

Abstract
We describe a family with a novel, inherited AXIN2 mutation (c.1989G>A) segregating in an autosomal dominant pattern with oligodontia and variable other findings including colonic polyposis, gastric polyps, a mild ectodermal dysplasia phenotype with sparse hair and eyebrows, and early onset colorectal and breast cancers. This novel mutation predicts p.Tyr663X, which is a truncated protein that is missing the last three exons, including the DIX (Disheveled and AXIN interacting) domain. This nonsense mutation is predicted to destroy the inhibitory action of AXIN2 on WNT signaling. Previous authors have described an unrelated family with autosomal dominant oligodontia and a variable colorectal phenotype segregating with a nonsense mutation of AXIN2, as well as a frameshift AXIN2 mutation in an unrelated individual with oligodontia. Our report provides additional evidence supporting an autosomal dominant AXIN2-associated ectodermal dysplasia and neoplastic syndrome.
Keywords: AXIN2, polyposis, ectodermal dysplasia, oligodontia
Introduction
Several inherited colorectal cancer syndromes result from germline mutations in tumor suppressor or mismatch repair genes. These include familial adenomatous polyposis (FAP) resulting from germline APC mutations, MYH-associated polyposis resulting from biallelic germline MYH mutations, and Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome, resulting from mutations in one of four mismatch repair genes, MSH2, MLH1, MSH6, or PMS2 [Lindor, 2009; Lynch, 2008]. These syndromes often include extracolonic manifestations. However, not all familial colon cancer can be attributed to these or other known, rare conditions, suggesting that other uncharacterized cancer syndromes and genes remain to be described.
Because mutations in several genes of the canonical WNT pathway (i.e., APC, CTNNB1 which encodes B-catenin) have been implicated in colorectal tumorigenesis, other genes in this pathway may play a role in these uncharacterized families, including AXIN2. Mutations in genes of the WNT signaling pathway that prevent the degradation of B-catenin lead to tumorigenesis by upregulating WNT signaling. Within the WNT pathway, AXIN2 acts as a negative regulator by contributing to the assembly of the B-catenin degradation complex [Polakis, 2007]. Somatic AXIN2 mutations have been described in a variety of human cancers, including colorectal cancers[Liu et al., 2000; Salahshor and Woodgett, 2005]. AXIN2 has also been independently implicated in tooth agenesis and oral clefts [Callahan et al., 2009; Letra et al., 2009; Mostowska et al., 2006].
Of particular interest is the description of a four-generation Finnish kindred in which both oligodontia and a variable colorectal phenotype segregated with a nonsense mutation in AXIN2 [Lammi et al., 2004]. In this family, the oligodontia phenotype (defined as congenital absence of six or more permanent teeth, third molars excluded) was entirely penetrant in mutation carriers. No other ectodermal findings such as those involving the nails, hair, or skin were described. The colorectal phenotype in this family was variable. Six of the seven family members with oligodontia had colorectal neoplasms, ranging from polyposis to colorectal malignancy with no polyps. Lammi et al. [2004] also screened oligodontia patients for AXIN2 mutations and identified a de novo germline mutation in a 13-year-old boy with oligodontia. Of note, the germline mutation in this patient was a 1-bp insertion in exon 7 that was identical to a frameshift mutation described in a colorectal cancer tissue [Liu et al., 2000].
We describe an unrelated family with an inherited AXIN2 mutation segregating in an autosomal dominant pattern with oligodontia and variable other findings including colonic polyposis, gastric polyps, a mild ectodermal dysplasia phenotype with sparse hair and eyebrows, and early onset colorectal and breast cancers. This report provides additional clinical description of and support for an autosomal dominant AXIN2-associated ectodermal dysplasia and neoplastic syndrome.
Clinical Report
A 35-year-old woman presented for evaluation of a possible ectodermal dysplasia syndrome due to a history of oligodontia, including the congenital absence of greater than ten secondary teeth, and a family history of oligodontia (Figs 1A and 1B). The proband was the product of a full-term, uncomplicated pregnancy with a birth weight of 3232 g. Her past medical history was notable for intermittent non-bloody diarrhea, gastric reflux, peptic ulcer disease, iron deficiency anemia, polycystic ovary disease, decreased fertility, fibromyalgia, insulin resistance, fundic gland polyps, Sjogren disease, anxiety, and depression. She reported normal body hair, normal nails, and normal sweating. Previous evaluations included a normal colonoscopy and upper endoscopy exams revealing multiple fundic gland polyps at ages 33 and 35, normal capsule endoscopy at age 34, and normal rheumatology and celiac sprue evaluations.
Figure 1.
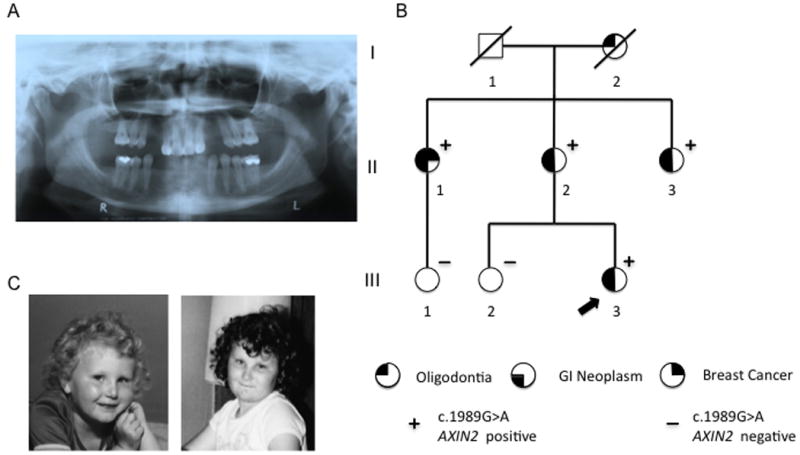
Oligodontia phenotype and pedigree. (A) Panoramic radiograph demonstrating proband's congenital absence of teeth 2, 7, 10, 15, 18, 23, 24, 25, 26, 27, and 31. (B) Autosomal dominant inheritance of oligodontia, c.1989A>G, and variable colorectal phenotype. (C) Photographs of the proband at ages 4 and 8 years showing sparse eyebrows, slightly upslanting palpebral fissures, and thin upper vermillion.
At the time of her visit, physical exam was notable for hypognathia, malar hypoplasia, broad nasal bridge, very sparse eyebrows, fine scalp hair, and slightly upslanting palpebral fissures. Oropharyngeal exam was noted for a high palate, several missing teeth with increased spacing and conical shaping of several teeth (Fig 1A). She was otherwise noted to have normal body hair distribution, normal nails, and normal skin. Review of photographs from ages 4 years and 8 years showed sparse eyebrows, slightly upslanting palpebral fissures, and thin upper vermillion (Fig 1C).
Her family history was notable for oligodontia, absent eyebrows, sparse hair, colon polyps, early onset colon cancer, and early onset breast cancer (Figure 1B and Table 1). The proband's mother (II-2) had oligodontia with absence of most of her secondary teeth. She began colonoscopic screening at age 60 and was found to have >100 adenomatous polyps requiring a sigmoid colectomy at age 60 and a right hemicolectomy at age 62. She was evaluated in our clinic and was also noted to have soft skin with particularly scant body hair, minimal axillary hair, short eyelashes, and very sparse eyebrows, especially laterally.
Table I. Summary of Clinical Findings and Mutation Analysis.
| ID Number | Age | Ectodermal findings | GI Findings | Malignancies | AXIN2 c.1989G>A |
|---|---|---|---|---|---|
| I-2 | Deceased at 97 yrs | Oligodontia, absent eyebrows, sparse hair | Unknown | None | Not tested |
| II-1 | 68 yrs | Oligodontia | At least 5 adenomas No upper GI screening | Breast (44 y) Colon (50 y) Colon (59 y) |
Positive |
| II-2 | 65 yrs | Oligodontia, scant body hair, minimal axillary hair, short eyelashes, very sparse eyebrows | >100 adenomas No Upper GI screening | None | Positive |
| II-3 | 63 yrs | Oligodontia, absent eyebrows, sparse hair | Colon polyps (details unknown) |
None | Positive |
| III-1 | 39 yrs | Normal | Normal colonoscopy | None | Negative |
| III-2 | 38 yrs | Normal | No GI screening | None | Negative |
| III-3 | 35 yrs | Oligodontia, absent eyebrows, sparse hair | Normal colonoscopy Multiple fundic gland polyps | None | Positive |
The proband had two maternal aunts, including one (II-1) with oligodontia, breast cancer diagnosed at age 44, metachronous colon cancers diagnosed at ages 50 and 59, and five adenomatous polyps of the transverse and ascending colon at age 69. The other maternal aunt (II-3) had a reported history of colon polyps, oligodontia, absent eyebrows, and sparse hair. Confirmation of her history was not available. The index patient's sister (III-2) had no dental or other ectodermal findings and had not undergone endoscopic screening. One cousin (III-1) also had no dental or ectodermal findings and had normal colon screening at age 41. The proband's maternal grandmother (I-2) died at age 97 with a reported history of oligodontia, absent eyebrows, and sparse hair, and no known history of colon polyps or cancers.
Methods
Informed consent and blood samples were obtained from the patient, her sister (III-2), mother (II-2), one maternal aunt (II-1), and one maternal cousin (III-1) under the Institutional Review Board approved University of Michigan Cancer Genetics Registry. Bidirectional direct sequencing of known AXIN2 exons and flanking intronic regions was performed on the proband. Bidirectional sequencing of exon 7 and flanking intronic regions was performed in the other study participants. The proband's mother (II-2) also underwent clinical genetic testing of the APC gene including sequence analysis of exons 1-14 and the 5′ end of exon 15, protein truncation testing for mutations in exon 15, and gene dosage analysis (MLPA) to test for the presence of large deletions, duplications, and other genomic rearrangements. The proband's mother (II-2) also under went clinical testing for the p.Tyr165Cys and p.Gly382Asp mutations of the MYH genes by a PCR-based analysis (restriction enzyme digest).
The AXIN2 DNA sequence (NCBI reference sequence NG_012142.1) was cloned from the DLD-1 cell line (ATCC). The sequence was PCR amplified from cDNA using primers specific to AXIN2 and inserted into the pCR2.1-TOPO vector (Invitrogen). The c.1989G>A mutation was introduced by site-directed mutagenesis using the QuickChangeII site-directed mutagenesis kit (Stratagene). In vitro transcription and translation (TNT, Promega) was performed following manufacturer's instructions. Biotinylated proteins were visualized following separation by SDS-PAGE with streptavidin-conjugated HRP. HEK 293 cells were transiently transfected with either a wildtype or c.1989G>A AXIN2 construct using FugeneHD (Roche). Cell lysates were analyzed by immunoblotting, following resolution by SDS-PAGE, with a monoclonal AXIN2 antibody (Cell Signaling, 76G6).
Results
Genetic testing in the proband showed a heterozygous c.1989G>A gene alteration (p.Trp663X) (Fig 2). This is a novel mutation that introduces a stop codon at amino acid 663. The proband's mother (II-2) and maternal aunt (II-1) tested positive and her sister (III-2) and cousin (III-1) tested negative (Table I and Fig 1b). In vitro transcription and translation of the c.1989G>A construct and expression of the construct in HEK 293 cells produced an approximately 80 kDa protein representing a truncated AXIN2 product missing the last three exons (Fig 2b). The proband's mother (II-2) did not have a germline APC mutation identified and was negative for the MYH p.Tyr165Cys and p.Gly382Asp mutations.
Figure 2.
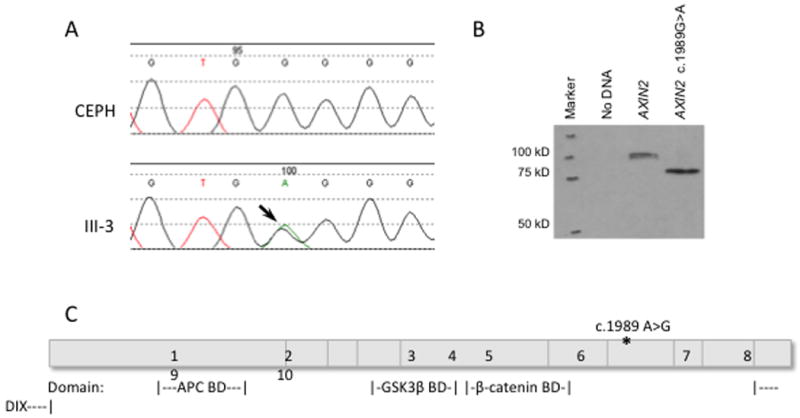
c.1989G>A mutation produces a truncated protein. (A) Sequencing of the proband's DNA (III-3) showed a heterozygous mutation, c.1989G>A, which encodes a premature stop codon. (B) HEK 293 cells were transfected with wildtype AXIN2 or AXIN2 c.1989G>A DNA. Immunoblotting of cell lysates showed that AXIN2 c.1989G>A produces an approximately 80 kDa protein representing a truncated AXIN2 product. (C) Schematic of the AXIN2 protein and important binding domains shows the site of the identified mutation.
Discussion
Our findings provide further evidence of an autosomal dominant multisystem ectodermal and neoplastic phenotype associated with a germline AXIN2 mutation. The novel mutation found in the family reported here predicts p.Tyr663X. This truncated protein is missing the last three exons, including the DIX domain. The DIX (Disheveled and AXIN interacting domain) is required for AXIN2 homodimerization. Dimerization allows AXIN2 to act as a scaffold for the assembly of protein complexes, in particular, the B-catenin destruction complex. Formation of this complex is important for the inhibition and negative feedback of WNT signaling. We predict that loss of the DIX domain will have functional consequences and destroy the inhibitory action of AXIN2 on WNT signaling.
Similar to the findings of Lammi et al., [2004] the oligodontia phenotype in the family reported here is highly penetrant. In addition, members of the family had a mild ectodermal dysplasia phenotype characterized by absent or sparse eyebrows and body hair. Furthermore, three of the four AXIN2 mutation carriers in the family had a colorectal neoplasia, including one with a modest number of adenomas (less than ten) who had two primary colon cancers in her 50's (II-1), one with polyposis reminiscent of FAP (II-2) and one with an unspecified number of colon polyps (II-3). The youngest mutation carrier in the family, the proband (III-3), had normal colonoscopic findings in her mid 30's, but was noted to have multiple fundic gland polyps. Given that upper GI screening has not been performed in other mutation carriers and that the proband's fundic gland polyps could be related to proton-pump inhibitor use, further exploration is needed to determine if fundic gland polyps are related to AXIN2 mutations [Choudhry et al., 2005].
The diagnosis of early onset breast cancer in one member of this family (II-1) is intriguing given that others have hypothesized that genetic variation influencing the expression of APC and AXIN2 influences breast cancer risk [Wang et al., 2008]. Supporting evidence includes the finding that AXIN2 maps to a chromosomal region at band 17q24 that has demonstrated allelic imbalance in breast cancer samples, and an increased rate of breast cancer in mouse models with upregulated WNT signaling [Zhang et al., 2010]. Additionally, AXIN2 is involved in epithelial to mesenchymal transitions in mammary epithelial cells, which is thought to be a critical in the development of invasive and metastatic breast cancer [Yook et al., 2006]. It is unclear if the observed development of early onset breast cancer in one member of this kindred is related to the germline AXIN2 mutation identified. Further studies are needed to substantiate a causal role.
AXIN2 mutations have also been associated with the development of oral clefts, such as cleft lip and cleft palate (CL/CP). A recent analysis of 75 families with CL/CP compared to controls found that there was a significantly higher incidence of cancer in families with CL/CP. In fact, the rates of breast and colon cancer in these families were significantly higher [Menezes et al., 2009]. Additional studies to examine the correlation of AXIN2 mutations with CL/CP associated with an increased risk of breast or colon cancer are needed to determine if a unique AXIN2-related syndrome of oral clefs associated with an increased risk for cancer exists.
This report describes a second family with an AXIN2 mutation cosegregating in an autosomal dominant pattern with oligodontia and early onset cancers. In addition, this study extends the phenotypic spectrum associated with germline heterozygous AXIN2 mutations to include other ectodermal dysplasia features and raises the possibility of an association with increased breast cancer risk. Further studies are needed to determine the role of AXIN2 mutations in inherited cancer predisposition and better characterize the phenotype associated with this newly recognized autosomal dominant AXIN2-associated ectodermal dysplasia and neoplastic syndrome. Similar to other inherited cancer predisposition syndromes, a careful family and personal medical history is critical to developing a complete differential. Specifically, cancer genetics professionals should inquire about the presence of ectodermal phenotypes when assessing patients with familial colorectal neoplasias. The co-occurrence of colorectal neoplasia, including polyposis and colorectal cancer, with oligodontia, other ectodermal dysplasia findings such as sparse hair and eyebrows, or oral clefts should prompt an evaluation for AXIN2 mutations.
Acknowledgments
This work was supported in part by the National Cancer Institute RO1 CA81488 and the University of Michigan's Cancer Center Support Grant 5 P30 CA465920.
References
- Callahan N, Modesto A, Meira R, Seymen F, Patir A, Vieira AR. Axis inhibition protein 2 (AXIN2) polymorphisms and tooth agenesis. Arch Oral Biol. 2009;54:45–49. doi: 10.1016/j.archoralbio.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry U, Boyce HW, Jr, Coppola D. Proton pump inhibitor-associated gastric polyps: a retrospective analysis of their frequency, and endoscopic, histologic, and ultrastructural characteristics. Am J Clin Pathol. 1998;110:615–621. doi: 10.1093/ajcp/110.5.615. [DOI] [PubMed] [Google Scholar]
- Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letra A, Menezes R, Granjeiro JM, Vieira AR. AXIN2 and CDH1 polymorphisms, tooth agenesis, and oral clefts. Birth Defects Res A Clin Mol Teratol. 2009;85:169–173. doi: 10.1002/bdra.20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor NM. Hereditary colorectal cancer: MYH-associated polyposis and other newly identified disorders. Best Pract Res Clin Gastroenterol. 2009;23:75–87. doi: 10.1016/j.bpg.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C, Christensen E, Schmidt SS, Roche PC, Smith DI, Thibodeau SN. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000;26:146–147. doi: 10.1038/79859. [DOI] [PubMed] [Google Scholar]
- Lynch PM. Standards of care in diagnosis and testing for hereditary colon cancer. Fam Cancer. 2008;7:65–72. doi: 10.1007/s10689-007-9159-3. [DOI] [PubMed] [Google Scholar]
- Menezes R, Marazita ML, Goldstein McHenry T, Cooper ME, Bardi K, Brandon C, Letra A, Martin RA, Vieira AR. AXIS inhibition protein 2, orofacial clefts and a family history of cancer. J Am Dent Assoc. 2009;140:80–84. doi: 10.14219/jada.archive.2009.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowska A, Biedziak B, Jagodzinski PP. Axis inhibition protein 2 (AXIN2) polymorphisms may be a risk factor for selective tooth agenesis. J Hum Genet. 2006;51:262–266. doi: 10.1007/s10038-005-0353-6. [DOI] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol. 2005;58:225–236. doi: 10.1136/jcp.2003.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Goode EL, Fredericksen ZS, Vierkant RA, Pankratz VS, Liu-Mares W, Rider DN, Vachon CM, Cerhan JR, Olson JE, Couch FJ. Association of genetic variation in genes implicated in the beta-catenin destruction complex with risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:2101–2108. doi: 10.1158/1055-9965.EPI-08-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li Y, Liu Q, Lu W, Bu G. Wnt signaling activation and mammary gland hyperplasia in MMTV-LRP6 transgenic mice: implication for breast cancer tumorigenesis. Oncogene. 2010;29:539–549. doi: 10.1038/onc.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]