

SUMMARY
Septation of the single tubular embryonic outflow tract into two outlet segments in the heart requires the precise integration of proliferation, differentiation and apoptosis during remodeling. Lack of proper coordination between these processes would result in a variety of congenital cardiac defects such as those seen in the retinoid X receptor α knockout (Rxra−/−) mouse. Rxra−/− embryos exhibit lethality between embryonic day (E) 13.5 and 15.5 and harbor a variety of conotruncal and aortic sac defects making it an excellent system to investigate the molecular and morphogenic causes of these cardiac malformations. At E12.5, before the embryonic lethality, we found no qualitative difference between wild type and Rxra−/− proliferation (BrdU incorporation) in outflow tract cushion tissue but a significant increase in apoptosis as assessed by both TUNEL labeling in paraffin sections and caspase activity in trypsin-dispersed hearts. Additionally, E12.5 embryos demonstrated elevated levels of transforming growth factor β2 (TGFβ2) protein in multiple cell lineages in the heart. Using a whole-mouse-embryo culture system, wild-type E11.5 embryos treated with TGFβ2 protein for 24 hours displayed enhanced apoptosis in both the sinistroventralconal cushion and dextrodorsalconal cushion in a manner analogous to that observed in the Rxra−/−. TGFβ2 protein treatment also led to malformations in both the outflow tract and aortic sac. Importantly, Rxra−/− embryos that were heterozygous for a null mutation in the Tgfb2 allele exhibited a partial restoration of the elevated apoptosis and of the malformations. This was evident at both E12.5 and E13.5. The data suggests that elevated levels of TGFβ2 can (1) contribute to abnormal outflow tract morphogenesis by enhancing apoptosis in the endocardial cushions and (2) promote aortic sac malformations by interfering with the normal development of the aorticopulmonary septum.
Keywords: Apoptosis, Outflow tract, Retinoid X receptor, TGFβ2, Whole mouse culture
INTRODUCTION
Development of the outflow tract (OFT) in the heart is a complex process involving proliferation, differentiation, and apoptosis of multiple cell lineages of myocardial, endothelial, mesenchymal, neural crest and epicardial origins (DeRuiter et al., 1992; Markwald et al., 1977; Ya et al., 1998). The intricate processes associated with the remodeling of cushion tissue into valvular and septal structures involves several cell lineage-dependent events including apoptosis in outflow tract cushion tissue, myocardialization in the OFT and atrioventricular (AV) canal, and expansion of the inter-ventricular septum and compact zone. Although remodeling has been described on a morphogenic level, the molecular mechanisms that integrate proliferation, differentiation and apoptosis for proper remodeling of the tubular heart into the four-chambered organ remain largely unknown.
Proper growth and maturation of the heart requires a variety of molecular signals including retinoic acid (RA) (Chien et al., 1993; Fishman and Chien, 1997; Kubalak and Sucov, 1999). Too much or too little RA causes a spectrum of cardiovascular malformations including OFT and aortic arch anomalies (Lammer et al., 1985; Pexieder et al., 1995; Taylor et al., 1980; Wilson and Warkany, 1949; Wilson et al., 1953), AV canal malformations (Wilson and Warkany, 1949; Wilson et al., 1953), looping disturbances (Dickman and Smith, 1996; Smith et al., 1997) and ventricular septal defects (Smith et al., 1998; Wilson and Warkany, 1949; Wilson et al., 1953). Still, the mechanistic links between the teratogenic effect of RA and the resultant anomalies remain clouded by the fact that RA activates multiple signaling pathways.
A variety of gene-targeted mice have been generated to investigate the roles of RA during development. Mutations in several of the RA receptor subtypes lead to specific cardiac malformations and provide an avenue for understanding both the events leading up to embryonic lethality and for investigating mechanistic links between RA signaling and cardiac malformations (Kastner et al., 1994; Kastner et al., 1997; Sucov et al., 1994). Previous studies have documented the wide range of OFT abnormalities in the embryonic day (E) 13.5–15.5 retinoid X receptor α knockout (Rxra−/−) mouse (Gruber et al., 1996). The significance of RXRα in heart development is underscored by the fact that embryos heterozygous for Rxra also displayed cardiac malformations (Gruber et al., 1996). Defects were observed in several compartments of the heart, including the aortic sac, conotruncus, atrioventricular canal and ventricular myocardium (Gruber et al., 1996). To date, no candidate downstream genes have been uncovered that offer an explanation of how these cardiac malformations occur.
We report that maldevelopment of the aorticopulmonary (AoP) septum is evident by E11.5 in the Rxra−/−. By E12.5, malformations of the endocardial cushions in the OFT became apparent. Associated with the malformed cushions is an increased level of apoptosis. In normal development, the onset of apoptosis in OFT cushion tissue occurs between E11.5 and E12.5 (for a review, see (Poelmann et al., 2000). Malformations in this region were evident by E12.5, coincident with the apoptosis and at least 1 day before the embryonic lethality in mutant mice. Concomitant with these morphologic changes was elevated levels of transforming growth factor β2 (TGFβ2) protein in the Rxra−/− embryonic heart. When we exposed wild-type whole mouse embryos in culture to TGFβ2 protein, apoptosis was enhanced in the OFT. TGFβ2 treatment also lead to abnormal development of the OFT cushions and AoP septum. Accordingly, lowering TGFβ2 levels by intercrossing the TGFβ2 heterozygous mouse into the Rxra−/− background partially restored the apoptosis and decreased the developmental defects. These results suggest that elevated levels of TGFβ2 can increase apoptosis in the OFT, which in turn perturb cardiac development. This study is the first report that identifies a potential downstream target molecule in the retinoic acid signaling pathway proven to be critical during cardiac morphogenesis.
MATERIALS AND METHODS
Transgenic mice and embryos
Colonies of Rxra and Tgfb2 heterozygous animals were bred and maintained on the C57Bl6 background as described previously (Sanford et al., 1997; Sucov et al., 1994). Genotyping was performed by PCR analysis as previously described (Gruber et al., 1996; Sucov et al., 1994). Timed pregnant females were sacrificed, the embryos removed and bisected just below the heart. Embryos were staged according to Kaufman (Kaufman, 1992) with the day of plug being embryonic day (E) 0.5. The lower torso was processed for genotyping by PCR analysis. The upper torso of the embryo was fixed in 2% paraformaldehyde in phosphate-buffered saline (PBS) for 1 hour at room temperature. Embryos were then rinsed with PBS, dehydrated in a graded ethanol series, cleared in xylenes and embedded in Paraplast Xtra (Oxford, St Louis, MO). Specimens were sectioned at a thickness of 5 μm and mounted on Superfrost-Plus slides (VWR, West Chester, PA) for histological and immunohistochemical analysis.
Gene expression analyses
RNase protection analysis and semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) were performed on single hearts that had only their atria removed (Gruber et al., 1996; Kubalak et al., 1994). Preparation of RNA and generation of the MLC2a probe was as previously described (Kubalak et al., 1994). The TGFβ1 and TGFβ2 plasmids were kind gifts from Harold Moses and probes were generated as described (Derynck et al., 1986; Miller et al., 1989). For RT-PCR, RNA was prepared as described previously (Kubalak et al., 1994). PCR amplification of a 132 bp fragment corresponding to wild-type TGFβ2 was as described by Sanford et al. (Sanford et al., 1997). Amplification conditions were optimized to acquire semi-quantitative expression levels of TGFβ2 and GAPDH (as control). The conditions for TGFβ2 were 30 cycles at 95°C for 30 seconds, 50°C for 50 seconds and 72°C for 1 minute. PCR primers for GAPDH were as described by Kondo et al. (Kondo et al., 1998) and the conditions were 20 cycles at 94°C for 30 seconds, 54°C for 30 seconds and 72°C for 45 seconds. Relative expression levels of TGFβ2 were normalized to GAPDH using densitometry (NIH Image 1.62) of the respective PCR products. Numbers represent the -fold expression relative to wild type.
Whole-mouse-embryo culture
Preparation of rat serum was performed as described (Cockroft, 1990). Whole mouse embryos were cultured as described previously (Gruber et al., 1998; Hertig et al., 1999). Embryos were incubated in culture for 1 hour before the addition of reagents and then treated either with vehicle [4 mM HCl, 0.1% bovine serum albumin (BSA)] or with TGFβ2 protein (R&D Systems, Minneapolis, MN) at a final concentration of 0.1, 10 or 30 ng/ml. The culture continued for 18 hours at which time the media was changed to fresh, reagent-containing, pre-equilibrated media and allowed to continue for an additional 6 hours. The embryos were then fixed in 2% paraformaldehyde-PBS for 1 hour at room temperature, washed in PBS and embedded as described above.
Immunohistochemistry
Protein expression was examined by first rehydrating the sections through xylenes and graded ethanols, then rinsing in PBS. Sections were blocked in 10% normal goat serum for 1 hour, washed in PBS, and incubated with antibodies against either TGFβ1 (1:100), TGFβ2 (1:300) or TGFβ3 (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA). They were then washed in PBS and incubated with FITC-conjugated goat anti-rabbit secondary antibody for 2.5 hours at room temperature. All primary and secondary antibodies were diluted in 0.1% BSA-PBS. After a final wash in PBS, slides were mounted with Slowfade AntiFade (Molecular Probes, Eugene, OR) and analyzed using a confocal laser-scanning microscope (BioRad MRC-1024, Cambridge, MA).
BrdU incorporation
Timed pregnant mice at E12.5–14.5 were injected intraperitoneally with 100 μg/gm body weight of 5-bromo-2′-deoxyuridine (BrdU, Sigma) in 200 μl of sterile 0.9% NaCl 1 hour before sacrificing (Porteus et al., 1994). Embryos were dissected from the uterus and processed as described above for paraffin-wax embedding and sectioning in the transverse orientation. After rehydrated through graded ethanols, sections were washed in TBSA-BSAT (10 mM Tris-HCl, 150 mM NaCl, 0.02% sodium azide, 1% BSA, 0.1% Triton X-100). They were then incubated with 4 N HCl for 30 minutes, followed by neutralization with 100 mM sodium tetraborate for 5 minutes, and then incubated in 0.1% Triton X-100 for 20 minutes. Specimens were co-stained with monoclonal anti-BrdU (Developmental Studies Hybridoma Bank, Iowa City, IA) and polyclonal atrial myosin light chain 2 (MLC2a, 1:500) (Kubalak et al., 1994) in TBSA-BSAT, for 16 hours at room temperature. After a wash in TBSA-BSAT, they were incubated with FITC-conjugated anti-mouse secondary antibody and Cy5-conjugated anti-rabbit at room temperature for 2.5 hours. After a final rinse in TBSA-BSAT, the sections were mounted with Slowfade AntiFade mounting media and analyzed by laser-scanning confocal microscopy.
TUNEL staining
TUNEL assays were performed using the ApopTag kit (Oncor, Gaithersburg, MD) according to the manufacturer’s recommendations with minor modifications in order to co-stain for MLC2a. After incubation in the TdT enzyme reaction mixture, sections were exposed to fluorescein-conjugated anti-digoxigenin combined with anti-MLC2a for 30 minutes at room temperature. Slides were then washed for 15 minutes in PBS. After a 1 hour incubation with anti Cy5-conjugated anti-rabbit secondary antibody in 1% BSA-PBS, the slides were mounted as described above.
Quantitation of apoptotic cells in the sinistroventralconal cushion (SVCC) and the dextrodorsalconal cushion (DDCC) was estimated by scoring the number of apoptotic nuclei in serial sections 40 μm apart from the most anterior to the most posterior aspects of the cushions. Because in Rxra−/− hearts it was frequently difficult to discern valve primordia from other neighboring cushion tissues, all sections in the series that contained cushion tissue were routinely included in the TUNEL analysis in both wild-type and mutant embryos. Apoptotic nuclei in the myocardium and endocardium of the OFT were excluded from the quantification. Positive nuclei were normalized to the total number of cushion cells counted from adjacent Hematoxylin and Eosin sections. Data are expressed as the number of positive apoptotic nuclei as a percentage of total cushion cells. Differences were considered significant when P<0.05 using a two-way analysis of variance.
Caspase activity assay
Caspase activity was performed using the Caspatag Fluorescein Caspase Activity Kit (Intergen Co., Purchase, NY) according to the manufacturer’s recommendations with minor modifications to assay single embryonic hearts. Briefly, cardiac cells from E12.5 embryonic hearts (atria removed) were isolated by incubation and trituration in 0.05% trypsin for 20 minutes at 37°C. Activated caspases were then labeled and quantitated by measuring the intensity of the fluorescein label using a fluorescence spectrophotometer (SLM Aminco, Urbana, IL). A portion of the final cell suspension was removed for protein determination in order to normalize the caspase activity to total protein using the Micro BCA Protein Assay Kit (Rockford, IL). Each litter was assayed separately and results are expressed as -fold change relative to wild-type embryos within each individual litter. Statistics were performed on -fold change values with differences considered significant when P<0.05 using a one-way analysis of variance.
RESULTS
Rxra−/− embryos display abnormal development of the aortic sac and outflow tract before the onset of embryonic lethality
The window of lethality in Rxra gene-targeted mice is between E13.5 and E15.5. The cause of death has been attributed to embryonic heart failure (Dyson et al., 1995). Approximately 67% of mutant embryos display OFT and aortic arch malformations, including abnormal development of the aorticopulmonary (AoP) septum, hypoplastic or absent conotruncal ridges, shortened conotruncal septum and double outlet right ventricle (Gruber et al., 1996). These malformations suggest a perturbation of the development of mesenchymal tissues in the OFT. Therefore, we evaluated OFT morphogenesis with an emphasis on the development of the conotruncal cushions in the mutant from the onset of heart tube formation (E7.5–8.5) through the final stages of septation (E13.5–14.5). No abnormalities were observed in the overall morphology of the OFT in Rxra−/− embryos between E7.5 and 10.5. The formation of the heart tube (E7.5–8.5) and the onset (E9.0–9.5) and extent of mesenchymal cell seeding in OFT cushion tissue was similar between wild type and mutant (data not shown), such that by E10.5, the morphology of the OFT ridges were indistinguishable between the two genotypes (Fig. 1A,B). Furthermore, by E11.5 the extent of transformed epithelial cells was equivalent between wild-type and mutant hearts and resulted in a normal overall shape of the DDCC and SVCC in the mutant (Fig. 1D,F,H) when compared with the wild type (Fig. 1C,E,G).
Fig. 1.
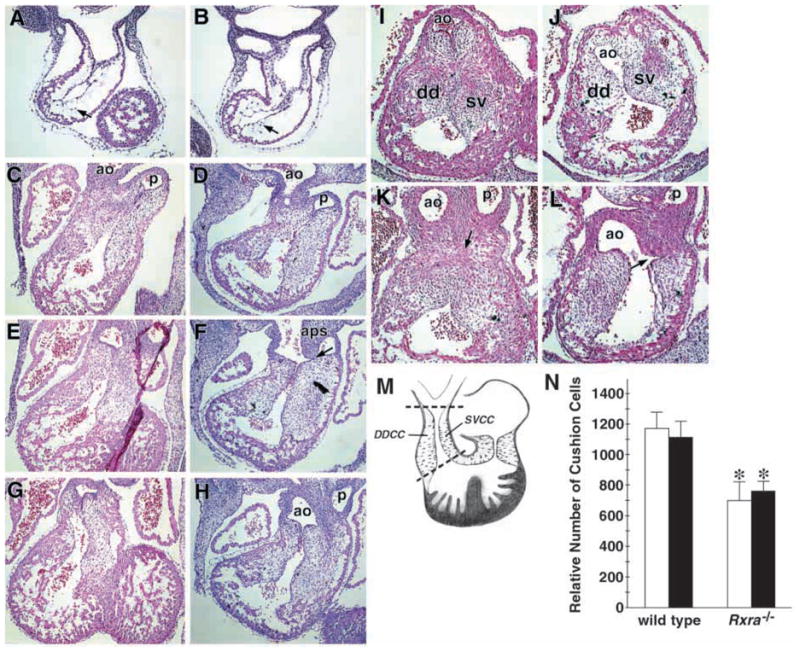
Developmental abnormalities in the Rxra−/− embryo are evident at E11.5 in the aortic sac and at E12.5 in outflow tract cushion tissue. Embryos at various times during embryogenesis were analyzed for morphological malformations of the heart. Transverse sections of E10.5 wild-type (A) and Rxra−/− (B) hearts show that the early stages of epithelial-to-mesenchymal cell transformation in outflow tract cushion tissue occurs in a similar manner between wild type and mutant. Arrows indicate mesenchymal cells that have migrated into the cardiac jelly. (C–H) Transverse sections of E11.5 embryonic hearts. Note throughout the outflow tract (OFT), the extent of mesenchymal seeding in the cushions is similar in the wild type (C,E,G) and Rxra−/− (D,F,H). However, at this age, the aorticopulmonary (AoP) septum in the mutant shows signs of abnormal development, such as incomplete fusion with the sinistroventralconal cushion (F, arrow). (I–L) Transverse sections of E12.5 embryonic hearts. The OFT cushions have begun to fuse in the wild type (K, arrow) while they have become noticeably hypoplastic and malformed in the Rxra−/−. Further evidence of an eventual AoP window is also observed in the Rxra−/− (L, arrow). dd, dextrodorsalconal cushion; sv, sinistroventralconal cushion. (M,N) Quantitation of the relative number of cells was performed by counting nuclei in serial sections 40 μm apart beginning at the most cranial aspects of the OFT cushions and continuing down to the most caudal aspects of the cushions (between broken lines, M). (N) Cells from the dextrodorsalconal cushion (DDCC in M; white bars) and sinistroventralconal cushion (SVCC in M; black bars) were counted separately. Both OFT cushions were significantly smaller in the Rxra−/−. *P<0.05. Error bars represent the s.e.m.
Despite the apparent normal development of the OFT cushions at E11.5, it was at this age in the Rxra−/− that the first indications of abnormal development of the AoP septum were evident. Normally, at this stage, the AoP septum has descended far enough towards the heart to fuse with the OFT cushions. This process is pivotal for normal septation of the OFT at the level of the semilunar valve primordia (Fig. 1C,E,G). However, in the E11.5 Rxra−/−, the AoP septum fails to make proper contact with the OFT cushions and incompletely fuses with the conotruncal ridges (Fig. 1F, arrow).
By E12.5 in the wild type, the opposing DDCC and SVCC ridges have made contact with each other and, in the majority of specimens studied have begun to fuse (Fig. 1I,K, arrow), indicating that septation of the OFT is well under way by E12.5. At this age in the Rxra−/−, the development of both OFT cushions was compromised. Not only was there a lack of fusion of the cushions (Fig. 1J,L), we also frequently observed evidence of abnormal interactions between the AoP septum and the conotruncal ridges, as was suggested at E11.5. Indeed, by E12.5 all of the Rxra−/− embryos examined had malformed OFT cushions (11/11) and of those, 64% (7/11) displayed a lack of any fusion between the AoP septum and the SVCC and DDCC suggestive of an imminent AoP window (Fig. 1L, arrow). The remaining embryos displayed incomplete fusion resulting in irregular and hypoplastic ridges (Fig. 1J,L). To evaluate the relative sizes of the DDCC and SVCC in wild-type and mutant embryos, we quantitated the number of cushion cells in sections every 40 μm apart, spanning from the most distal to the most proximal aspects of each of the cushions (Fig. 1M). Confirming what we observed visually, the relative number of cushion cells in both the DDCC and SVCC was significantly less in the mutant (669±116 and 761±69, respectively) when compared with the wild type (1170±107 and 1113±105, respectively, P<0.01) (Fig. 1N). Therefore, by E12.5, the Rxra−/− is characterized by an underdeveloped AoP septum and hypoplastic OFT cushions.
By E13.5 in the wild type, the DDCC and SVCC have nearly completely fused (Fig. 2A) and OFT morphogenesis proceeds into the final stages of septation. In the mutant, the two cushions are so significantly malformed and undersized that the lack of fusion may simply reflect they are physically too far apart to make proper contact with each other (Fig. 2D). The varying degrees of fusion and hypoplastic nature of the OFT ridges would explain the wide range and severity of the OFT defects typically seen in the Rxra−/− (Gruber et al., 1996). Collectively, the data demonstrate that the morphological manifestations of a lack of RXRα protein in the OFT and aortic sac are evident between E11.5–12.5, at least 1 day before the embryonic lethality in these mice.
Fig. 2.
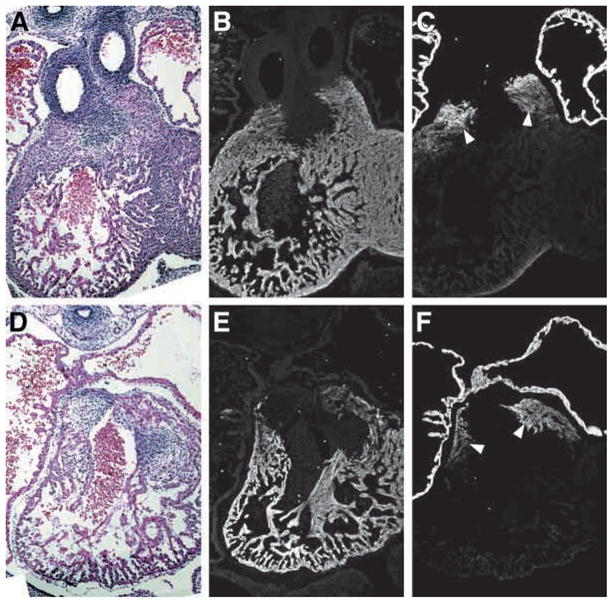
MLC2v and MLC2a are expressed in a normal manner and mark the developing outflow tract in both wild-type and Rxra−/− embryos. Adjacent transverse sections of E13.5 wild-type (A–C) and Rxra−/− (D–F) embryonic hearts. Both MLC2v (B,E) and MLC2a (C,F) are expressed in their respective chambers in both wild-type and mutant embryos. Note that MLC2a is also expressed in a subset of OFT myocardium (C,F, arrowheads) and that this expression pattern is identical in both wild-type and mutant embryos.
MLC2a and MLC2v mark the developing outflow tract and myocardializing myocytes in both wild type and Rxra−/− embryos
The OFT myocardium is unique in that myocytes co-express the atrial and ventricular isoforms of MLC2 throughout the course of cardiogenesis (Franco et al., 1999). Expression of MLC2v (ventricular myosin light chain 2) is restricted to ventricular and OFT myocardium in both the wild type (Fig. 2B) and Rxra−/− (Fig. 2E) at E13.5. Similarly, expression of MLC2a (atrial myosin light chain 2) protein was maintained in the OFT, but downregulated in the ventricles in both wild-type (Fig. 2C) and Rxra−/− embryos (Fig. 2F). This contrasts to message levels of MLC2a, which remain elevated in the ventricles of the mutant embryo (Dyson et al., 1995). These data suggest that some degree of post-transcriptional regulation is maintained within the myocardium of the Rxra−/− during cardiogenesis and reinforces the importance of evaluating protein expression, as opposed to transcript levels, when characterizing the null phenotype.
Myocardialization is the process of myocyte migration that contributes to the formation of the muscular portion of the outlet segments (van den Hoff et al., 1999) and begins at E12.5–13.5 in the mouse (Waller et al., 2000). These specialized myocytes have been reported to co-express MLC2a and MLC2v (Franco et al., 1999). To determine if myocardialization is occurring in the Rxra−/− mice, sections were selected from E13.5 embryos in order to visualize the area between the primordia of the aortic and pulmonary outlets. Identification of this region in wild-type embryos is straightforward as the two outlet cushions are well fused at this stage of development and the condensed mesenchyme is readily identified on histological sections (Fig. 3A, broken line). However, in the Rxra−/−, identification of the corresponding region is complicated by the frequent occurrence of unfused, hypoplastic conotruncal ridges (Fig. 3B). Therefore, serial sections 40 μm apart were evaluated to ensure the analysis of any potential regions of myocardialization. Interestingly, MLC2a and MLC2v were not co-expressed in all myocardializing myocytes in either wild-type or mutant embryos at E13.5. The lateral myocardial cuffs of the OFT expressed both MLC2a and MLC2v (Fig. 3C–F asterisks). However, the midline of this region in the wild type (Fig. 3C, arrowheads) and the corresponding region in the mutant (Fig. 3F, arrowheads) expresses only MLC2v. Importantly, myocytes along the endocardial cushion-myocardial interface manifest the finger-like phenotype characteristic of myocardializing myocytes. This was particularly evident in the wild type (Fig. 3G, arrows). In the Rxra−/−, even though the OFT cushions were not fused, a few myocytes could still be identified with the same finger-like phenotype projecting into the OFT cushions (Fig. 3H, arrows).
Fig. 3.
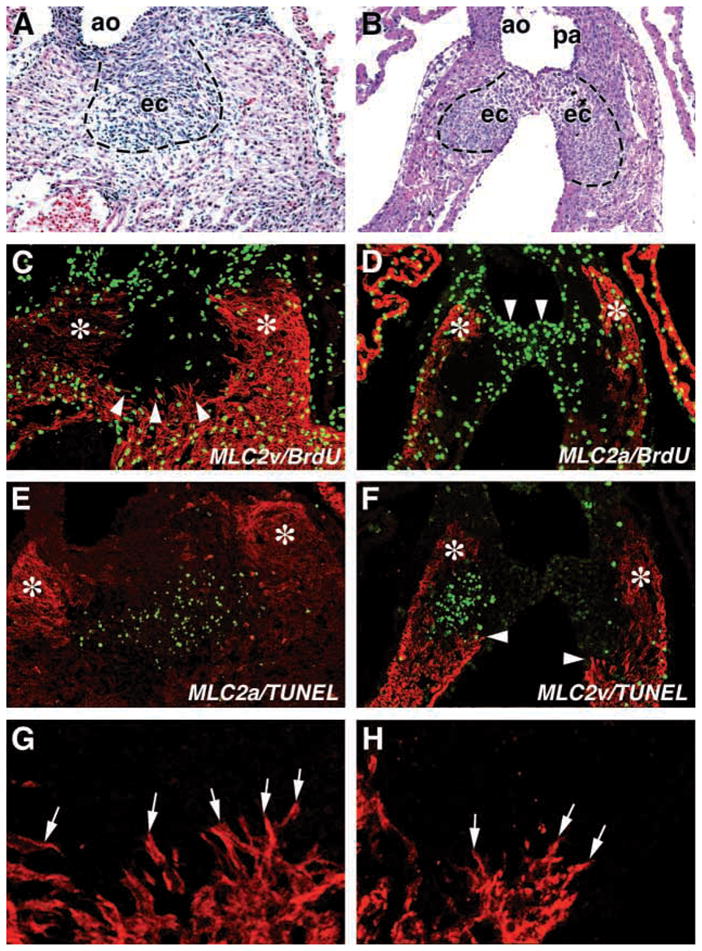
Delineation of outflow tract proliferation, apoptosis and myocardialization in E13.5 wild-type and Rxra−/− embryos. (A,B) Transverse sections of the outflow tracts of E13.5 wild-type and Rxra−/− embryonic hearts, respectively. Broken line depicts the boundary between endocardial cushion tissue (ec) and myocardializing myocytes. ao, aortic outlet. pa, pulmonary outlet. (C) Adjacent section to A immunostained for MLC2v (red) and BrdU (green). Note less cells are BrdU-positive in the myocardial cuffs (asterisks) and endocardial cushion tissue compared with the ventricular myocardium. Myocardializing myocytes are readily identified (arrowheads) and were more easily visualized at higher magnification (G, arrows). (D) Adjacent section to B immunostained for MLC2a (red) and BrdU (green). Similar to wild-type hearts, less cells are BrdU-positive in the myocardial cuffs (asterisks) and endocardial cushion tissue compared with the ventricular myocardium in the Rxra−/−. (E) Adjacent section to C immunostained for MLC2a and apoptotic cells using the TUNEL assay. Apoptosis was largely confined to the endocardial cushion tissue between the myocardial cuffs (asterisks) in the outflow tract. (F) Adjacent section to D immunostained for MLC2v and apoptotic cells using the TUNEL assay. Apoptosis was again largely confined to the endocardial cushion cells between the myocardial cuffs (asterisks). Myocardializing myocytes were also identifiable in the Rxra−/− (arrowheads) and were more easily visualized at higher magnification (H, arrows).
Enhanced apoptosis precedes the decreased proliferation in Rxra−/− embryos
As indicated above, the initial manifestation of a morphologic defect in the OFT of mutant embryos was the decrease in size and abnormal shape of the cushions at E12.5 (Fig. 1J,L). This effect is even more evident by E13.5 (Fig. 2D) when the two cushions are rarely seen in contact with each other. To evaluate whether the undersized cushions are the result of a decrease in proliferation, an increase in programmed cell death, or a combination of both, we evaluated the proliferation status by analyzing BrdU incorporation and determined the level of apoptosis by performing the TUNEL assay. We could not detect a qualitative difference in cell proliferation between wild type and Rxra−/− embryos until E14.5, i.e. well into the embryonic lethality of most Rxra−/− embryos (data not shown). Additionally, the pattern of BrdU incorporation was also similar between wild-type and mutant embryos. Notably, within the heart, the degree of proliferation varied depending on the cell type examined. For example, the endocardial cushions and myocardializing myocytes flanking the cushions proliferate more slowly than myocytes within the ventricles. This was true for both wild-type (Fig. 3C) and mutant (Fig. 3D) embryos. Mesenchymal cells destined to become valvular tissue displayed a higher proliferative capacity than the adjacent cushion mesenchyme involved in conal septum formation in both wild-type (data not shown) and mutant embryonic hearts (Fig. 3D, arrowheads). Collectively, these data suggest that the reduced cushion sizes in the mutant were not the result of an overt decrease in proliferation.
At E13.5 in the wild-type heart, apoptosis is predominantly confined to OFT cushion tissue lying between the aortic and pulmonary outlet valve primordia where the two OFT cushions have fused (Fig. 3E) (Okamoto et al., 1981; Ya et al., 1998). Atrioventricular cushion tissue and the myocardial crest of the interventricular septum also contain apoptotic nuclei which is normal at this stage of development (Poelmann et al., 2000). The sites of cell death in the mutant heart were confined to these same regions (Fig. 3F, and data not shown). Thus, ectopic apoptosis was not found in the Rxra−/− endocardium, epicardium or the myocardium of either the ventricles or atria (data not shown).
As E12.5 mutant embryos already display hypoplastic cushions (Fig. 1J,L) before any overt decrease in proliferation, we set out to evaluate whether this could be explained by a higher degree of apoptosis (Fig. 4). To quantitate this, we counted the number of apoptotic nuclei in serial sections 40 μm apart from the most cranial to the most caudal aspect of the OFT cushions (Fig. 1M). Fig. 4 shows representative images from a single wild-type embryo (Fig. 4A,C,E) and a single mutant (Fig. 4B,D,F). In wild-type embryos, 3.9% of cells in the DDCC and 5.5% of cells in the SVCC were apoptotic, while in mutant embryos this was elevated to 10.6% and 9.8%, respectively (Fig. 4G; P<0.05). Additionally, the region of the enhanced apoptosis in the mutant extended more cranial and caudal than in wild-type cushion, particularly in the SVCC (Fig. 4A–F). To confirm what we observed using the TUNEL assay, we analyzed caspase activity in trypsin-dispersed embryonic heart cells. Caspase activity has been reported to be a good indicator of apoptotic activity in the developing OFT (Watanabe et al., 1998). Consistent with the TUNEL assay, we observed significantly elevated caspase activity in mutant E12.5 hearts (Fig. 4H). Not surprisingly, as there were no apparent morphological differences between E11.5 wild-type and mutant OFT cushions, there were also no differences in the observed programmed cell death. In fact, there was essentially no OFT apoptosis for either genotype at E11.5 (data not shown). Hence, we conclude that elevated apoptosis is responsible for the hypoplastic phenotype of the OFT cushions observed in Rxra−/− mouse.
Fig. 4.

Programmed cell death in the outflow tract is enhanced in the E12.5 Rxra−/− embryo. Serial sections from a representative wild-type (A,C,E) and Rxra−/− (B,D,F) outflow tract showing TUNEL-positive nuclei and co-immunostained with MLC2a to demarcate the OFT myocardium. More cells in the mutant were apoptotic than in the wild type (arrowheads). (G) The relative numbers of cushion cells were counted from the adjacent histological sections and the data are expressed as the relative number of apoptotic cells as a percent of the total cells. Cells from the dextrodorsalconal cushion (DDCC, white bars) and sinistroventralconal cushion (SVCC, black bars) were counted separately. Both OFT cushions in the Rxra−/− exhibited significantly more apoptosis than in the wild type. *P<0.05, compared with wild type DDCC. **P<0.01, compared with wild type SVCC. Error bars indicate the s.e.m. (H) Caspase activity assays confirmed the results obtained with the TUNEL technique in that mutant hearts demonstrated enhanced apoptosis. Results are expressed as -fold change relative to wild type. Three separate litters were assayed and -fold changes were calculated separately for each litter. Each litter had at least one wild-type and one mutant embryo. *P<0.05, compared with wild type (wt) and heterozygous (het) embryos. Error bars indicate the s.e.m.
TGFβ2 transcript and protein levels are elevated prior to the developmental defects in Rxra−/− embryos
The transforming growth factor β family of proteins have been implicated in retinoic acid signaling pathways in several different model systems (Bavik et al., 1996; Chen et al., 1996; Jakowlew et al., 1992; Tsuiki and Kishi, 1999). The cardiac expression pattern reported for TGFβ2 suggests that it may play a role in cardiogenesis during the E11.5–E13.5 window of development (Dickinson et al., 1990). In E13.5 cardiac tissue, where the cardiovascular abnormalities are evident, we found TGFβ2 mRNA expression elevated in the mutant, whereas TGFβ1 mRNA levels remained unchanged (Fig. 5A,B). TGFβ2 protein was elevated in the mutant (Fig. 5C) in parallel with TGFβ2 mRNA. Upregulation of TGFβ2 was also evident in E12.5 embryos (i.e. 1 day before the lethality in Rxra−/− embryos) (Fig. 5D). Note the expression of TGFβ2 in the wild type is slightly higher at E12.5 than at E13.5, consistent with the findings of Dickson et al. (Dickson et al., 1993). Interestingly, the distribution of the elevated TGFβ2 appeared to encompass several cell types, including ventricular and atrial myocardium, endocardium and epicardium and, to a lesser extent cells within the endocardial cushions. TGFβ1 and TGFβ3 in the mutant did not appear different from wild type (data not shown). These results demonstrate that increased TGFβ2 expression coincides with elevated apoptosis and precedes diminished proliferation in Rxra−/− embryos.
Fig. 5.

TGFβ2 mRNA and protein expression is elevated in the Rxra−/− embryo. (A) RNase protection assays showed that message levels for TGFβ2 were elevated in the mutant (KO). WT, wild type. (B) Message levels for TGFβ1 were not altered while MLC2a was elevated in the mutant. EF1α was used as a loading control. (C) Immunohistochemical analysis on transverse serial sections of E13.5 OFT regions demonstrated that TGFβ2 protein was also elevated in the mutant. (D) E12.5 OFT also shows elevated TGFβ2 in the mutant heart.
TGFβ2 promotes apoptosis and malformations in the developing outflow tract in cultured whole mouse embryos
To test whether TGFβ2 could induce apoptosis and malformations in the OFT, wild-type E11.5 whole mouse embryos were cultured for 24 hours in the absence and presence of 0.1–30 ng/ml TGFβ2. Outflow tract apoptosis was quantitated in the DDCC and SVCC as described above. TGFβ2 treatment of wild-type embryos significantly enhanced apoptosis in the OFT cushions in a concentration-dependent manner (Fig. 6). Interestingly, after 24 hours in culture, apoptosis in the OFT was no different in wild-type than in mutant embryos. When compared with non-cultured embryos (Fig. 4G), this appeared to reflect an increase in apoptosis in the wild type but not in the mutant. The finding that apoptosis in the controls was elevated was actually less surprising than finding apoptosis in the mutant relatively unchanged. It has been well established that the ability of whole embryos to develop in culture becomes increasingly difficult from E12.5 onwards (Tam, 1998). Thus, we would expect apoptosis to begin to increase in the embryo around this embryonic age. The observation that apoptosis in the mutant did not increase to the extent that it did in the wild type may reflect that TGFβ2 expression is elevated as a result of placing the embryo in culture. Nonetheless, TGFβ2 treatment was still able to significantly increase apoptosis in both the DDCC and the SVCC in the OFT (Fig. 6).
Fig. 6.
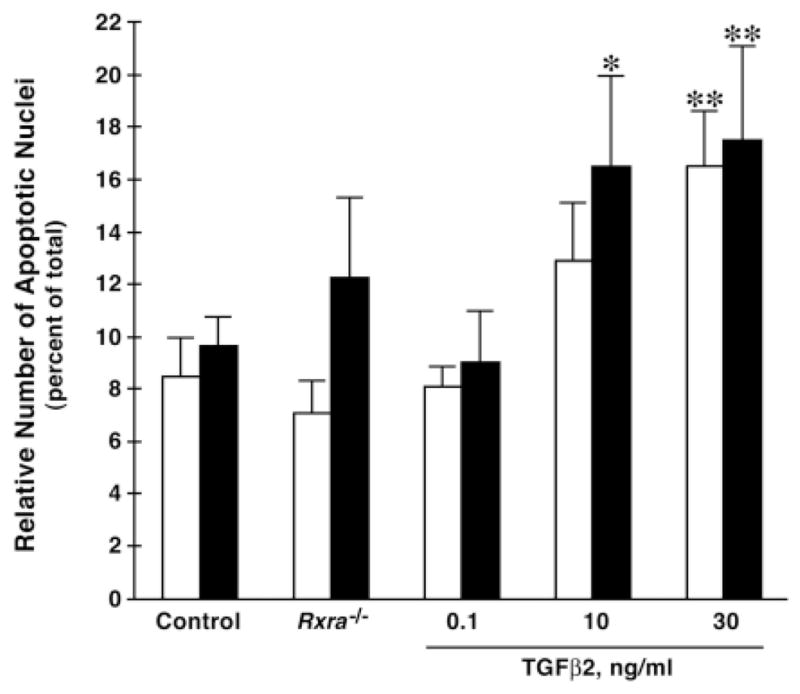
TGFβ2 treatment enhances outflow tract apoptosis in whole mouse embryo culture. Embryonic day 11.5 wild-type embryos were treated with 0.1–30 ng/ml TGFβ2 protein and placed in whole embryo culture for 24 hours, then processed as described in Materials and Methods. Wild-type (Control) or mutant (Rxra−/−) E11.5 embryos were also cultured for 24 hours as comparative controls. TGFβ2 treatment enhanced outflow tract apoptosis in a concentration-dependent manner. Apoptosis was quantitated using the TUNEL assay as described in Fig. 5. Dextrodorsalconal cushion (white bars). Sinistroventralconal cushion (black bars). *P<0.05, compared with control DDCC. **P<0.05, compared with the respective DDCC and SVCC Control and 0.1 ng/ml TGFβ2. Error bars indicate s.e.m.
In addition to the enhanced apoptosis in the OFT, morphological abnormalities were found that appeared similar to those in the Rxra−/− (Fig. 7). Control embryos dissected at E12.5 are shown in Fig. 7A–D. Vehicle-treated wild type embryos cultured at E11.5 for 24 hours retained much of their capacity to remodel the OFT (Fig. 7E–H). In these embryos, the AoP septum descended into the OFT and fused with the SVCC and DDCC to the extent that a clear separation between the aortic and pulmonary outlets could be detected (Fig. 7A,E, respectively). Cultured E11.5 Rxra−/− embryos displayed smaller cushions and abnormal development of the AoP septum (Fig. 7I–L, arrow). Wild-type embryos treated with TGFβ2 exhibited OFT malformations similar to those found in the Rxra−/− (Fig. 7M–P); that is, both the SVCC and the DDCC were irregular and hypoplastic. Additionally, the AoP septum had not descended to the same extent as in the wild-type or untreated embryos so that it frequently never reached either the SVCC or the DDCC (Fig. 7M, arrow).
Fig. 7.

TGFβ2-treated whole mouse embryos in culture reveal outflow tract malformations that are similar to Rxra−/− embryos. Each group of four images represents serial sections 40 μm apart through the OFT of a representative embryo. (A–D) OFT of a wild-type embryo dissected at E12.5. The orifices of the aortic and pulmonary outlets are well separated. (E–H) OFT of a wild-type embryo dissected at E11.5 and cultured for 24 hours. Similar to the E12.5 wild type, the orifices of the aortic and pulmonary outlets are well separated. (I–L)) OFT of an Rxra−/− embryo dissected at E11.5 and cultured for 24 hours. Rxra−/− embryos in culture retained the malformed outflow tract phenotype. Thus, the AoP septum has not descended into the OFT to the same extent as in the wild type and, as a result, the beginning of an AoP septum defect is apparent (I, arrow). Note the cushions are also underdeveloped and malformed. (M–P) OFT of a wild-type embryo dissected at E11.5 and cultured for 24 hours in the presence of 10 ng/ml TGFβ2. The phenotype is similar to the Rxra−/− in that the AoP septum has not formed properly (M, arrow) and the cushions are underdeveloped and malformed. ao, aortic outlet. p, pulmonary outlet.
Heterozygosity for Tgfb2 partially rescues the enhanced apoptosis and outflow tract malformations in Rxra−/− embryos
To further evaluate the effect of TGFβ2 on apoptosis, we questioned whether lowering the levels of TGFβ2 in the Rxra− would restore the programmed cell death and rescue some of the OFT malformations in the mutant. To accomplish this, we crossed the Tgfb2 gene-targeted mouse (generously provided by Dr Tom Doetschman) onto the Rxra− background. Embryos heterozygous for Tgfb2 express an intermediate amount of TGFβ2 compared with wild-type and knockout Tgfb2 embryos, as determined by RT-PCR (Fig. 8A). We then analyzed the level of apoptosis and extent of the OFT malformations in E12.5 and E13.5 Rxra−/− embryos that were also heterozygous for the Tgfb2 allele (Fig. 8B–D). Wild-type or heterozygous Rxra embryos were normal whether they were either wild type or heterozygous for Tgfb2 (data not shown). Littermate Rxra−/− embryos (wild type for Tgfb2) showed the typical abnormal development of the AoP septum and malformed cushions (Fig. 8B, Rxra−/−/Tgfb2+/+, arrow). Embryos that were Rxra−/− and heterozygous for Tgfb2 demonstrated a partial rescue of the AoP septum phenotype as indicated by the extent of septation of the aortic sac (Fig. 8B, Rxra−/−/Tgfb2+/−, arrow); that is, the AoP septum had extended into the OFT beyond the most cranial aspect of the pulmonary valves. At E13.5 when septation should be nearly finished, the septum still had not completely divided the OFT/aortic sac in the Rxra−/−/Tgfb2+/−, but had developed further than in the Rxra−/− alone, indicating a partial rescue of the Rxra−/− phenotype (Fig. 8C)
Fig. 8.

Heterozygosity for Tgfb2 partially restores the outflow tract and apoptotic phenotypes in the Rxra−/−. (A) RT-PCR demonstrating the intermediate expression of TGFβ2 message in the E12.5 Tgfb2+/−(HT) embryo when compared with wild type (WT) and knockout (KO) Tgfb2. Numbers represent the -fold change compared with the wild-type sample in the first lane and are normalized to the levels of GAPDH as described in Materials and Methods. Expression levels of TGFβ2 in the Tgfb2+/− were intermediate between wild type and knockout. (B) OFT histology of E12.5 wild-type, Rxra−/−/Tgfb2+/+ and Rxra−/−/Tgfb2+/− embryos. The typical malformed AoP septum (aps) is evident in the Rxra−/−/Tgfb2+/+ (arrow) as well as the irregular and hypoplastic endocardial cushions. In the Rxra−/−/Tgfb2+/− embryo, the AoP septum has migrated far enough into the OFT that there is a distinct separation between the aortic outlet and the pulmonary valve primordia (arrow). (C) OFT histology of E13.5 wild-type, Rxra−/−/Tgfb2+/+ and Rxra−/−/Tgfb2+/− embryos. In Rxra−/−/Tgfb2+/− embryos, the AoP septum has descended into the OFT and fused with the SVCC, separating the aortic outlet from the semilunar valve primordia (compare arrows). (D) Quantitation of apoptosis in E12.5 and E13.5 embryos from Rxra/Tgfb2 intercrossed mice. Apoptosis was quantitated as described in Materials and Methods. For E12.5: white bars, dextrodorsalconal cushion; black bars, sinistroventralconal cushion. For E13.5, the single bar represents the total apoptotic nuclei in both cushions as the cushions are fused in the wild type at this age. Rxra wild type (R-WT), Rxra−/− (R-KO), Tgfb2 wild type (T-WT), Tgfb2 heterozygous (T-HT). The levels of apoptosis in both the E12.5 and E13.5 Rxra−/−/Tgfb2+/− embryos are intermediate between the wild-type and Rxra−/−/Tgfb2+/+ embryos. In particular, note that the relative number of apoptotic nuclei in the Rxra−/−/Tgfb2+/− is no longer significantly different from that in Rxra−/−/Tgfb2+/+. Five different litters are represented in each of the two age groups. N, the number of embryos within each genotype combination.
Quantitation of the apoptosis in Rxra−/−/Tgfb2+/− embryos correlated with the partial restoration of the defects in the mutant (Fig. 8D); that is, the percentage of apoptotic nuclei in the Rxra−/−/Tgfb2+/− was intermediate between the wild type and Rxra−/−. The degree of apoptosis was inversely related to the cushion size (data not shown), as depicted above (compare Fig. 1M with Fig. 4G). However, the DDCC and SVCC were still irregular in intercrossed embryos. These results demonstrate the importance of programmed cell death during remodeling of cushion tissue. Furthermore, our results indicate that TGFβ2 is involved in the regulation of apoptosis in the OFT. Increased levels of endogenous TGFβ2 in the Rxra−/− results in elevated levels of apoptosis, which can be decreased by crossing into the TGFβ2-deficient mouse.
DISCUSSION
Early during the development of the mouse heart, endothelial cells in the OFT transform into mesenchyme and migrate into the cardiac jelly forming the cellularized outflow tract cushions. Simultaneously, in the aortic sac, the AoP septum extends in a ventrocaudal direction towards the OFT cushions to contribute to the supravalvular portion of the outlet conduits. At E11.5–12.5 in the mouse the DDCC and SVCC cushions begin to fuse in the region between the two outlet valve primordia. Concomitant with fusion, and in this same midline region of cushion tissue, neural crest cells that have migrated into the OFT ridges undergo apoptosis (Poelmann et al., 1998). Myocytes that surround the apoptotic zone then begin to migrate in from the lateral myocardial cuffs, a process termed myocardialization (Lamers et al., 1995), and, over the ensuing days of development, form the muscular region of the outlet septum. While the above morphological processes have been well documented and extensively studied using various in vitro culture systems (DeRuiter et al., 1992; Markwald et al., 1977; van den Hoff et al., 1999; van den Hoff et al., 2000; Ya et al., 1998), the molecular cues that regulate these events in vivo are not well understood. In the current study, we show that Rxra−/− embryos display elevated levels of TGFβ2 mRNA and protein. We also show, using a whole-mouse-embryo culture system and a transgenic approach, that elevated TGFβ2 can contribute to malformations in both the OFT and aortic sac.
Effect of enhanced apoptosis on OFT development in the Rxra−/−
Each mutant E12.5 embryo examined displayed irregular and hypoplastic conotruncal ridges. We conclude that this is not a result of a decrease in proliferation as we could not detect differences in BrdU incorporation until E14.5 when the embryos were already showing the typical signs of lethality in these mice (Sucov et al., 1994). The hypoplastic phenotype could also be the result of a decrease in the number of cells that undergo epithelial-mesenchymal transformation. This, too, is unlikely as there were no obvious morphological differences between wild-type and mutant OFT regions through E11.5. Additionally, OFT cushion explants from E9.5 mutant embryos displayed equivalent epithelial-mesenchymal transformation and migration of mesenchymal cells as their wild-type littermates in three-dimensional collagen gel cultures (S. W. K., P. J. Gruber and K. R. Chien, unpublished). Indeed, at E11.5 in the Rxra−/−, the sizes of the cushions and relative density of cushion cells were similar to the wild type. Alternatively, enhanced apoptosis could lead to hypoplastic cushions by eliminating cells that would otherwise proliferate in maturing cushions. Neither wild-type nor Rxra−/− embryonic cushions displayed any significant cell death at E11.5. However, at E12.5 apoptosis was significantly elevated in the Rxra−/− over that in the wild type. Because, in normal development, apoptosis in the OFT progressively increases between E12.5 and E13.5, the enhanced apoptosis at E12.5 in the mutant may represent premature differentiation of cells predestined for programmed cell death. Chambon and coworkers have suggested this concept for the ventricular myocyte population in the Rxra−/− embryo (Kastner et al., 1997). The additional apoptotic cells in the OFT of the Rxra−/− may be surrendering to this same fate. Alternatively, the enhanced apoptosis may represent a subpopulation of cells that would have otherwise not undergone programmed cell death. The exact identity of the apoptotic cell population is currently under investigation. Nonetheless, our results suggest that enhanced apoptosis, and not decreased proliferation, is the major contributing factor to the development of the irregular and hypoplastic cushions in the Rxra−/−.
The precise role of apoptosis during OFT development has not been established. Programmed cell death would not contribute to the overall structure of the OFT cushions per se, but instead may occur as a prerequisite to myocardialization (Poelmann et al., 2000). One line of thinking is that apoptosis of neural crest cells triggers the activation of latent TGFβ2 and, in turn, signals adjacent OFT myocytes to begin myocardializing the apoptotic zone (Poelmann et al., 2000). Conversely, our data support the notion that TGFβ2 might be involved in a signaling cascade that would initiate, rather than respond to, the apoptosis. This does not rule out the possibility that cell lysis may activate latent TGFβ2, particularly if a positive feedback loop exists between activated TGFβ2 and programmed cell death. With regard to myocardialization, the fact that myocyte migration was not enhanced in either the Rxra−/− or in TGFβ2-treated whole mouse embryos suggests that TGFβ2 may not be the stimulus that triggers myocyte migration during septation of the OFT. In fact, even though TGFβ2 was elevated, myocardialization seemed to be blunted in the Rxra−/−. Interestingly, TGFβ2-deficient mice also lack a muscular outlet septum (Sanford et al., 1997). As fusion of the cushion ridges was also lacking in the Rxra−/−, it is intriguing to speculate that TGFβ2, in combination with cushion fusion, may be required to trigger myocardialization. In either case, we propose that TGFβ2 alone is not sufficient. Nevertheless, the temporal and spatial relationship between apoptosis and myocardialization leaves open the possibility that lysis of apoptotic cells somehow signals adjacent myocytes to begin their migration.
Identification of the cell type(s) that undergo apoptosis in wild-type embryos and enhanced apoptosis in the Rxra−/− remains to be determined. The current line of thinking is that all cells destined to undergo programmed cell death in OFT cushion tissue are neural crest cells (Poelmann et al., 1998; Poelmann et al., 2000). Our data do not discount this possibility, because the spatial distribution of apoptotic cells in the mutant is confined to the central region of the cushion ridges where the majority of neural crest cells are thought to reside. This would still be consistent with the sentiment that most dying cells in the mutant could be of neural crest origin rather than, for example, recently transformed epithelial cells (which would be found adjacent to the endocardium). However, the boundaries of apoptosis extend more cranial and caudal than in the wild type, suggesting that other cell types may be subject to programmed cell death in the Rxra−/−. It is intriguing to speculate that if lysis of neural crest cells is a prerequisite for myocardialization to occur (Poelmann et al., 2000), then the fact that myocardialization is not enhanced in the Rxra−/− may mean that the additional cells undergoing apoptosis in the mutant are not neural crest cells.
The developing OFT cushions also participate in the formation of the interventricular septum (Bartelings and Gittenberger-de Groot, 1991). At the inner curvature, the convergence of the outflow septum, interventricular septum, primary atrial septum, spina vestibuli and atrioventricular cushion tissue [collectively termed the central mesenchymal mass (Kirby and Waldo, 1995)] culminates in the formation of a normally septated four-chambered heart (Kirby and Waldo, 1995; Webb et al., 1998; Wessels et al., 1996). Therefore, it is not surprising that the Rxra−/− has a high frequency (94%) of membranous ventricular septal defects (Gruber et al., 1996) because apoptotic nuclei were found in the caudal aspect of the SVCC adjacent to the inner curvature. The lack of expansion of the cushion ridges in combination with the abnormal development of the ventricular myocardium would prevent the normal interactions between the outflow cushions and the interventricular septum and result in a membranous ventricular septal defect as seen in nearly every Rxra−/− (Gruber et al., 1996).
Contribution of TGFβ2 to OFT and aortic sac development in the Rxra−/−
The transforming growth factor β family of proteins has been shown to promote epithelial-mesenchymal cell transformation in culture. TGFβ1 and TGFβ3 can promote transformation of endothelial cells in cultured chick atrioventricular cushion explants (Potts and Runyan, 1989; Potts et al., 1991). Data that support a role for TGFβ2 in cushion development are limited, particularly in OFT cushion tissue. In the atrioventricular canal, it appears that TGFβ2 and TGFβ3 may substitute for each other in promoting epithelial-mesenchymal transformation (Potts and Runyan, 1989). Perhaps the best data supporting a role for TGFβ2 during OFT development have come from TGFβ2 knockout mice, which display hyperplastic OFT cushions (Sanford et al., 1997). This would imply that the lack of TGFβ2 either decreases programmed cell death, or allows for a certain degree of cell division above normal. Surprisingly, however, it has been recently shown that TGFβ2 knockout mice display an elevated level of apoptosis in the OFT, while proliferation was not determined (Bartram et al., 2001). Embryos heterozygous for TGFβ2 were apparently normal (Bartram et al., 2001). Based on the phenotype of the TGFβ2 mutant then, elevated TGFβ2 should result in decreased cushion size. Our data are consistent with this concept because the OFT cushions were hypoplastic both in the E12.5 Rxra−/− and in TGFβ2-treated whole mouse embryos in culture. We propose that the effects of elevated TGFβ2 on cushion development are due to enhancing apoptosis rather than decreasing proliferation.
Rxra−/− embryos that were also heterozygous for Tgfb2 showed a partial restoration of the apoptosis and partial rescue of the developmental defects in the OFT. The reason for only a partial rescue may indicate that the precise level of activated TGFβ2 in the Rxra−/−/Tgfb2+/− varies between embryos. Alternatively, other molecules may also be involved, such that lowering TGFβ2 alone may not be sufficient to rescue fully the defects in the Rxra−/−. Unfortunately, there is no reliable method to determine the precise levels of activated TGFβ2 in an individual embryo. At present we are confident that TGFβ2 plays a role in OFT remodeling; the extent of that contribution beyond modulating apoptosis is currently under investigation.
Normal development of the AoP septum results in the supravalvular formation of separate aortic and pulmonary outlets. If the AoP septum is either absent or incompletely formed, a spectrum of defects can be observed that ranges from persistent truncus arteriosus (no septum) to AoP window (incomplete septum). These defects are found in the Rxra−/− (Gruber et al., 1996), as well as Splotch mice (Franz, 1989) and neural crest ablation studies (Nishibatake et al., 1987). However, very little information has been reported regarding the molecular regulation of the development of the AoP septum. Our data suggest that TGFβ2 is involved in this process. Both Rxra−/− embryos and TGFβ2-treated whole mouse embryos displayed an abnormal development of the AoP septum. Importantly, in Rxra−/− embryos that were also heterozygous for Tgfb2, the AoP septum had descended further down into the OFT and, in each embryo, demonstrated partial fusion with the OFT ridges. This supports the notion that TGFβ2 contributes to the aortic sac defects in the Rxra−/−. The apparent partial rescue suggests that either there are other molecules working in concert with TGFβ2, or the precise level of activated TGFβ2 in the Rxra−/−/Tgfb2+/− depicts the degree of rescue.
Taken together, the results suggest that disturbances in TGFβ2 signaling disrupts the integrative processes of differentiation, apoptosis and proliferation within the OFT endocardial cushions and the aortic sac. Elevated TGFβ2 results in enhanced apoptosis in OFT cushion tissue with minimal effects on proliferation until after the heart begins to fail. The increased apoptosis effectively removes cells from the cushions prematurely and results in hypoplastic cushions. As a result, the malformed DDCC and SVCC are prevented from achieving normal fusion and can not contribute to the septation of the OFT. The role TGFβ2 plays in myocardialization may be indirect as myocyte migration appears to be decreased both in the Rxra−/− where TGFβ2 is elevated and in TGFβ2-knockout mice where TGFβ2 is absent (Sanford et al., 1997). Enhanced TGFβ2 also attenuates the development of the AoP septum so that it can no longer interact properly with the OFT cushions. Consequently, defects that range from an AoP window to persistent truncus arteriosus would be observed depending on the degree of extension of the AoP septum toward the OFT.
We conclude therefore, that one role for Rxrα during cardiogenesis is to maintain proper levels of TGFβ2 expression in the OFT and aortic sac. TGFβ2, in turn, modulates the degree of apoptosis during remodeling of the OFT cushions. Whether TGFβ2 directly or indirectly promotes apoptosis of cushion cells remains to be determined. As TGFβ2 receptors (type II) appear to be ubiquitously expressed in the heart and are unchanged in the Rxra−/− (S. W. K., unpublished), the effects of elevated TGFβ2 in the mutant may be more dependent on the levels of activated protein than on the expression of the receptors. Additionally, whether RXRα directly or indirectly regulates the TGFβ2 expression during murine cardiogenesis is not known. In human osteoblasts and keratinocytes, RXRα heterodimerizes with vitamin D receptors to directly downregulate the TGFβ2 promoter and inhibit cell growth in culture (Wu et al., 1999). Therefore, it is possible that RXRα directly modulates TGFβ2 expression in the embryo. However, an indirect regulatory mechanism is also plausible. Lack of RXRα in one cell may effect expression of TGFβ2 in the neighboring cell. Alternatively, RXRα may indirectly regulate TGFβ2 levels, owing to the potential contributions of TGFβ2 protein by maternal sources or by the placenta. It has been shown in TGFβ1 null mice that maternal sources of TGFβ1 can rescue defects in the TGFβ1 null embryo (Letterio et al., 1994). Similar studies have not been carried out for TGFβ2 null mice. Studies that have examined the maternal influence of TGFβ2 on embryogenesis have primarily focused on the effects of TGFβ2 on the developing fetal-maternal placenta (Ando et al., 1998; Gorivodsky et al., 1999; Schilling and Yeh, 2000), rather than the maternal contribution of TGFβ2 to the embryo. It has been shown that expression of TGFβ2 in the fetal placenta protects the embryo from immune rejection by the mother (Gorivodsky et al., 1999). Thus, a decrease in TGFβ2 expression has been correlated with pregnancy loss (Gorivodsky et al., 1999) while the effects of elevated expression have not been examined. Maternal TGFβ1 has been reported to cross the placenta and can be found in the embryo (Letterio et al., 1994). However, whether TGFβ2 crosses the placenta and contributes to the development of the embryo is not known. Studies designed to evaluate the embryonic effects of a teratogenic insult on the placenta have thus far concentrated on the levels of TGFβ2 mRNA and protein within embryonic target organs and not on potential circulating levels of growth factor from placental tissue (Ivnitsky et al., 2001). Even assuming there are maternal contributions to the levels of TGFβ2 exposed to the embryo, these contributions are probably minimal as we still observe differences in the levels of expression of TGFβ2 protein between wild type and Rxra null mutants, even though both have a TGFβ2 heterozygous mother. Additionally, mRNA levels were also elevated in the mutant heart. Our current work and that by Dickson et al. show that by E12.5 in the wild-type embryo, the levels of TGFβ2 total protein have decreased in the heart to near undetectable levels, i.e. normal levels in the wild type (Dickson et al., 1993). This suggests that altered levels of TGFβ2 in the Rxra−/− are largely determined by expression patterns within the embryo proper.
The role of RXRα-related placental defects in the mutant must also be considered. Chambon and co-workers reported that the placenta of the Rxra−/− was found to be abnormal from E14.5 (Sapin et al., 1997). This suggests that placental defects may contribute only to the lethality from that day forwards. They also documented that premature differentiation of cardiac myocytes in the Rxra−/−, with regard to sarcomeric organization, is first observed at E9.5, before the overt placental defects (Kastner et al., 1997). In our study, we observe malformations in the aortic sac as early as E11.5, also before overt placental defects. In a recent report describing the PPARγ null mouse, Barak et al. have described signs of abnormal development of the placenta in the E9.5 Rxra−/− (Barak et al., 1999). However, the authors also stated that the defects were not nearly as severe as those found in PPARγ null embryos. PPARγ null embryos die at E10.0 because of severe placenta-related developmental defects (Barak et al., 1999). Indeed, no malformations other than the thin myocardium in the Rxra−/− are manifest until E11.5 (our current study) or later (Dyson et al., 1995; Gruber et al., 1996; Sucov et al., 1994). Because RXRα heterodimerizes with PPARγ (Kliewer et al., 1994), one may expect to see an overlap in phenotype between these two knockout mice. However, Barak et al. have noted that the cardiac phenotype (thinning of the ventricular myocardium) in PPARγ null embryos ‘markedly exceeds’ that observed in the Rxra null (Barak et al., 1999) and, importantly, no other malformations in the heart were noted. This could indicate that the thin ventricular myocardium phenotype in the Rxra−/− is due to inadequate PPARγ/RXRα signaling in the placenta, while the outflow tract and aortic sac phenotypes might occur via a distinct heterodimer pair such as RAR/RXR. Therefore, while sites of minimal placentation defects were observed at E9.5 (Barak et al., 1999), overt defects that would effect the function of the placenta in the Rxra−/− were not apparent until E14.5 (Sapin et al., 1997). Moreover, it has been documented that the fetal-maternal circulatory system in the mouse does not become functional, with exchange of nutrients, until around E12.5 (Muntener and Hsu, 1977). Collectively, these data suggest that at least the initial defects in the outflow tract and aortic sac of the Rxra−/− occur independently of influences by the placenta.
Finally, identification of the cell types responding to the elevated TGFβ2 remains to be established. Because during development of the murine ventricular myocardium, RXRα functions in a non-cell autonomous manner (Chen et al., 1998; Tran and Sucov, 1998), it is possible that more than one cell type may be involved in the RXRα/TGFβ2 signaling pathway that leads to the elevated OFT apoptosis. Candidate cell types include the neural crest cells, the mesenchymal cells of the cushions, as well as the endocardium, epicardium and myocardium. We are currently investigating which cell lineages are responding to the elevated TGFβ2 in the Rxra−/−. Determining the regional locations where elevated TGFβ2 becomes activated would help to answer this question, but the issue is complicated by the fact that presently available antibodies for immunohistochemistry can reliably detect only the latent form of the protein. A recent report in the chick heart holds promise for identifying regions where TGFβ2 becomes activated (McCormick, 2001). Identification of similar regions in the mouse will be important in order to predict what cell lineages may be under the influence of TGFβ2.
Acknowledgments
We are grateful to Andy Wessels and Peter Gruber for invaluable discussions and critical review of the manuscript, Thomas Doetschman for the TGFβ2 gene-targeted mouse, and Harold Moses for the use of the TGFβ1 and TGFβ2 plasmids. The BrdU monoclonal antibody developed by S. Kaufman was obtained from the Developmental Studies Hybridoma Bank under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA. The authors also thank Ms Nan Fox for her excellent technical assistance in the husbandry and genotyping of the RXRα and TGFβ2 mouse colonies. This work was supported by grants from the American Heart Association, South Carolina Affiliate and the National Institutes of Health grant HL63714 to S. W. K.
References
- Ando N, Hirahara F, Fukushima J, Kawamoto S, Okuda K, Funabashi T, Gorai I, Minaguchi H. Differential gene expression of TGF-beta isoforms and TGF-beta receptors during the first trimester of pregnancy at the human maternal-fetal interface. Am J Reprod Immunol. 1998;40:48–56. doi: 10.1111/j.1600-0897.1998.tb00388.x. [DOI] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- Bartelings MM, Gittenberger-de Groot AC. Morphogenetic considerations on congenital malformations of the outflow tract. Part 1: Common arterial trunk and tetralogy of Fallot. Int J Cardiol. 1991;32:213–230. doi: 10.1016/0167-5273(91)90329-n. [DOI] [PubMed] [Google Scholar]
- Bartram U, Molin DGM, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- Bavik C, Ward S, Chambon P. Developmental abnormalities in cultured mouse embryos deprived of retinoic acid by inhibition of yolk-sac retinol binding protein synthesis. Proc Natl Acad Sci USA. 1996;93:3110–3114. doi: 10.1073/pnas.93.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRα gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takeshita A, Ozaki K, Kitano S, Hanazawa S. Transcriptional regulation by transforming growth factor beta of the expression of retinoic acid and retinoid X receptor genes in osteoblastic cells is mediated through AP-1. J Biol Chem. 1996;271:31602–31606. doi: 10.1074/jbc.271.49.31602. [DOI] [PubMed] [Google Scholar]
- Chien KR, Zhu H, Knowlton KU, Miller-Hance W, van-Bilsen M, O’Brien TX, Evans SM. Transcriptional regulation during cardiac growth and development. Annu Rev Physiol. 1993;55:77–95. doi: 10.1146/annurev.ph.55.030193.000453. [DOI] [PubMed] [Google Scholar]
- Cockroft DL. Postimplantation Mammalian Embryos: A Practical Approach. IRL: Oxford Press; 1990. Dissection and culture of postimplantation embryos; pp. 15–40. [Google Scholar]
- DeRuiter M, Poelmann R, VanderPlas-de Vries I, Mentink M, Gittenberger-de Groot A. The development of the myocardium and endocardium in mouse embryos. Anat Embryol. 1992;185:461–473. doi: 10.1007/BF00174084. [DOI] [PubMed] [Google Scholar]
- Derynck R, Jarrett JA, Chen EY, Goeddel DV. The murine transforming growth factor-beta precursor. J Biol Chem. 1986;261:4377–4379. [PubMed] [Google Scholar]
- Dickinson ME, Kobrin MS, Silan CM, Kingsley DM, Justice MJ, Miller DA, Ceci JD, Lock LF, Lee A, Buchberg AM, et al. Chromosomal localization of seven members of the murine TGF-beta superfamily suggests close linkage to several morphogenetic mutant loci. Genomics. 1990;6:505–520. doi: 10.1016/0888-7543(90)90480-i. [DOI] [PubMed] [Google Scholar]
- Dickman ED, Smith SM. Selective regulation of cardiomyocyte gene expression and cardiac morphogenesis by retinoic acid. Dev Dyn. 1996;206:39–48. doi: 10.1002/(SICI)1097-0177(199605)206:1<39::AID-AJA4>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Dickson MC, Slager HG, Duffie E, Mummery CL, Akhurst RJ. RNA and protein localisations of TGFβ2 in the early mouse embryo suggest an involvement in cardiac development. Development. 1993;117:625–639. doi: 10.1242/dev.117.2.625. [DOI] [PubMed] [Google Scholar]
- Dyson E, Sucov HM, Kubalak SW, Schmid-Schonbein GW, DeLano FA, Evans RM, Ross J, Chien KR. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor α−/− mice. Proc Natl Acad Sci USA. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman MC, Chien KR. Fashioning the vertebrate heart: earliest embryonic decisions. Development. 1997;124:2099–2117. doi: 10.1242/dev.124.11.2099. [DOI] [PubMed] [Google Scholar]
- Franco D, Markman MMW, Wagenaar GTM, Ya J, Lamers WH, Moorman AFM. Myosin light chain 2a and 2v identifies the embryonic outflow tract myocardium in the developing rodent heart. Anat Rec. 1999;254:135–146. doi: 10.1002/(SICI)1097-0185(19990101)254:1<135::AID-AR17>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Franz T. Persistant truncus arteriosus in the Splotch mouse. Anat Embryol. 1989;180:457–464. doi: 10.1007/BF00305120. [DOI] [PubMed] [Google Scholar]
- Gorivodsky M, Torchinsky A, Zemliak I, Savion S, Fein A, Toder V. TGF beta 2 mRNA expression and pregnancy failure in mice. Am J Reprod Immunol. 1999;42:124–133. [PubMed] [Google Scholar]
- Gruber PJ, Kubalak SW, Pexieder T, Sucov HM, Evans RM, Chien KR. RXRα deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J Clin Invest. 1996;98:1332–1343. doi: 10.1172/JCI118920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber PJ, Kubalak SW, Chien KR. Down-regulation of atrial markers during cardiac chamber morphogenesis is cell-autonomous in murine embryos. Development. 1998;125:4427–4438. doi: 10.1242/dev.125.22.4427. [DOI] [PubMed] [Google Scholar]
- Hertig CM, Kubalak SW, Wang Y, Chien KR. Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J Biol Chem. 1999;274:37362–37369. doi: 10.1074/jbc.274.52.37362. [DOI] [PubMed] [Google Scholar]
- Ivnitsky I, Torchinsky A, Savion S, Shepshelovich J, Orenstein H, Toder V, Fein A. TGFβ2 in embryos with inborn anomalies: Effect of maternal immunopotentiation. Am J Reprod Immunol. 2001;45:41–51. doi: 10.1111/j.8755-8920.2001.450107.x. [DOI] [PubMed] [Google Scholar]
- Jakowlew AM, Cubert J, Danielpour D, Sporn MB, Roberts AB. Differential regulation of the expression of transforming growth factor-β mRNAs by growth factors and retinoic acid in chicken embryo chondrocytes, myocytes, and fibroblasts. J Cell Pysiol. 1992;150:337–385. doi: 10.1002/jcp.1041500222. [DOI] [PubMed] [Google Scholar]
- Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P. Genetic analysis of RXRα developmental function: convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell. 1994;78:987–1003. doi: 10.1016/0092-8674(94)90274-7. [DOI] [PubMed] [Google Scholar]
- Kastner P, Messaddeq N, Mark M, Wendling O, Grondona JM, Ward S, Ghyselinck N, Chambon P. Vitamin A deficiency and mutations of RXR-Alpha, RXR-Beta and RAR-Alpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development. 1997;124:4749–4758. doi: 10.1242/dev.124.23.4749. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. The Atlas of Mouse Develoment. New York, NY: Academic Press; 1992. [Google Scholar]
- Kirby ML, Waldo KL. Neural Crest and Cardiovascular Patterning. Circ Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, Kusachi S, Ninomiya Y, Yoshioka H, Oohashi T, Doi M, Murakami T, Moritani H, Kumashiro H, Tsuji T. Expression of type XVII collagen alpha-1 chain mRNA in the mouse heart. Jpn Heart J. 1998;39:211–220. doi: 10.1536/ihj.39.211. [DOI] [PubMed] [Google Scholar]
- Kubalak SW, Sucov HM. Retinoids in Heart Development. In: Harvey RP, Rosenthal N, editors. Heart Development. San Diego, CA: Academic Press; 1999. pp. 209–219. [Google Scholar]
- Kubalak SW, Miller-Hance WC, O’Brien TX, Dyson E, Chien KR. Chamber specification of atrial myosin light chain-2 expression precedes septation during cardiogenesis. J Biol Chem. 1994;269:16961–16970. [PubMed] [Google Scholar]
- Lamers WH, Virágh S, Wessels A, Moorman AFM, Anderson RH. Formation of the tricuspid valve in the human heart. Circulation. 1995;91:111–121. doi: 10.1161/01.cir.91.1.111. [DOI] [PubMed] [Google Scholar]
- Lammer E, Chen D, Hoar R, Agnish N, Benke P, Braun J, Curry C, Fernoff P, Grix A, Jr, Lott I, et al. Retinoic acid embryopathy. New Engl J Med. 1985;313:837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Geiser AG, Kulkarni AB, Roche NS, Sporn MB, Roberts AB. Maternal rescue of transforming growth factor-β1 null mice. Science. 1994;264:1936–1938. doi: 10.1126/science.8009224. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–120. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- McCormick KM. TGFβ2 activation status during cardiac morphogenesis. Dev Dyn. 2001;222:17–25. doi: 10.1002/dvdy.1167. [DOI] [PubMed] [Google Scholar]
- Miller DA, Lee A, Pelton RW, Chen EY, Moses HL, Derynck R. Murine transforming growth factor-β2 cDNA sequence and expression in adult tissues and embryos. Mol Endocrinol. 1989;3:1108–1114. doi: 10.1210/mend-3-7-1108. [DOI] [PubMed] [Google Scholar]
- Muntener M, Hsu YC. Development of trophoblast and placenta of the mouse. A reinvestigation with regard to the in vitro culture of mouse trophoblast and placenta. Acta Anat. 1977;98:241–252. [PubMed] [Google Scholar]
- Nishibatake M, Kirby ML, Van Mierop LH. Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation. 1987;75:255–264. doi: 10.1161/01.cir.75.1.255. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Akimoto N, Satow Y, Hidaka N, Miyabara S. Role of cell death in conal ridges of the developing human heart. In: Pexieder T, editor. Mechanisms of Cardiac Morphogensis and Teratogenesis. Vol. 5. New York, NY: Raven Press; 1981. pp. 127–137. [Google Scholar]
- Pexieder T, Blanc O, Pelouch V, Ostadalova I, Milerova M, Ostadal B. Late fetal development of retinoic acid induced transposition of the great arteries. In: Clark MR, Takas A, editors. Developmental Mechanisms of Heart Disease. Armonk, NY: Futura Publishing Company; 1995. pp. 297–307. [Google Scholar]
- Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: differentiation and apoptosis. Dev Dyn. 1998;212:373–384. doi: 10.1002/(SICI)1097-0177(199807)212:3<373::AID-AJA5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Molin D, Wisse LJ, Gittenberger-de Groot AC. Apoptosis in cardiac development. Cell Tiss Res. 2000;301:43–52. doi: 10.1007/s004410000227. [DOI] [PubMed] [Google Scholar]
- Porteus MH, Bulfone A, Liu JK, Peulles L, Lo CC, Rubenstein JL. DLX-2, MASH-1 and MAP-2 expression and bromodeoxyuridine incorporation define molecularly distinct cell populations in the embryonic mouse forebrain. J Neurosci. 1994;14:6370–6383. doi: 10.1523/JNEUROSCI.14-11-06370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts JD, Runyan RB. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor β. Dev Biol. 1989;134:392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- Potts JD, Dagle J, Walder JA, Weeks DL, Runyan RB. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor β3. Proc Natl Acad Sci USA. 1991;88:1516–1520. doi: 10.1073/pnas.88.4.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenbergerdegroot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapin V, Dolle P, Hindelang C, Kastner P, Chambon P. Defects of the chorioallantoic placenta in mouse RXRalpha null fetuses. Dev Biol. 1997;191:29–41. doi: 10.1006/dbio.1997.8687. [DOI] [PubMed] [Google Scholar]
- Schilling B, Yeh J. Transforming growth factor-beta(1), –beta(2), –beta(3) and their type I and II receptors in human term placenta. Gynecol Obstet Invest. 2000;50:19–23. doi: 10.1159/000010272. [DOI] [PubMed] [Google Scholar]
- Smith SM, Dickman ED, Thompson RP, Sinning AR, Wunsch AM, Markwald RR. Retinoic acid directs cardiac laterality and the expression of early markers of precardiac asymmetry. Dev Biol. 1997;182:162–171. doi: 10.1006/dbio.1996.8474. [DOI] [PubMed] [Google Scholar]
- Smith SM, Dickman ED, Power SC, Lancman J. Retinoids and their receptors in vertebrate embryogenesis. J Nutr. 1998;128:467S–470S. doi: 10.1093/jn/128.2.467S. [DOI] [PubMed] [Google Scholar]
- Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. RXRα mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8:1007–1018. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- Tam PPL. Postimplantation mouse development: Whole embryo culture and micro-manipulation. Int J Dev Biol. 1998;42:895–902. [PubMed] [Google Scholar]
- Taylor IM, Wiley MJ, Agur A. Retinoic acid-induced heart malformations in the hamster. Teratology. 1980;21:193–197. doi: 10.1002/tera.1420210210. [DOI] [PubMed] [Google Scholar]
- Tran CM, Sucov HM. The RXRalpha gene functions in a non-cell-autonomous manner during mouse cardiac morphogenesis. Development. 1998;125:1951–1956. doi: 10.1242/dev.125.10.1951. [DOI] [PubMed] [Google Scholar]
- Tsuiki H, Kishi K. Retinoid-induced limb defects 2: Involvement of TGF-β2 in retinoid-induced inhibition of limb bud development. Reprod Toxicol. 1999;13:113–122. doi: 10.1016/s0890-6238(98)00070-7. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, Wessels A. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–490. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJB, van den Eijnde SM, Viragh S, Moorman AFM. Programmed cell death in the developing heart. Cardiovasc Res. 2000;45:603–620. doi: 10.1016/s0008-6363(99)00401-0. [DOI] [PubMed] [Google Scholar]
- Waller BR, McQuinn T, Phelps AL, Markwald RR, Lo CW, Thompson RP, Wessels A. Conotruncal anomalies in the Trisomy 16 mouse: An immunohistochemical analysis with emphasis on the involvement of neural crest. Anat Rec. 2000;260:279–293. doi: 10.1002/1097-0185(20001101)260:3<279::AID-AR65>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Choudhry A, Berlan M, Singal A, Siwik E, Mohr S, Fisher SA. Developmental remodeling and shortening of the cardiac outflow tract involves myocyte programmed cell death. Development. 1998;125:3809–3820. doi: 10.1242/dev.125.19.3809. [DOI] [PubMed] [Google Scholar]
- Webb S, Brown NA, Anderson RH. Formation of the atrioventricularseptal structures in the normal mouse. Circ Res. 1998;82:645–656. doi: 10.1161/01.res.82.6.645. [DOI] [PubMed] [Google Scholar]
- Wessels A, Markman MW, Vermeulen JL, Anderson RH, Moorman AF, Lamers WH. The development of the atrioventricular junction in the human heart. Circ Res. 1996;78:110–117. doi: 10.1161/01.res.78.1.110. [DOI] [PubMed] [Google Scholar]
- Wilson J, Warkany J. Aortic-arch and cardiac anomolies in offspring of vitamin A deficient rats. Am J Anat. 1949;83:113–155. doi: 10.1002/aja.1000850106. [DOI] [PubMed] [Google Scholar]
- Wilson J, Roth C, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- Wu Y, Craig TA, Lutz WH, Kumar R. Identification of 1 alpha,25-dihydroxyvitamin D3 response elements in the human transforming growth factor beta 2 gene. Biochemistry. 1999;38:2654–2660. doi: 10.1021/bi981944s. [DOI] [PubMed] [Google Scholar]
- Ya J, Vandenhoff MJB, Deboer PAJ, Tesinktaekema S, Franco D, Moorman AFM, Lamers WH. Normal development of the outflow tract in the rat. Circ Res. 1998;82:464–472. doi: 10.1161/01.res.82.4.464. [DOI] [PubMed] [Google Scholar]