+ buffers. To test whether membrane proteins also redistribute during Ca2+ increases occurring under physiological conditions, we performed experiments using primary neuroendocrine cells (bovine chromaffin cells) that were stimulated by high potassium treatment. Reminiscent of physiological stimulation, high potassium depolarizes the plasma membrane that in turn mediates opening of voltage-gated calcium channels, thereby mimicking a physiological Ca2+ response.
After potassium treatment, cells were fixed and immunostained. As shown in Figure 5, after 30 s treatment with high potassium SNAP25 redistribution can be observed, documented by a strong decrease in plasmalemmal immunostaining in the equatorial plane. As expected from Figure 2, the effect was less prominent for the less Ca2+-sensitive proteins (Figure 5; Supplementary Figure S8).
Figure 5.

Membrane protein redistribution after depolarization-induced calcium channel opening. (A, B) Bovine chromaffin cells were treated for 30 s with low or high potassium Ringer solution at 37°C, fixed and immunostained for syntaxin (A) or SNAP25 (B). Confocal micrographs from equatorial sections in the immunofluorescence (left) and in the brightfield (right) channels are shown. Depolarization-induced decrease of plasmalemmal immunofluorescence was analysed by linescans placed at the periphery of the cell (for details see Materials and methods). (C) High potassium values were related to corresponding low potassium values (set to 100%). Values are means±s.e.m. (n=4 independent experiments).
Hence, redistribution occurs at physiological Ca2+ increases and can be fast in the range of less than a minute. As in these experiments we studied the equatorial plane of the cells, the data also show that Ca2+-dependent remodelling is not restricted to the basal plasma membrane but occurs at the entire plasmalemma.
Clustering reduces the availability of proteins for protein–protein interactions
The reduction of immunoreactivity may be explained by lack of accessibility due to epitope crowding while the function of the protein is not changed. However, it is also possible that crowding and/or associated conformational changes directly affect the biological activity. To examine this possibility, we used the SNAREs SNAP25 and syntaxin as examples. During exocytosis, these two membrane proteins form a complex with the vesicle-bound SNARE synaptobrevin, which generates a tight connection between the membranes leading to membrane fusion. The amount of active SNAREs in the plasma membrane can be conveniently measured by incubating membrane sheets with a fluorescently labelled soluble fragment of synaptobrevin (Lang et al, 2002). Therefore, we incubated membrane sheets with labelled synaptobrevin and determined to which extent binding is influenced by Ca2+. As shown in Figure 6, synaptobrevin binding diminished with increasing Ca2+, indicating that the clustering state reciprocally correlates with the fraction of active syntaxin and SNAP25. To differentiate further whether this reduction is caused by a reduced availability of SNAP25, we directly measured the available SNAP25 pool by incubating the sheets with a soluble and labelled syntaxin fragment. Under these conditions, SNAP25 forms a binary complex with syntaxin (Lang et al, 2002). Again, we found that binding of recombinant soluble syntaxin to SNAP25 was reduced to about 60% in the presence of 1.35 μM Ca2+ (10 min incubation at 37°C, data not shown).
Figure 6.

Ca2+ inhibits SNARE reactivity. (A, B) Freshly prepared membrane sheets were incubated for 5 min with 10 μM Alexa594-labelled soluble synaptobrevin 2 in the presence of variable Ca2+ concentrations, washed, fixed and binding of synaptobrevin to membrane sheets was quantified by fluorescence microscopy. (A) (Left) TMA-DPH phospholipid staining visualizing the basal plasma membrane; (right) membrane-bound synaptobrevin. (B) For each Ca2+ concentration incorporated synaptobrevin was quantified and intensity was related to the value obtained in the absence of Ca2+ (set to 100%). Values are given as means±s.e.m. (n=4–5 experiments, 12–66 membrane sheets were analysed for one condition in one experiment). (C) Illustration of the biochemical reaction underlying synaptobrevin 2 binding to the plasma membrane. Synaptobrevin binding depends on both syntaxin 1 and SNAP25 (Lang et al, 2002), which form an acceptor complex (not shown for clarity) that in turn interacts with soluble synaptobrevin, resulting in a so-called cis-SNARE complex (right).
Hence, we conclude that Ca2+-mediated clustering of membrane proteins can indeed alter the activity of membrane proteins, as demonstrated here for the Ca2+-dependent reduction of plasma membrane-resident SNARE activity.
Discussion
In the present study, we show that physiological concentrations of Ca2+ lead to rapid redistribution of a variety of structurally different membrane proteins, which is characterized by stronger clustering accompanied by epitope masking.
Several lines of evidence support the view that Ca2+ acts directly on membrane proteins involving an electrostatic mechanism that is solely dependent on the overall negative charge. First, secondary Ca2+ effects such as exocytosis, endocytosis, protease activation, actin-cortex remodelling or membrane recruitment of cytosolic factors are not involved as Ca2+-modulation of membrane proteins is also evident on membrane sheets. Second, specific binding of Ca2+ to a Ca2+ binding site can be excluded as the tested membrane proteins contain no specific Ca2+ binding sites. Third, Ca2+ acts independent of the domain structure or the nature of membrane anchoring. Fourth, Ca2+-mediated effects correlate with the availability of negative charges in the protein, indicating electrostatics as underlying mechanism. Fifth, although shown only in the case of SNAP25 in bovine chromaffin cells, the data document that the effect can be fast in the range of 30 s. The high speed of the reaction is in line with the idea of protein electrostatics rather than protein–protein interactions or enzymatic activities. Hence, we conclude that Ca2+ directly acts on membrane proteins via fast electrostatic mechanisms.
For syntaxin and SNAP25, previous studies have shown that they are tightly packed into dense protein clusters in large copy numbers (Sieber et al, 2007; Knowles et al, 2010; Rickman et al, 2010). Ca2+ could increase the clustering state of these proteins by several mechanisms. First, rather loosely associated proteins could be packed more tightly if Ca2+ compensates the many accumulating negative charges in a protein cluster. In addition, compensation of negative charges by Ca2+ could decrease the unbinding rate from a highly negatively charged protein cluster, leading to a longer residence time of a protein within the cluster. Second, Ca2+ may lead to higher-order aggregates composed of several individual clusters and third, Ca2+ may increase the fraction of clustered proteins. In addition, it remains to be established if Ca2+-induced clustering is primarily homophilic or reflects the segregation of membrane proteins into areas that are in general more enriched in or depleted from membrane proteins. In any case, these mechanisms would diminish the protein accessibility for immunostaining and increase the size of the immobile fraction in FRAP experiments.
Presently, we can only speculate if the redistribution of membrane proteins by Ca2+ constitutes a biologically relevant mechanism involved in the regulation of membrane proteins, particularly their organization in microdomains.
As outlined above, the average intracellular Ca2+ concentration is in the range of about 100 nM. However, inward Ca2+ leakage causes even in resting cells Ca2+ microdomains (Rizzuto and Pozzan, 2006) and some studies in non-neuronal cells revealed a strong concentration gradient from the bulk cytoplasm to the subplasmalemmal area from 30 to 80 nM in HeLa cells (Nagai et al, 2004), from 63 to 376 nm in bovine endothelial cells (Isshiki et al, 2002) or even from 0.2 to 2 μM in a smooth muscle cell line ((Marsault et al, 1997); possibly caused by hot spots involving flickering channels rather than representing a homogenous distribution (Rizzuto and Pozzan, 2006)). Hence, it is possible that in the subplasmalemmal area of some cell types, Ca2+ exceeds the threshold for the Ca2+ effect, and therefore would provide some membrane proteins by default in a more clustered state.
In any case, as shown in this study, in resting PC12 and bovine chromaffin cells, proteins are present in a state in which they are still sensitive to Ca2+. It has been observed that after a strong stimulus, excitable cells remain refractory to a second stimulus and it is likely that high Ca2+ would drive syntaxin and SNAP25 into a more inactive state. However, the timing of clustering is relatively slow compared with the timing of nerve cell physiology and it is possible that Ca2+-induced clustering becomes noted only when cells are suffering. Hence, it needs to be established if the here observed redistribution has any regulatory role.
In conclusion, we show that the domain structure of biological membranes, in addition to being mediated by specific protein–lipid and protein–protein interactions, is sensitive to submicromolar concentrations of the second messenger Ca2+. Mg2+ may also participate in the compensation of charges. However, as the charge density of the smaller Mg2+ ion is higher than for Ca2+, the energy for removing coordinated water molecules, a prerequisite for strong interactions with negatively charged amino acids or lipids, is accordingly higher, providing an explanation for the inefficiency of Mg2+. For this reason, and because Mg2+ is not undergoing large intracellular fluctuations, we propose that Ca2+ is the only divalent ion that may have the potential to regulate biological processes by a mechanism involving redistribution of membrane proteins.
Materials and methods
Cell culture
PC12 cells (clone 251; Heumann et al, 1983) were maintained, propagated and transfected as described (Lang et al, 1997; Zilly et al, 2006). In transfection experiments, 20 μg of the corresponding plasmid was added to an electroporation cuvette. Cells were plated onto 25 mm polylysine-coated glass coverslips and used 24–48 h later for experiments. Bovine chromaffin cells were isolated, maintained and plated onto coverslips essentially as previously described (Nagy et al, 2005).
Plasmids
Plasmids were used for overexpression of constructs under the control of the CMV promoter. For syntaxin 1A, syntaxin 1A C-terminally fused to a monomeric variant of enhanced GFP (syntaxin 1A-mGFP; Sieber et al, 2006) was used. For SNAP25, SNAP25B was fused N-terminally to mGFP (Halemani et al, 2010) or to EYFP (using the pEYFP-C1 vector (Clontech)). The coding sequence was verified by sequencing using for SNAP25B the rat sequence as reference.
Antibodies
Mouse monoclonal antibodies were used for detection of syntaxin 1A/B (HPC-1 (Barnstable et al, 1985)), SNAP25 (Cl. 71.1, recognizes AA20-40 (cat111001, SynapticSystems, Germany)), synaptophysin (Cl. 7.2 (cat101001, SynapticSystems)) and human transferrin receptor (cat13-6800, Zymed). In addition, commercially available rabbit polyclonal antibodies were used for detection of Munc18–1 (raised against a peptide, cat116002, SynapticSystems) and SNAP23 (raised against a peptide, cat111202, SynapticSystems). As secondary antibodies, goat anti-rabbit-Cy3 and goat anti-mouse-Cy3 (Dianova, Hamburg, Germany) were used.
Recombinant proteins
Constructs encoding for syntaxin 1A-171Cys (1–262) (Lang et al, 2002) and synaptobrevin 2–28Cys (1–96) (Margittai et al, 2001) were expressed as His6-tagged fusion proteins and purified by Ni2+-agarose beads. The His6-tags were removed by thrombin cleavage, and proteins were purified by ion-exchange chromatography.
The proteins were dialyzed against a buffer containing 120 mM KGlu, 20 mM KAc, 20 mM HEPES (pH 7.2) and 1 mM DTT. The labelling was performed as previously described (Pabst et al, 2002). In brief, the protein was mixed with freshly prepared DTT to a final concentration of 10 mM and incubated for 30 min at room temperature. Excess DTT was removed using a PD-10 column (GE Healthcare). The elution buffer contained 120 mM KGlu, 20 mM KAc, 20 mM HEPES (pH 7.2). The eluted protein was mixed with Alexa594-maleimide (Molecular Probes) at a molecular ratio of 1:5 and rotated for 2 h at room temperature. Excess of non-reacted dye was deactivated using 10 mM DTT. The labelled protein was separated from non-reacted dye using a sephadex G75 superfine (GE Healthcare) gelfiltration column.
Ionomycin treatment of PC12 cells
Cells were plated on glass coverslips and grown in six-well plates for 1 day. Then, the cells were washed twice with starving medium (DMEM with 4.5 g/l glucose (Cambrex, NJ) supplemented with 4 mM L-glutamine (Cambrex) and 60 μg/ml Penicillin and Streptomycin (Cambrex)) and then equilibrated in starving medium in the cell culture incubator for 2 h at 37°C. For experiments, the six-well plates were removed from the cell incubator and placed in a standard 37°C incubator. The medium was replaced by 1.5 ml starving medium containing 20 mM HEPES (pH 7.2), 20 μM ionomycin (Calbiochem) and additional 3.2 mM CaCl2 (together with the calcium of the DMEM medium leading to a total of 5 mM calcium). In control experiments, Ca2+ was chelated by 10 mM EGTA. After 5 min incubation, membrane sheets were prepared by sonication, fixed in 4% paraformaldehyde in PBS and immunostained as described below.
For recovery experiments, normal medium was replaced by Ringer solution (130 mM NaCl, 4 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 48 mM D(+)glucose, 10 mM HEPES pH 7.4) containing 20 μM ionomycin (for stimulation) or 20 μM ionomycin and 10 mM EGTA (for controls). After 5 min at RT, cells were briefly washed three times with medium, incubated at 37°C in medium for the times as indicated and membrane sheets were generated. For ionomycin treatments applied for FRAP and TIRF microscopy experiments see below.
Generation, treatments and immunofluorescence of PC12 membrane sheets
For Figures 2A, B, 3 and 6, Supplementary Figures S3, S4 and S5 untransfected or transfected PC12 cells grown on glass coverslips were sonicated in ice-cold KGlu buffer (containing 120 mM KGlu, 20 mM KAc, 2 mM EGTA, 4 mM MgCl2, 2 mM ATP and 20 mM HEPES pH 7.2) by applying a 100-ms ultrasound pulse. Membrane sheets were then either directly fixed in 4% paraformaldehyde in PBS (150 mM NaCl, 10 mM Na2HPO4, 10 mM NaH2PO4 (pH 7.2)) for 1 h at room temperature or fixed after an incubation with BSA-KGlu (KGlu containing 3% BSA), or BSA-KGlu containing a Ca2+ buffer. Where indicated, solutions also contained recombinant proteins. For Supplementary Figure S3, a protease inhibitor cocktail (cocktail complete/Mini/EDTA-free (cat1836170, Roche) applied at working concentration in addition to 1 mM 1,10-Phenanthroline (catP1294, Sigma) and 1.46 μM Pepstatin A (cat4265, Sigma)) was added where indicated. For Figure 3 and Supplementary Figure S3 for no Ca2+ 2 mM EGTA in the KGlu buffer was replaced by 2 mM DPTA.
For Figure 2C and D and Supplementary Figure S2 untransfected PC12 cells were sonicated in ice-cold KGlu buffer without MgCl2 and ATP. Incubations were performed for times as indicated in the same buffer with 3% BSA containing a Ca2+ buffer or 2 mM MgCl2 and 2 mM ATP where indicated.
For Ca2+ buffering, Ca2+ chelator systems based on EGTA (ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid) or DPTA (1,3-Diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid) were used. For Ca2+ ranging from 100 to 1350 nM calcium/EGTA was applied (KD EGTA=150 nM (Tsien and Pozzan, 1989)). For instance, for 850 nM Ca2+ CaEGTA and K2EGTA were mixed at a ratio of 8.5:1.5 (with a 2-mM EGTA working concentration) and for 225 nM Ca2+ a ratio of 6:4 was used. For Ca2+ in the range of 9–1500 μM calcium/DPTA was used (KD DPTA=81 μM (Heinemann et al, 1994)). For instance, a 4:6 CaDPTA:K2DPTA ratio yielded 54 μM Ca2+ (with a 2-mM DPTA working concentration).
In Figure 6, during incubation the concentration of Alexa-labelled synaptobrevin was 10 μM and membranes were briefly washed before fixation. After fixation, paraformaldehyde was quenched for 20 min in PBS containing 50 mM NH4Cl, and washed two times in PBS for 10 min each. For immunostaining, coverslips were incubated for 1 h with the primary antibodies (diluted 1:200 to 1:400 in BSA-KGlu or 1:100 to 1:200 in PBS containing 3% BSA). Subsequently, sheets were washed three times in PBS for 10 min each, followed by a 1-h incubation with secondary antibodies (diluted 1:200 in BSA-KGlu or 1:200 to 1:400 in PBS containing 3% BSA). Before imaging, membrane sheets were washed three times in PBS for 10 min. For Figure 3 and Supplementary Figures S1 and S3, to 4% PFA and all subsequently applied solutions 2 mM EGTA was added. Imaging was performed at room temperature in PBS containing TMA-DPH (1-(4-tri-methyl-ammonium-phenyl)-6-phenyl-1,3,5-hexatriene p-toluenesulfonate, catT204, Molecular Probes, Eugene, OR), in order to judge the continuity and shape of the imaged membrane. In experiments comparatively studying immunostaining intensities, membrane sheets were selected and first imaged in the TMA-DPH channel and then in the Cy3 channel.
Fluorescence microscopy and quantitation of fluorescence intensity
Membrane sheets were imaged using a Zeiss Axiovert 100 TV fluorescence microscope with a × 100 1.4 NA plan achromate objective (Zeiss, Oberkochen, Germany) and a back-illuminated CCD camera (512 × 512-NTE chip, 24 × 24 μm2 pixel size; Princeton Scientific Instruments) with a × 2.5 Optovar magnifying lens. TMA-DPH fluorescence was detected using Zeiss filter set 02 (excitation filter G 365, BS 395 and emission LP 420). The following filter sets were used for the detection of EGFP, EYFP, Cy3 and Alexa594 fluorescence (all from AHF Analysentechnik AG, Tübingen, Germany): for EGFP and EYFP, excitation filter BP 480/40, BS 505 and emission filter BP 527/30; for Cy3, excitation filter BP 525/30, BS 550 LP and emission filter BP 575/30; for Cy3 (if co-analysed with EGFP or EYFP) and Alexa594 excitation filter BP 565/30, BS Q595 LP and emission filter HQ 645/75. Images were acquired and analysed using the program Metamorph (Universal Imaging Corporation, Downingtown, PA). In some experiments, we used an Olympus IX81 fluorescence microscope with a × 60 1.49 NA Apochromat objective with a × 1.6 magnifying lens (Olympus, Tokyo, Japan) and an EMCCD camera (ImagEM C9100-13, 512 × 512-chip, 16 × 16 μm2 pixel size; Hamamatsu Photonics, Hamamatsu, Japan) with a × 2 magnifying lens. For detection of TMA-DPH and Cy3, the triple band fluorescence filter set U-M3DAFIC3/HC (Olympus) was used in combination with a 150-W Xenon-lamp integrated into the MT20-I-fluorescence illumination system (Olympus). Alternatively, for imaging a Zeiss Axio Observer D1 fluorescence microscope equipped with a × 100 1.4 NA plan apochromate objective and a cooled digital 12 bit CCD camera (Sensicam QE, 6.45 × 6.45 μm2 pixel size, PCO AG) was used. Cy3 fluorescence was detected using filter set F36-503 TRITC (BrightLine HC 543/22, BS 562 and Bright Line HC 593/40 (AHF Analysentechnik AG)) and for TMA-DPH detection we used filter set F11000 (excitation filter D 350/50, 400 DCLP and emission filter E 420 LP (AHF Analysentechnik AG)). Images thus obtained were analysed using the program ImageJ.
Comparative quantitation of fluorescence intensity was done essentially as previously described (Lang et al, 2002). In brief, membrane sheets were selected for analysis in the TMA-DPH image in order to avoid preferential selection and to be able to recognize also membrane sheets with only a very dim or no staining. The TMA-DPH image was also used as reference for defining a rectangular ROI on the membrane sheet area from which the mean fluorescence intensity was analysed. Next to the membrane sheet a background value was taken, which was subtracted from the mean fluorescence intensity value, and background corrected values were averaged. The number of membrane sheets analysed for one condition in one experiment are given in the respective figure legends. For one condition and experiment the average value was calculated and the mean±s.e.m. was determined from several independent experiments (for number of independent experiments see also the figure legend).
In Figure 6, values were fitted to a curve applying the equation 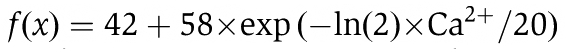 . For correlation coefficient analysis, the Pearson's correlation coefficient r was calculated within a defined ROI placed at the aligned green and the red channel of a membrane sheet. Correlation analysis was performed with the ImageJ Plugin ‘Just Another Colocalisation Plugin’ (Bolte and Cordelieres, 2006) and resulting r values were averaged for each condition in one individual experiment (for method see also Manders et al, 1992).
. For correlation coefficient analysis, the Pearson's correlation coefficient r was calculated within a defined ROI placed at the aligned green and the red channel of a membrane sheet. Correlation analysis was performed with the ImageJ Plugin ‘Just Another Colocalisation Plugin’ (Bolte and Cordelieres, 2006) and resulting r values were averaged for each condition in one individual experiment (for method see also Manders et al, 1992).
Stimulation and immunostaining of bovine chromaffin cells
Coverslips with bovine chromaffin cells were incubated for 30 s at 37°C in high or low potassium Ringer solution (low potassium: 130 mM NaCl, 4 mM KCl, 10 mM HEPES, 1 mM MgCl2, 5 mM CaCl2, 44 mM glucose, pH 7.4; high potassium: 50 mM NaCl, 80 mM KCl, 10 mM HEPES, 1 mM MgCl2, 5 mM CaCl2, 44 mM glucose, pH 7.4).
Cells were then fixed in paraformaldehyde in PBS for 1 h. Afterwards they were washed for 20 min using PBS supplemented with 50 mM NH4Cl and additionally twice for 10 min each with PBS. The cells were permeabilized using for 5 min PBS+0.2% Triton-X 100, followed by a treatment for 10 min with the same buffer supplemented with BSA (PBS-BSA-Triton). Permeabilized cells were incubated for 1 h with the primary antibodies (diluted 1:200 in PBS-BSA-Triton). After three washing steps for 10 min each (in PBS-BSA-Triton), the cells were incubated for 1 h with the secondary antibody (diluted 1:200 in PBS-BSA-Triton). For the final washing steps, the cells were incubated three times for 10 min each with PBS. The samples were embedded in Mowiol (6 g Glycerol (AR No. 4094, Merck, Darmstadt, Germany), 2.4 g Mowiol 4-88-Hoechst, Frankfurt, Germany, 6 ml water, 12 ml 200 mM Tris, pH 7.2 buffer) (provided by Department of Nanobiophotonics, Max-Planck Institute for Biophysical Chemistry, Göttingen, Germany), which contained DAPI (Molecular probes, Invitrogen) at a dilution of 1:1000 to counterstain nuclear DNA in chromaffin cells.
Confocal microscopy
For confocal microscopy, an Olympus FluoView1000 laser scanning microscope with the following settings was used: objective: UPLANSAPO × 60 NA 1.35; DAPI was excited by the 405-nm laser line and emission was collected between 425 and 475 nm; Cy3 was excited at 543 nm and emission was collected between 555 and 655 nm; frame size was 512 × 512 pixels, pixel size 207 nm and sampling speed was 80.0 μs/pixel. To avoid preferential selection of brightly stained cells during imaging, we selected in the transmission mode a region with chromaffin cells that was centred. Using the live mode in the transmission setting, we focussed to the equatorial layer of the cell. Then images were recorded in the brightfield and the fluorescence channels.
Analysis of bovine chromaffin cells
Images were analysed using ImageJ. In order to avoid preferential selection of very bright or dim immunostainings, cells were chosen for analysis randomly in the brightfield channel. Cells already dead before the treatment were identified using the DAPI staining as reference. Then, in the Cy3 fluorescence channel a linescan of 5 pixel width was placed along the edge of the cell in a circular manner so as to record the average fluorescence intensity along the plasma membrane. The mean background fluorescence intensity outside of the cell was subtracted from the intensity recording. For one condition in one experiment, values were averaged for 19–34 cells and the average obtained for fluorescence under high potassium condition was normalized to the average value obtained for low potassium treatment.
FRAP experiments and TIRF microscopy
Experiments were performed 1–2 days after transfection. Cells carrying coverslips were mounted into a microscopy chamber filled with 1 ml low calcium Ringer solution (130 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 48 mM D(+)glucose, 10 mM HEPES pH 7.4) and a cell was selected. After performing one FRAP measurement or taking a TIRF micrograph, we applied the same buffer with 20 μM ionomycin and 5 mM CaCl2 (for stimulation) or 20 μM ionomycin, 5 mM CaCl2 and 10 mM EGTA (for controls). After 5 min, a second FRAP measurement was performed or TIRF micrograph was recorded.
FRAP was performed using a confocal laser scanning microscope from Olympus (Olympus FluoView1000) applying a UPLANSAPO × 60 NA 1.35 objective (pixel size was adjusted to 207 nm for SNAP25 and 103 nm for syntaxin). FRAP experiments were performed at RT, using for bleaching an ∼3 μm × 3 μm ROI at the basal plasma membrane. In addition, after the measurement ROIs outside from the cell (for background analysis) and next to the ROI used for bleaching (control ROI) were defined. The FRAP experiment included a pre-bleach phase (three frames at 1 Hz for SNAP25 and 0.5 Hz for syntaxin using the 488-nm laser line at laser intensities <1%), then bleaching with a 405-nm laser at full intensity and finally a post-bleaching phase in which 76 frames were monitored at 1 Hz for SNAP25 and 0.5 Hz for syntaxin.
Intensity traces from the three ROIs were imported into Microsoft Excel and further processed (in some cases bleaching occurred not exactly at the defined ROI and the ROI for analysis was adjusted manually). First, the background was subtracted and then each time point of the background corrected post-bleaching sequence was normalized to the averaged pre-bleach values (average of the three pre-bleach frames) using the following equation:
After this procedure all traces obtained from individual cells measured the same day were averaged (three to six cells were averaged per day and construct).
In SigmaPlot, the obtained averaged traces were fitted as described (Ficz et al, 2005), applying a hyperbola function with off set:
where t½ corresponds to the half-maximum recovery time, y0 is the offset of the curve and maxrec is the maximal amplitude of the recovery curve. From the values obtained for t½ the apparent lateral diffusion coefficients were determined according to Ficz et al (2005) using a correction factor for the ROI geometry (Ficz, 2005). Experiments exhibiting a z drift of the focal plane, identified by an intensity variation of >10% (comparing the average of the first three pre-bleach values to the average of the final three post-bleach values) in the control ROI, were excluded from the analysis.
For TIRF microscopy, a motorized Olympus IX81 microscope (Olympus) was used equipped with an EMCCD camera (16 μm × 16 μm pixel size, ImagEM C9100-13, Hamamatsu Photonics, Hamamatsu, Japan) and a × 60 NA 1.49 Apochromat objective in combination with a × 1.6 and an additional × 2 magnifying lens. For imaging, the 488-nm laser band from an Argon laser was used in combination with the Olympus filter U-MTIR488-HC. Images were analysed using the program ImageJ.
Protein charge distribution analysis
In order to analyse the percentage of negatively and positively charged amino-acid side chains within the cytoplasmic regions, we used reference protein sequences identified in the protein database as outlined in Supplementary Table S1. Using uniprot.org as reference, the cytoplasmic regions were determined. Within these stretches the number of Asp (D), Glu (E), Arg (R) and Lys (K) amino acids were calculated, since they represent the set of polar and charged amino acids at pH 7.4. The percentage of negatively (Asp and Glu) or positively (Arg and Lys) charged amino acids within the cytoplasmic region was calculated and the difference between the two values was determined (Supplementary Table S1) and plotted versus the relative residual immunofluorescence after treatment of the plasma membranes with 850 nM or 54 μM Ca2+.
Supplementary Material
Acknowledgments
We thank Dr Jochen J Sieber for providing plasmids, Dagmar Diezmann for excellent technical assistance, Professor Dr Erwin Neher and Ina Herford for providing bovine chromaffin cells and Dr Gottfried Mieskes for assistance in handling data. This work was supported by grants of the Deutsche Forschungsgemeinschaft including funding from the SFB 406 (TL and RJ) and the SFB645 (TL).
Footnotes
The authors declare that they have no conflict of interest.
References
- Barnstable CJ, Hofstein R, Akagawa K (1985) A marker of early amacrine cell development in rat retina. Brain Res 352: 286–290 [DOI] [PubMed] [Google Scholar]
- Barszczewski M, Chua JJ, Stein A, Winter U, Heintzmann R, Zilly FE, Fasshauer D, Lang T, Jahn R (2008) A novel site of action for alpha-SNAP in the SNARE conformational cycle controlling membrane fusion. Mol Biol Cell 19: 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B (2005) Control of synaptic strength, timing by the release-site Ca2+ signal. Nat Neurosci 8: 426–434 [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- Dupuy AD, Engelman DM (2008) Protein area occupancy at the center of the red blood cell membrane. Proc Natl Acad Sci USA 105: 2848–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G (2005) Protein Dynamics in the Nucleus: Implications for Gene Expression. Dissertation, University of Göttingen, Göttingen, Germany [Google Scholar]
- Ficz G, Heintzmann R, Arndt-Jovin DJ (2005) Polycomb group protein complexes exchange rapidly in living Drosophila. Development 132: 3963–3976 [DOI] [PubMed] [Google Scholar]
- Frick M, Schmidt K, Nichols BJ (2007) Modulation of lateral diffusion in the plasma membrane by protein density. Curr Biol 17: 462–467 [DOI] [PubMed] [Google Scholar]
- Halemani ND, Bethani I, Rizzoli SO, Lang T (2010) Structure, dynamics of a two-helix SNARE complex in live cells. Traffic 11: 394–404 [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G (1994) Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371: 513–515 [DOI] [PubMed] [Google Scholar]
- Heinemann C, Chow RH, Neher E, Zucker RS (1994) Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+. Biophys J 67: 2546–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann R, Kachel V, Thoenen H (1983) Relationship between NGF-mediated volume increase, ‘priming effect’ in fast, slow reacting clones of PC12 pheochromocytoma cells. Role of cAMP. Exp Cell Res 145: 179–190 [DOI] [PubMed] [Google Scholar]
- Hurley JH, Misra S (2000) Signaling, subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomol Struct 29: 49–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki M, Ying YS, Fujita T, Anderson RG (2002) A molecular sensor detects signal transduction from caveolae in living cells. J Biol Chem 277: 43389–43398 [DOI] [PubMed] [Google Scholar]
- Jacobson K, Mouritsen OG, Anderson RG (2007) Lipid rafts: at a crossroad between cell biology, physics. Nat Cell Biol 9: 7–14 [DOI] [PubMed] [Google Scholar]
- Kai M, Sakane F, Jia YJ, Imai S, Yasuda S, Kanoh H (2006) Lipid phosphate phosphatases 1, 3 are localized in distinct lipid rafts. J Biochem (Tokyo) 140: 677–686 [DOI] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W (2010) Single secretory granules of live cells recruit syntaxin-1, synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci USA 107: 20810–20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SR, Liu Y (1997) Characterization of the palmitoylation domain of SNAP-25. J Neurochem 69: 1864–1869 [DOI] [PubMed] [Google Scholar]
- Lang T (2003) Imaging SNAREs at work in ‘unroofed’ cells--approaches that may be of general interest for functional studies on membrane proteins. Biochem Soc Trans 31: 861–864 [DOI] [PubMed] [Google Scholar]
- Lang T, Margittai M, Holzler H, Jahn R (2002) SNAREs in native plasma membranes are active, readily form core complexes with endogenous, exogenous SNAREs. J Cell Biol 158: 751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Wacker I, Steyer J, Kaether C, Wunderlich I, Soldati T, Gerdes HH, Almers W (1997) Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein, evanescent-wave microscopy. Neuron 18: 857–863 [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K (2010) Lipid rafts as a membrane-organizing principle. Science 327: 46–50 [DOI] [PubMed] [Google Scholar]
- Low SH, Vasanji A, Nanduri J, He M, Sharma N, Koo M, Drazba J, Weimbs T (2006) Syntaxins 3, 4 are concentrated in separate clusters on the plasma membrane prior to the establishment of cell polarity. Mol Biol Cell 17: 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA (1992) Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA, confocal microscopy. J Cell Sci 103: 857–862 [DOI] [PubMed] [Google Scholar]
- Margittai M, Fasshauer D, Pabst S, Jahn R, Langen R (2001) Homo-, heterooligomeric SNARE complexes studied by site-directed spin labeling. J Biol Chem 276: 13169–13177 [DOI] [PubMed] [Google Scholar]
- Marsault R, Murgia M, Pozzan T, Rizzuto R (1997) Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J 16: 1575–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Murray D (2005) Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438: 605–611 [DOI] [PubMed] [Google Scholar]
- Mulgrew-Nesbitt A, Diraviyam K, Wang J, Singh S, Murray P, Li Z, Rogers L, Mirkovic N, Murray D (2006) The role of electrostatics in protein-membrane interactions. Biochim Biophys Acta 1761: 812–826 [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A (2004) Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA 101: 10554–10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sorensen JB (2005) Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell 16: 5675–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst S, Margittai M, Vainius D, Langen R, Jahn R, Fasshauer D (2002) Rapid, selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J Biol Chem 277: 7838–7848 [DOI] [PubMed] [Google Scholar]
- Pike LJ (2009) The challenge of lipid rafts. J Lipid Res 50 (Suppl): S323–S328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Medine CN, Dun AR, Moulton DJ, Mandula O, Halemani ND, Rizzoli SO, Chamberlain LH, Duncan RR (2010) t-SNARE protein conformations patterned by the lipid microenvironment. J Biol Chem 285: 13535–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Kusumi A (2004) Role of the membrane skeleton in creation of microdomains. Subcell Biochem 37: 233–245 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants, functional consequences. Physiol Rev 86: 369–408 [DOI] [PubMed] [Google Scholar]
- Salaün C, Gould GW, Chamberlain LH (2005) The SNARE proteins SNAP-25, SNAP-23 display different affinities for lipid rafts in PC12 cells. Regulation by distinct cysteine-rich domains. J Biol Chem 280: 1236–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber JJ, Willig KI, Heintzmann R, Hell SW, Lang T (2006) The SNARE motif is essential for the formation of syntaxin clusters in the plasma membrane. Biophys J 90: 2843–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber JJ, Willig KI, Kutzner C, Gerding-Reimers C, Harke B, Donnert G, Rammner B, Eggeling C, Hell SW, Grubmuller H, Lang T (2007) Anatomy, dynamics of a supramolecular membrane protein cluster. Science 317: 1072–1076 [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569–572 [DOI] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GL (1972) The fluid mosaic model of the structure of cell membranes. Science 175: 720–731 [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J et al. (2006) Molecular anatomy of a trafficking organelle. Cell 127: 831–846 [DOI] [PubMed] [Google Scholar]
- Tsien R, Pozzan T (1989) Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol 172: 230–262 [DOI] [PubMed] [Google Scholar]
- Uhles S, Moede T, Leibiger B, Berggren PO, Leibiger IB (2003) Isoform-specific insulin receptor signaling involves different plasma membrane domains. J Cell Biol 163: 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are, how they behave. Nat Rev Mol Cell Biol 9: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly FE, Sorensen JB, Jahn R, Lang T (2006) Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol 4: e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.