

Abstract
2-(2-Pyridyldithio-3-butenyl) glycosides react with carbohydrate-based thiols in a two step process involving sulfenyl transfer followed by desulfurative 2,3-allylic rearrangement, promoted by either triphenylphosphine or silver nitrate, to give novel saccharide mimetics. In an alternative embodiment of the same chemistry anomeric thiols are coupled with carbohydrates derivatized in the form of 2-(2-pyridyldithio-3-butenyl) ethers. This new method of glycoligation does not require protection of hydroxyl groups and is compatible with the presence of acetamides, azides, trichloroethoxycarbamates, and thioglycosides. Variations on the general theme enable the preparation of mimetics of reducing and non-reducing oligosaccharides as well as of non-glycosidically linked systems.
Introduction
Peptide chemistry has benefitted enormously from the advent of chemical ligation and native chemical techniques enabling the assembly of larger peptides from two or more smaller segments using minimalist protecting group strategies;1 the synthesis of complex oligosaccharides would similarly be considerably advanced by methods for the linking together of smaller pre-assembled oligosaccharides, preferably in an unprotected form. The need for the synthesis of such oligosaccharides, however, is increasing as it becomes apparent that for optimal binding certain carbohydrate-protein interactions require longer oligosaccharides. A case in point is the β-(1→3)-glucan interaction with the lectin dectin 1 for which the minimal binding motif is the decasaccharide.2 While the synthesis of a β-(1→3)-glucodecaose has been reported,2c the assembly of even relatively short β-(1→3)-glucans3 is challenging4 and is frequently complicated by the unanticipated formation of α-glycosidic bonds depending on the substituent pattern in both the donor and the acceptor despite the reliance on neighboring group participation.5 The established adoption of non-chair conformations by some pyranoside residues in the growing chain,3,6 stereochemical matching and mismatching effects,7 and the conformation of the polymer go some way toward explaining the origin of these problems but do not lessen the challenges to be faced in such syntheses. Other oligosaccharides of interest include the β-(1→6)-glucans, which are critical components of yeast cell walls,8 and the mycobacterial polysaccharides 3-O-methyl-α-(1→4)-mannan and 6-O-methyl-α-(1→4)-glucan. These latter methylated glycans enhance the rate of fatty acid biosynthesis by the FA synthetase I from Mycobacterium smegmatis through association with the tetraenonic fatty acid products, for which at least a dodecasaccharide is required for optimal binding.9 Another oligosaccharide of interest is the adhesin poly-β-(1→6)-N-acetyl-D-glucosamine from Escherichia coli10 and Staphylococcus aureus11 implicated in the growth of biofilms of these organisms and for which oligomers up the undecamer have been assembled by stepwise and block organic syntheses.12 Yet another situation is presented by the glycan heparin of which a pentasaccharide motif suffices for association with antithrombin III, while an octadecasaccharide is the optimal chain length for the formation of a tertiary complex with both thrombin and antithrobin III.13
Ideally, large glycans can be constructed by the block assembly approach,14 unfortunately, with certain exceptions,15 such methods have enjoyed only modest success owing to the complexities of the structures involved and especially the need for very high levels of stereocontrol. As such, chemists have turned to alternative methodologies that enable the ligation of preformed oligosaccharides into larger glycan-like structures. Among these methods the copper-catalyzed variant of the Huisgen 1,3-dipolar cycloaddition of alkynes and azides, known as Click chemistry,16 because of the mildness of its reaction conditions and very broad functional group tolerance, has come to dominate the area of glycoconjugate and oligosaccharide mimetic chemistry.17 The rigid nature of the triazole unit introduced in this process, however, leaves room for the development of alternative ligation techniques. For this reason, and because of their functional group tolerance, the so-called thiol-ene and thiol-yne click reactions18 in which thiols undergo additions to alkenes or alkynes, respectively, resulting in thioether linkages have gained increased popularity in recent years albeit with most examples still involving the conjugation of sugars to peptides.19 The thiol-ene and thiol-yne reactions differ from the copper-catalyzed Huisgen 1,3-dipolar cycloaddition click process not only in the nature of the fundamental chemistry and the functional groups required for ligation, but also in terms of the physical nature of the linkage unit introduced. Thus, the planar triazole moiety capable of imitating either a trans- or a cis-amide with its extensive hydrogen bonding capabilities20 is replaced by the less-polar and more conformationally labile thioether unit, which may impact either positively or negatively on the binding of the construct to its biological target. In our laboratory we have been engaged in the development of the highly functional group respective dechalcogenative 2,3-sigmatropic rearrangement of allylic disulfides and selenosulfides as a means of the functionalization of thiols under mild protic conditions.21 This method, which is somewhat related to the thiol-ene and thiol-yne reactions in that it requires derivatization of one of the substrates with a thiol and results in a thioether linkage, has been successfully applied to the formation of glycopeptides mimetics22 and to the functionalization of proteins.23 Here, we describe the extension of this method to the formation of oligosaccharide mimetics and, out of necessity, describe syntheses of carbohydrate-based thiols that may also find application in the thiol-ene and thiol-yne reactions and in other conjugation processes relying on thiolate alkylations.24
Results and Discussion
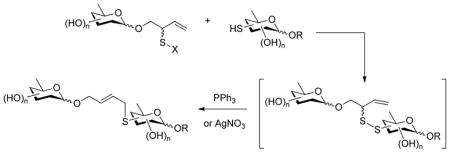
Application of the desulfurative rearrangement of allylic disulfides to the problem of the formation of oligosaccharide mimetics can be envisaged as involving the coupling of an allylic sulfenyl donor 1 affixed to the anomeric position to a carbohydrate based thiol 2 (Scheme 1, strategy a) or, conversely, through the coupling of an anomeric thiol 4 with a sulfenyl donor located elsewhere in the second partner 5 (Scheme 1, strategy b). Ultimately, the two strategies give mimetics 3 and 6 that are close structural analogs differing only in the placement of the oxygen and sulphur atoms in the linker. Additionally, the rearrangement step can be executed with the aid of either triphenylphosphine or silver nitrate (Scheme 1). Both strategies and both reagents have been investigated as described below.
Scheme 1.

Two Global Strategies for the Construction of Oligosaccharide Mimetics
The anomeric sulfenyl donor strategy was targeted first, necessitating the synthesis of anomeric allylic sulfenyl donors and of deoxy sugar thiols. With the direct glycosylation of 2-(2-pyridyldisulfenyl)-3-buten-1-ol giving only moderate yields as previously described,22b attention was focused on approaches involving construction of the allylic sulfenyl donor moiety after formation of the glycosidic bond. As described previously,21e,22b,25 readily the assembled glycoside 7 can be converted into a thiocarbonate 9 and subjected to [3,3]-sigmatropic rearrangement to afford the a secondary allylic thiol derivative 11, that can be converted to the sulfenyl donor 13 by saponification and installation of the disulfide moiety (Scheme 2). While this method requires several steps it has the advantage of using only simple robust chemistry and can generally be conducted in high overall yield.
Scheme 2.

The Allylic Xanthate Rearrangement Pathway to Mono and Disaccharyl Anomeric Sulfenyl Donors
Seeking to lower the temperature of the sigmatropic rearrangement step and at the same time to dissociate saponification of the acetate protecting groups from the unmasking of the allylic thiol, we briefly investigated a variant on this approach (Scheme 3). This modification features the use of a thionocarbamate rather than a thiocarbonate, such that differential saponification can be affected, the use of a palladium-catalyzed [3,3]-sigmatropic rearrangement of the allylic thiocarbonyl system related to ones developed earlier by ourselves25 and in other laboratories,26 and, recalling the work of Kahne on the release of esters under neutral conditions,27 cleavage of the rearranged thiocarbamate by a reductive cyclization process (Scheme 3). Although certainly longer than the approach employing the thermal sigmatropic rearrangement, the chemistry described in Scheme 3 may be advantageous for more complex and thermally sensitive substrates.
Scheme 3.

Alternative Approach to an Anomeric Sulfenyl Donor
A third more efficient approach is exemplified for the case of N-acetylglucosamine, which was converted to the oxazoline 19 by standard means28 and coupled with the acceptor 2022b in the presence of cupric chloride (Scheme 4). The subsequent steps of saponifcation and sulfenyl transfer took place under the standard conditions and led to the formation of the anomeric sulfenyl donor 22.
Scheme 4.

A More Direct Preparation of an Anomeric Sulfenyl Donor
With respect to the alternative strategy based on the use of anomeric thiols, a protocol for the synthesis of O-[2-(2-pyridyldisulfenyl)-3-butenyl] ethers was developed for the 3-position of the glucopyranosyl systems based on the allylic xanthate rearrangement. Thus, alkylation of diisopropylidene-D-glucofuranose with the mesylate derived from cis-2-butene-1,4-diol mono naphthylmethyl ether 23 gave a fully protected derivative 24 that was taken through a standard protocol to achieve rearrangement to the pyranose isomer 25 (Scheme 5). The anomeric acetate formed in this protocol was converted uneventfully to the corresponding methyl β-glycoside 27 by Schmidt’s trichloroacetimidate chemistry29 before the naphthylmethyl group was removed oxidatively, setting the stage for the application of the thiocarbonyl ester chemisty and conversion to an allylic sulfenyl donor (Scheme 5).
Scheme 5.

Synthesis of a 3-O-Sulfenyl Donor
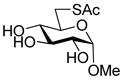
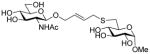
With respect to the synthesis of monosaccharyl thiols these were typically handled as their acetate esters to minimize problems of oxidation, with cleavage under Zemplen-like conditions immediately prior to use (Table 1, column 3). Depending on the synthetic route employed the hydroxyl groups of these thiol precursors were either free or protected in the form of acetate esters. In the latter case, the esters were cleaved concomitantly with the thioesters under Zemplen conditions prior to the ligation reaction. A 6-deoxyglucose-6-thiol 32 was prepared according to a literature route in the form of its derived thioacetate,30 while an analogous N-acetyl glucosamine 6-thioacetate 33 was assembled by selective Mitunsobu reaction31 at the 6-position without the need for protecting groups. A glucose-based 3-deoxy-3-mercapto sugar precursor 37 was prepared in a straightforward manner from peracetyl 3-thioglucopyranoside32 via the trichloroacetimidate 36, while the known phenylthio analog 38 was obtained directly from the anomeric acetate (Scheme 6). The 1-deoxy-mercapto-β-D-glucopyranose perester 39 was a commercial compound.
Table 1.
Ligation of Monosaccharyl Sulfenyl Donors and Thiols.
| Entry | Sulfenyl Donor | Thiol Precursor | Reagent | Rearrangement Time (h) | Product, % yield |
|---|---|---|---|---|---|
| 1 |
 13 |
 32 |
PPh3 | 12 |
 40, 57 |
| 2 |
 21 |
 32 |
PPh3 | 30 |
 41, 80% |
| 3 |
 21 |
 32 |
AgNO3 | 40 |
 41, 57% |
| 4 |
 21 |
 33 |
PPh3 | 30 |
42, 82% |
| 5 |
 21 |
 33 |
AgNO3 | 40 |
42, 82% |
| 6 |
 13 |
 37 |
AgNO3 | 24 |
43, 65% |
| 7 |
 31 |
 39 |
AgNO3 | 16 |
44, 70% |
| 8 |
 31 |
 32 |
AgNO3 | 16 |
 45, 70% |
| 9 |
 13 |
 38 |
AgNO | 24 |
46, 70% |
Scheme 6.

Synthesis of 3-Deoxy-3-mercapto Sugar Precursors
With a series of monomeric sulfenyl donors and thiols in hand, attention was turned to the ligation protocol. This was typically conducted in methanolic solution by admixture of the two components, with monitoring of the sulfenyl transfer step either by TLC or electrospray mass spectrometry, followed by promotion of the desulfurative rearrangement by addition of triphenylphosphine or silver nitrate. As we have discussed previously,21b,21c the use of trialkylphosphines in place of triarylphosphines generally results in the competing nucleophilic cleavage of the disulphide intermediate and related processes. For this reason the use of highly water soluble phosphines such as tris(carboxyethyl)phosphine, which are otherwise expected to increase the rearrangement rate, is precluded. The silver nitrate-based system is a later variant on the original phosphine-mediated rearrangement that was developed to circumvent the reliance on the insoluble phosphine.25 Examples of both methods are presented in the reactions set out in Table 1.
All of the couplings illustrated in Table 1 proceeded in moderate to good yield, whether promoted by triphenylphosphine or by silver nitrate and gave excellent trans-selectivity for the 2-butenyl linker. The examples set out in Table 1 further demonstrate that this ligation protocol is compatible with acetamide groups and thioglycosidic units in addition to hydroxyl groups and O-glycosidic bonds. The reaction sequence can be used to assemble mimics of either classical “head to tail”-linked oligosaccharides (Table 1, entries 1–7, 9) or can be used to provide mimics of non-glycosidically-linked disaccharides (Table 1, entry 8) which are of current interest.33 For the mimics of the classical head to tail oligomers, the ligation can be conducted according to either of the two design principles set out in Scheme 1, but at least for the mimics of the β-(1→3)-glucans the employment of the anomeric thiol (Scheme 1b) results in a shorter reaction time than the use of an anomeric sulfenyl donor (Scheme 1a) as is seen from a comparison of entries 6 and 7 (Table 1). We believe that this difference in reactivity is due to the steric hindrance about the thiol derived from precursor 37 (Table 1, entry 6) which retards both sulfenyl transfer and especially the critical desulfurative 2,3-sigmatropic rearrangement step. This retarding effect of steric bulk around the thiol component on the sigmatropic rearrangement is accord with predictions from computational studies.21e
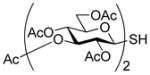
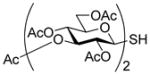
With proof of principle established at the level of monosaccharyl sulfenyl donors and thiols, attention was turned to the synthesis of a set of disaccharyl allylic disulfides and thiols. As exemplified in Schemes 7 and 8, and as is described fully in the experimental section (Schemes 9–14), these units were assembled by the combination of standard coupling methods with variations on the themes set out in Schemes 2–5 for the introduction of the allylic disulfide and thiol moieties. These syntheses were generally uneventful and featured, inter alia, the use of acetonitrile to direct glycosylations to the β-stereochemistry in the 2-azido-2-deoxyglucose series,34 the use of the sulfoxide glycosylation35 method and the activation of glycosyl sulfoxides in the presence of thioglycosides as described originally by the van Boom group,35b,36 the application of the Ley-type bisacetal protecting group for the 3- and 4-positions in the glucosamine series,37 and the employment of both diisopropylidene glucofuranose and a 4,6-O-benzylidene protected glucopyranosyl 3-ol3,38 as acceptors in the synthesis of laminaribiose derivatives. As in the monosaccharide series thiols were, with the exception of the laminaribiosyl thiol 65, generated and handled as thioacetates which were cleaved immediately prior to use typically with concomitant removal of any residual acetate esters. The various disaccharyl sulfenyl donors and thiols synthesized in this manner were coupled in methanol at room temperature leading to the results set out in Table 2.
Scheme 7.

Synthesis of a Disaccharyl Sulfenyl Donor.
Scheme 8.

Synthesis of Two Primary Disaccharyl Thiol Precursors
Scheme 9.

Preparation of 2-Azido-2-deoxyglucose-Based Monosaccharyl Units
Scheme 14.

Preparation of the Laminaribiosyl-3′-thiol Precursor 64
Table 2.
Ligation of Disaccharyl Sulfenyl Donors and Thiols.
| Entry | Sulfenyl Donor | Thiol Precursor | Reagent | Rearrangement Time (h) | Product, % yield |
|---|---|---|---|---|---|
| 1 |
 53 |
 56 |
AgNO3 | 54 |
66, 52% |
| 2 |
 62 |
 61 |
PPh3 | 60 |
67, 54% |
| 3 |
 14 |
64 |
AgNO3 | 24 |
68, 50% |
| 4 |
 63 |
 65 |
AgNO3 | 16 |
69, 55% |
| 5 |
 14 |
 65 |
AgNO3 | 16 |
70, 55% |
The results set out in Table 2 follow the pattern of moderate to good yield and excellent trans-selectivity established for the monosaccharides in Table 1. The results presented in Table 2 also bring the azide and trichloroethoxycarbamates into the range of functional groups tolerated by this ligation protocol and extend the type of linkages mimicked to include the head to head linkage of the non-reducing oligosaccharides (Table 2, entry 5) such as present in sucrose, trehalose, and the antibiotic everninomycin.39 As was the case with the monosaccharyl examples (vide supra) application of the protocol to a thiol located at the 3-position of a glucopyranose ring resulted in longer reaction times, presumably for steric reasons (Table 2, entry 3) than the employment of the alternative mode of operation with an anomeric thiol and an 3-O-allylic sulfenyl donor (Table 2, entry 4).
Overall, the results presented in Tables 1 and 2 demonstrate sulfenyl transfer to give allyl disulfides followed by desulfurative rearrangement provides a potentially useful means of combining short oligosaccharides into larger oligosaccharide mimetics. The ligation process takes place at room temperature in protic solvents and does not require the presence of protecting groups. It may be effectively promoted through the use of silver nitrate or triphenylphosphine, and tolerates the presence of various functional groups such as the azide, thioglycoside, and trichloroethoxycarbamate systems. Mimics of reducing and non-reducing oligosaccharides as well as of non-glycosidically linked systems can be produced by this facile ligation process.
Experimental Section
General Experimental
Unless otherwise stated 1H and 13C NMR spectra were carried out at 500 MHz and 125 MHz, respectively, in deuteriochloroform solution, with chemical shifts downfield from tetramethylsilane. Specific rotations were measured in chloroform solution unless otherwise stated. Unless otherwise stated extracts were dried over sodium sulfate and concentrated at ambient temperature under water aspirator vacuum. Column chromatography was conducted over silica gel unless otherwise stated.
General Procedure 1 for the Preparation of Allylic Thionocarbonates
A solution of phenyl chlorothionocarbonate (2.0 mmol) in CH2Cl2 (2 mL) was added to a solution of the alcohol (1.0 mmol), pyridine (15.0 mmol) and DMAP (0.1 mmol) in CH2Cl2 (10.0 mL) and the resulting dark-yellow solution was stirred at room temperature for 4 h. The reaction mixture was poured into H2O (20 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried, filtered, evaporated, and purified by column chromatography.
General Procedure 2 for the [3,3]-Sigmatropic Rearrangement of Allylic Thionocarbonates
A solution of allylic thionocarbonate (1 mmol) in toluene (10.0 mL) was heated at reflux for 12 h. Evaporation of the solvent and chromatographic purification of the crude products using EtOAc/hexanes as eluent afforded the products.
General Procedure 3 for Saponification of Phenoxycarbonylthioxybutenyl Groups and Installation of the Pyridyl Disulfide Moiety
To a solution of the glycosyl thiocarbonate (0.5 mmol) in MeOH (2.5 mL) was added, dropwise at 0 °C a freshly prepared solution of 1M KOH (2.3 mmol, 1.5 eq per group to be saponified). The resulting mixture was stirred for 0.5 h then neutralized by careful addition of Amberlyst-15 resin and then filtered. The filtrate was added dropwise to a solution of 2,2′-dipyridyl disulfide (0.7 mmol) in MeOH at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 4 h. The solvent was removed in vacuo and the residue was purified by silica gel column chromatography to give the desired products.
General Procedure 4 for the Preparation of Cyclic Bisacetals
Camphorsulfonic acid (0.34 mmol) was added to a stirred solution of the glycoside (3.4 mmol) in MeOH (32 mL) at room temperature. Then, trimethyl orthoformate (18.5 mmol) and butane-2,3-dione (4.0 mmol) were added and the mixture was heated at reflux for 14–16 h. The mixture was then basified to pH = 8 by addition of triethylamine and the solvents were removed under reduced pressure. The residue was purified by silica gel column chromatography to give the desired bisacetals.
General Procedure 5 for the Preparation and Isolation of Tosylates
p-Toluenesulfonyl chloride (4.86 mmol) and tetramethylethylenediamine (4.86 mmol) were added sequentially to a stirred solution of the glycoside (0.24 mmol) in acetonitrile (25 mL) at room temperature. The mixture was stirred for 2–5 h then, poured into ice and extracted twice with CH2Cl2. The combined organic layers were dried, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography.
General Procedure 6 for the Introduction of the 6-Acetylthio Group from the Corresponding Tosylates
To a solution of tosylate (2.12 mmol) in DMF (28 mL) was added potassium thioacetate (4.24 mmol). The reaction mixture was heated to 80 °C (50 °C in case of the disaccharide) for 18–36 h then concentrated in vacuo. The residue was taken up in CH2Cl2 and washed with water. The aqueous phase was extracted twice with CH2Cl2. The combined organic layers were dried, filtered and concentrated and the 6-acetylthio derivatives were isolated by silica gel column chromatography.
General Procedure 7 for the Oxidation of Thioglycosides to Glycosyl Sulfoxides
To a stirred solution of thioglycoside (1.06 mmol) in CH2Cl2 (30 mL) was added dropwise at −80 °C a freshly prepared solution of m-chloroperoxybenzoic acid (1.04 mmol) in CH2Cl2 (3.8 mL). The resulting mixture was stirred at −80 °C for 0.5–1.5 h then quenched by addition of saturated aqueous NaHCO3. The resulting mixture was allowed to warm to room temperature and the aqueous phase was extracted twice with CH2Cl2. The combined organic layers were dried, filtered, concentrated and purified by silica gel column chromatography to yield the desired glycosyl sulfoxides.
General Procedure 8 for Glycosylation under NIS/TfOH Conditions
The glycosyl donor (0.56 mmol) and glycosyl acceptor (0.73 mmol) were stirred in CH2Cl2 (4 mL) at room temperature in presence of activated 4Å powdered molecular sieves for 0.5 h before the reaction mixture was cooled to −35 °C. Then, N-iodosuccinimide (1.12 mmol) and trifluoromethanesulfonic acid (0.34 mmol) were added sequentially and the resulting mixture was stirred for 2 h. The reaction was quenched by addition of 20% Na2S2O3 and allowed to warm to room temperature and the aqueous phase was extracted twice with CH2Cl2. The combined organic layers were dried, filtered, concentrated and purified by silica gel column chromatography to yield the desired product.
General Procedure 9 for Deprotection of 2-(Phenyloxycarbonylthioxy)-3-butenyl Disaccharides with Trifluoroacetic Acid
To a stirred solution of the disaccharide (0.14 mmol) and thioanisole (1.35 mmol) in CH2Cl2 (8 mL) was added an aqueous solution of trifluoroacetic acid, TFA/H2O (19:1) (4mL), at room temperature. The resulting mixture was stirred for 0.5 to 1 h at room temperature, taken up in toluene and then evaporated. The deprotected disaccharide was isolated by silica gel column chromatography.
General Procedure 10 for Acetonide Removal from Derivatives of 1,2;5,6-Diisopropylideneglucofuranose Derivatives and the Subsequent Installation of Acetate Groups
The acetonide protected glucofuranoside (1.0 mmol) was dissolved in 80% acetic acid (10.0 mL) and heated with stirring to 95 °C for 8 h. After cooling the solvents were removed under reduced pressure, and the reaction mixture was azeotroped with toluene (2 × 30 mL). The crude product was dried under vacuo and dissolved in acetic anhydride (10.0 mmol), pyridine (10.0 mmol) and DMAP (0.1 mmol) and stirred at room temperature for 12 h. The solvents were evaporated under vacuum and the crude product was partitioned between EtOAc (30.0 mL) and water. The organic part was washed with brine (50 mL), dried, and evaporated to dryness.
General Procedure 11 for the Preparation of Glycosyl Trichloroacetimidates
To a stirred solution of the anomeric acetate (1.0 mmol) in DMF (10.0 mL), NH2NH2.AcOH (1.5 mmol) was added, after which the reaction mixture was stirred at room temperature for 3–4 h before it was diluted with EtOAc (100 mL) and washed with brine (50 mL). The organic portion was separated and dried to give the hemiacetal, which was dissolved in CH2Cl2 (100 mL) and treated with trichloroacetonitrile (10.0 mmol), followed by DBU (0.1 mmol). The reaction mixture was stirred at room temperature for 12 h before the solvents were evaporated and the crude product was purified by column chromatography using EtOAc/hexanes as eluent.
General Procedure 12 for Glycosylation with Trichloroacetaimidates
The trichloroacetimidate (1.2 mmol), alcohol (1.0 mmol) and activated 4 Å molecular sieves were mixed in CH2Cl2 (10 mL) and stirred at room temperature for 0.5 h before TMSOTf (0.125 mmol) was added. Stirring was continued at room temperature for 12 h before triethylamine (0.2 mmol) was added and the reaction mixture was filtered. The solvents were evaporated and the crude product was purified by column chromatography using EtOAc/hexanes as eluent.
General Procedure 13 for the Deprotection of Naphthylmethyl Groups
The protected pyranoside (1.0 mmol) was dissolved in a mixture of ~9:1 CH2Cl2 and water (10 mL) and DDQ (1.3 mmol) was added. The reaction mixture was stirred at room temperature for 3–4 h until TLC showed the starting material has been consumed. The reaction mixture was diluted with EtOAc (100 mL) and washed with saturated aqueous NaHCO3 (50 mL). The combined organic part was dried and evaporated to dryness. The crude product was purified by column chromatography using ethyl acetate/hexanes as eluent.
General Procedure 14 for Triphenylphosphine-promoted Rearrangement of Allylic Disulfides
A solution of acetylthio sugar (0.20 mmol) in MeOH (1.7 mL) was sparged with nitrogen before a freshly prepared solution of 1M NaOMe in degassed MeOH (0.2 mL) was added. The resulting mixture was stirred for 0.5 h, quickly neutralized by addition of dry Amberlyst IR 120 resin, filtered and then directly added to a stirred solution of the allylic sulfenyl donor (0.24 mmol) in MeOH (2 mL) at room temperature. The resulting mixture was stirred at room temperature until TLC showed complete consumption of the thiol (14 h). Then, triphenylphosphine (0.24 mmol) was added at room temperature and the resulting mixture was stirred for an additional 16 h. The mixture was evaporated in vacuo and subjected to chromatographic purification eluting with dichloromethane/methanol (15:1).
General Procedure 15 for Silver Nitrate Promoted Rearrangement of Allylic Disulfides
A stirred solution of protected thiol in degassed MeOH (1 mmol, 2.0 mL, 0.5 M) was treated with metallic sodium (2–3 equiv) and stirred under a N2 atmosphere for 4–6 h until the saponification was complete (monitored by ESI mass spectrometry and TLC). The reaction mixture was acidified with Amberlyst 15 resin and filtered. The resin was washed with MeOH (3 × 5 mL). The combined washings and the filtrate were concentrated to a final volume of 1–2 mL and transferred to a stirred solution of disulfide (1.0 mmol, 20 mL, 0.05 M) in MeOH. The reaction mixture was stirred at room temperature under an atmosphere of nitrogen until complete disulfide exchange was visible on TLC (usually less than an hour). The reaction mixture was then treated with solid silver nitrate (2.0 equiv) and stirred in the dark for 16 h. After completion of the reaction (monitored by ESI mass spectrometry), NaCl (10 equiv) was added and the reaction mixture was stirred for 3–4 h. The reaction mixture was diluted with MeOH and centrifuged to remove the black precipitate. The solvent was then concentrated to afford the crude product which was purified by column chromatography
4-(Phenyloxythionocarbonyloxy)-2Z-butenyl tetra-O-acetyl-β-D-glucopyranoside (9) was prepared in 90% yield from 7 by the literature procedure.22b It had spectral data identical with the literature values.22b
2-(Phenyloxycarbonylthioxy)-3-butenyl tetra-O-acetyl-β-D-glucopyranoside (11) was prepared in 90% yield from 9 by the literature procedure.22b It had spectral data identical with the literature values.22b
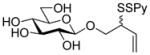
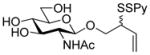
2-(2-Pyridyldithio)-3-butenyl β-D-glucopyranoside (13) was prepared in 76% yield as an approximately 1.1:1 mixture of isomers from 11 by general procedure 3 in the form of a white foam. 1H NMR (CD3OD) δ: 8.35–8.36 (m, 1H), 7.92–7.94 (m, 1H), 7.19–7.36 (m, 1H), 5.79–5.88 (m, 1H), 5.23 (dd, J = 17.0, 9.0 Hz, 1H), 5.13–5.16 (m, 1H), 4.27 (dd, J = 14.5, 8.0 Hz, 1H), 4.10–4.18 (m, 1H), 3.75–3.86 (m, 3H), 3.62–3.66 (m, 1H), 3.17–3.32 (m, 8H); 13C NMRδ: 160.6, 148.8, 138.0, 134.22, 134.17, 121.1, 120.41, 120.38, 118.4, 118.2, 103.4, 103.3, 76,88, 76.86, 73.9, 70.42, 70.38, 70.10, 70.07, 61.6, 61.5, 54.3, 54.2; ESIHRMS calcd for C15H21NO6S2Na [M+Na]+: 398.0708, found: 398.0717.
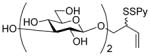
4-Hydroxy-2Z-butenyl hepta-O-acetyl-β-D-laminaribioside (8)
To a stirred solution of peracetyl laminaribiosyl bromide38 (698 mg, 1.0 mmol) in CH2Cl2 (10.0 mL) was added cis-butene-1,4-diol (1.76 g, 20.0 mmol), Ag2CO3 (411 mg, 1.5 mmol), CaSO4 (1.0 g) and a catalytic amount of I2. The reaction mixture was shielded from light and stirred at room temperature for 12 h, then was diluted with CH2Cl2 (50.0 mL) and filtered through a pad of Celite®. The filtrate was washed with saturated NaHCO3 (50.0 mL), and the combined organic portion was dried over Na2SO4 and evaporated to dryness. The crude product was purified by column chromatography over silica gel (EtOAc/hexanes) to give the title compound as colorless liquid in 80% yield. [α]23D −43.5 (c 1.5); 1H NMR δ: 5.84–5.79 (m, 1H), 5.60–5.55 (m, 1H), 5.11 (t, J = 9.5 Hz, 1H), 5.04 (t, J = 9.5 Hz, 1H), 4.98 (t, J = 8.0 Hz, 1H), 4.93 (t, J = 10.0 Hz, 1H), 4.87 (t, J = 8.5 Hz, 1H), 4.57 (d, J = 8.0 Hz, 1H), 4.41 (d, J = 8.0 Hz, 1H), 4.35 (dd, J = 12.5 Hz, J = 4.0 Hz, 1H), 4.31 (dd, J = 12.5 Hz, J = 5.5 Hz, 1H), 4.21 (dd, J = 13.0, 8.0 Hz, 1H), 4.17 (s, 2H), 4.16 (s, 2H), 4.02 (dd, J = 7.5, 2.5 Hz, 1H), 3.86 (t, J = 9.5 Hz, 1H), 3.67–3.64 (m, 2H), 2.12 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H × 2), 1.96 (s, 3H); 13C NMR δ: 171.1, 170.7, 170.6, 169.6, 169.5, 169.4, 169.2, 133.6, 126.9, 101.2, 99.3, 79.1, 73.2, 72.8, 72.1, 71.9, 71.2, 68.6, 68.3, 64.0, 62.5, 61.9, 58.7, 21.1, 20.9, 20.8, 20.7, 20.7, 20.6, 20.5; ESIHRMS: calcd for C30H42O19Na+ [M+Na]+: 729.2218, found: 729.2210.
4-(Phenyloxythionocarbonyloxy)-2Z-butenyl hepta-O-acetyl-β-D-laminaribioside (10)
Following the general procedure 1, and eluting with 75% EtOAc/hexanes the title compound was obtained in 88% yield. [α]23D −11.0 (c 1); 1H NMR δ: 7.43 (t, J = 8.0 Hz, 2H), 7.30 (t, J = 7.5 Hz, 1H), 7.11–7.09 (m, 2H), 5.88–5.84 (m, 1H), 5.82–5.77 (m, 1H), 5.15–5.11 (m, 2H), 5.09–5.06 (m, 2H), 5.04–4.99 (m, 1H), 4.96 (t, J = 10.0 Hz, 1H), 4.89 (t, J = 9.5 Hz, 1H), 4.58 (d, J = 8.5 Hz, 1H), 4.44 (d, J = 8.5 Hz, 1H), 4.38–4.33 (m, 3H), 4.19–4.18 (m, 2H), 4.03 (dd, J = 12.5, 2.0 Hz, 1H), 3.87 (t, J = 9.5 Hz, 1H), 3.68–3.66 (m, 2H), 2.14 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H); 13C NMR δ: 195.1, 170.9, 170.7, 170.6, 169.6, 169.5, 169.4, 169.1, 153.6, 130.9, 129.8, 126.9, 126.2, 122.1, 101.2, 99.6, 79.2, 73.2, 72.7, 72.2, 71.9, 71.3, 69.5, 68.4, 68.3, 64.4, 62.3, 61.9, 21.2, 21.0, 20.8, 20.8, 20.7, 20.7, 20.6; ESIHRMS calcd for C37H46O20SNa+ [M+Na]+:865.2201, found: 865.2190.
2-(Phenyloxycarbonylthioxy)-3-butenyl tetra-O-acetyl-β-D-laminaribioside (12)
Following the general procedure 2, and eluting with 75% EtOAc/hexanes the title compound was obtained as an approximately 1:1 mixture of stereoisomers in 95% yield. 1H NMR δ: 7.39–7.36 (m, 2H), 7.24–7.23 (m, 1H), 7.16–7.13 (m, 2H), 5.94–5.82 (m, 1H), 5.36 (dd, J = 17.0, 5.0 Hz, 1H), 5.22 (dd, J = 11.5, 10.5 Hz, 1H), 5.14–5.10 (m, 1H), 5.07–4.99 (m, 2H), 4.96-4.91 (m, 1H), 4.88 (t, J = 9.0 Hz, 1H), 4.58 (dd, J = 8.0, 6.0 Hz, 1H), 4.43 (t, J = 8.0 Hz, 1H), 4.35 (dd, J = 12.5, 4.5 Hz, 1H), 4.20–4.16 (m, 3H), 4.14–4.09 (m, 1H), 4.07–4.02 (m, 1H), 3.89–3.85 (m, 1H), 3.74 (dd, J = 10.5, 6.5 Hz, 1H), 3.68–3.65 (m, 2H), 2.14 (s, 3H), 2.12 (s, 3H), 2.07 (s, 3H X 2), 2.06 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H X 3), 1.97 (s, 3H), 1.96 (s, 3H); 13C NMR δ: 170.9, 170.7, 170.6, 169.6, 169.5, 169.4, 169.2, 169.0, 168.9, 151.3, 133.8, 133.5, 129.7, 126.5, 121.4, 119.1, 119.0, 101.5, 101.2 (2C), 100.7, 79.0, 78.9, 73.2, 72.7, 72.6, 72.2, 71.9, 71.7, 71.3, 70.3, 68.5, 68.4, 68.3, 62.3, 62.2, 61.9, 48.8, 48.0, 21.2, 21.1, 20.9, 20.8, 20.7, 20.7, 20.6, 20.5; ESIHRMS calcd for C37H46O20SNa+ [M+Na]+: 865.2201, found: 865.2220.
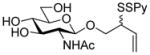
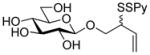
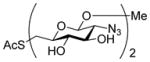
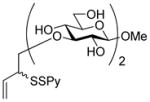
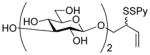
2-(2-Pyridyldithio)-3-butenyl β-D-laminaribioside (14)
Following the general procedure 3, and eluting with MeOH/CH2Cl2 the title compound was obtained as an approximately 1:1 mixture of stereoisomers in 70% yield. 1H NMR (CD3OD) δ: 8.36 (d, J = 7.0 Hz, 1H), 7.95 – 7.92 (m, 1H), 7.79 (t, J = 9.0 Hz, 1H), 7.20 (dd, J = 9.0, 6.0 Hz, 1H), 5.88–5.78 (m, 1H), 5.27–5.20 (m, 1H), 5.16–5.13 (m, 1H), 4.55 (d, J = 10.0 Hz, 1 H), 4.33 (dd, J = 15.0, 10.0 Hz, 1H), 4.16–4.09 (m, 1H), 3.89–3.86 (m, 3H), 3.85–3.80 (m, 1H), 3.78–3.77 (m, 1H), 3.69–3.61 (m, 2H), 3.56–3.51 (m, 1H), 3.42–3.36 (m, 4H), 3.33–3.25 (m, 6H); 13C NMR (CD3OD) δ: 160.6, 148.8, 137.9, 134.2, 134.1, 121.1, 120.4, 118.5, 118.3, 104.1, 102.9, 102.9, 86.7, 77.0, 76.6, 76.5, 74.3, 73.2, 70.4, 70.1, 68.8, 68.7, 61.4, 54.2, 54.1; ESIHRMS calcd for C21H31NO11S2Na [M+Na]+: 560.1236, found: 560.1220.
4-(4-Nitrophenyloxythionocarbonyloxy)-2Z-butenyl tetra-O-acetyl-β-D-glucopyranoside (15)
Alcohol 722b (209.2 mg, 0.5 mmol), pyridine (687.5 μL, 8.5 mmol), and DMAP (12.2 mg, 0.01 mmol) were dissolved in dry CH2Cl2 (4.0 mL), and 4-nitrophenyl chlorothionoformate (119.7 mg, 0.55 mmol) in CH2Cl2 (1.0 mL) was added dropwise. The reaction mixture was stirred at room temperature for 5 h and then diluted with CH2Cl2 (10 mL), washed with 2N HCl and brine. The organic layer was dried and concentrated and purified by column chromatography over silica gel (eluent: EtOAc/Hexanes = 1/2) to give the title compound (209.8 mg, 70%) as a colorless oil. [α]D −12.2° (c, 2.0); 1H NMR (400 Hz) δ: 8.28–8.32 (m, 2H), 7.26–7.30 (m, 2H), 5.82–5.85 (m, 2H), 5.19 (t, J = 9.6 Hz, 1H), 5.06–5.12 (m, 3H), 4.97–5.02 (m, 1H), 4.56 (d, J = 7.2 Hz, 1H), 4.41–4.45 (m, 1H), 4.30–4.34 (m, 1H), 4.24 (dd, J = 12.0, 4.8 Hz, 1H), 4.13 (dd, J = 12.0, 2.4 Hz, 1H), 3.67–3.71 (m, 1H), 2.03 (s, 3H), 2.06 (s, 3H), 1.98 (s, 3H), 2.00 (s, 3H); 13C NMR δ:193.7, 170.8, 170.5, 169.6, 169.5, 157.7, 146.2, 11.3, 125.6, 123.5, 99.8, 73.0, 72.1, 71.4, 70.0, 68.5, 64.9, 62.1, 21.0, 20.9, 20.8; ESIHRMS calcd for C25H29NO14SNa [M+Na]+: 622.1206, found: 622.1212.
4-[N-(2-Azidoethyl)-N-(benzyl)thionocarbamoyloxy]-2Z-butenyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (16)
The thionocarbonate 15 (196 g, 0.33 mmol), N-(2-azidoethyl) benzylamine40 (86.3 mg, 0.49 mmol), and DMAP (80.0 mg, 0.65 mmol) were dissolved in dry CH2Cl2 (2 mL), and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with CH2Cl2 (5 mL), washed with 2N HCl and brine. The organic layer was dried and concentrated and purified chromatographically (eluent: EtOAc/Hexanes 1/2) to give the title compound (152.1 mg, 73%) as a colorless oil. [α]D −2.9° (c, 0.6); 1H NMR (CDCl3) δ: 7.30–7.36 (m, 4H), 7.14 (d, J = 7.0 Hz, 1H), 5.65–5.83 (m, 2H), 5.16–5.20 (m, 3H), 5.04–5.12 (m, 2H), 4.97–5.03 (m, 2H), 4.81 (s, 1H), 4.51–4.59 (m, 1H), 4.32–4.33 (m, 1H), 4.24–4.29 (m, 1H), 4.12–4.17 (m, 1H), 3.90 (t, J = 6.5 Hz, 1H), 3.65–3.72 (m, 2H), 3.57 (t, J = 6.5Hz, 1H), 3.37 (t, J = 6.0 Hz, 1H), 2.04 (s, 3H), 2.08 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H); 13C NMR δ: 189.6, 188.7, 170.9, 170.5, 169.62, 169.57, 136.2, 136.1, 129.9, 129.8, 129.1, 129.0, 128.2, 128.1, 128.0, 127.7, 127.6, 127.3, 126.4, 99.8, 99.7, 73.1, 72.10, 72..06, 71.5, 68.5, 67.5, 67.1, 65.0, 64.9, 62.1, 57.1, 53.7, 52.0, 49.4, 48.8, 46.9, 21.0, 20.9, 20.84, 20.83; ESIHRMS calcd for C28H36N4O11SNa [M+Na]+: 659.1999, found: 659.1979.
2-[N-(2-Azidoethyl)-N-(benzyl)carbamoylthioxy]-3-butenyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (17)
The thionocarbamate 16 (91.4 mg, 0.15 mmol) and PdCl2(CH3CN)2 (3.9 mg, 0.015 mmol) were dissolved in CH2Cl2 (1.5 mL), and the reaction mixture was stirred at 40 °C for 20 h before it was concentrated and purified by column chromatography over silica gel (eluent: EtOAc/Hexanes = 1/2) to give the title compound (90.4 mg, 99%) as a complex mixture of diastereomers and rotamers in the form of colorless oil. 1H NMR δ: 7.22–7.37 (m, 5H), 5.85–5.95 (m, 1H), 5.36 (dd, J = 17.0, 6.0 Hz, 1H), 5.18–5.23 (m, 2H), 5.06–5.12 (m, 1H), 5.01 (t, J = 9.0 Hz, 1H), 4.64 (d, J = 7.5 Hz, 2.5H), 4.57 (d, J = 7.5 Hz, 0.5H), 4.35–4.39 (m, 0.5H), 4.24–4.31 (m, 1.5H), 4.12–4.16 (m, 1.5H), 4.01 (dd, J = 10.0, 8.0 Hz, 0.5 H), 3.84 (dd, J = 11.0, 4.0 Hz, 0.5 H), 3.70–3.76 (m, 1.5H), 3.44 (br. s, 4H), 2.09 (s, 3H), 2.08 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H); 13C NMR δ:170.9, 170.5, 169.6, 169.4, 134.8, 134.6, 129.1, 128.2, 127.5, 118.4, 118.3, 100.7, 100.5, 73.03, 72.95, 72.8, 72.1, 71.42, 71.37, 71.3, 68.7, 68.6, 62.2, 62.1, 52.8, 49.5, 47.9, 46.8, 46.5, 46.4, 21.0, 20.9, 20.85, 20.83; ESIHRMS calcd for C28H36N4O11SNa [M+Na]+: 659.1999, found: 659.1969.
2-[N-(2-Azidoethyl)-N-(benzyl)carbamoylthioxy]-3-butenyl β-D-glucopyranoside (18)
Compound 17 (46.0 mg, 0.07 mmol) was dissolved in dry MeOH (1 mL), and NaOMe (1.5 μL, 25%, 0.007 mmol) was added. The reaction mixture was stirred at room temperature for 30 minutes, checked by TLC, and once the starting material had disappeared, was neutralized with Amberlyst-15 resin, filtered, concentrated, purified by column chromatography over silica gel (eluent: CH2Cl2/MeOH 5/1) to give the title compound (25.5 mg, 75%) as a complex mixture of diastereomers and rotamers in the form of a colorless oil. 1H NMR (CD3OD) δ: 7.09–7.35 (m, 5H), 6.00–6.05 (m, 1H), 5.38 (d, J = 17.0 Hz, 1H), 5.16–5.19 (m, 1H), 4.68 (s, 2H), 4.38–4.43 (m, 1H), 4.34 (t, J = 8.5 Hz, 1H), 4.06–4.10 (m, 1H), 3.80–3.88 (m, 2H), 3.64–3.69 (m, 1H), 3.50 (t, J = 6.0 Hz, 2H), 3.43 (br. s, 4H), 3.34–3.37 (m, 1H), 3.27–3.31 (m, 4H), 3.20 (t, J = 8.5 Hz, 1H); 13C NMR δ: 135.4, 135.2, 128.8, 128.7, 128.0, 127.65, 127.61, 117.0, 116.9, 103.8, 103.3, 76.93, 76.86, 76.8, 73.9, 73.8, 71.9, 71.3, 70.5, 70.4, 61.7, 61.6; ESIHRMS calcd for C20H28N4O7SNa [M+Na]+: 491.1576, found: 491.1587.
2-(2-Pyridyldithio)-3-butenyl β-D-glucopyranoside (13)
Compound 18 (50.2 mg, 0.11 mmol) was dissolved in dry THF (1 mL), PPh3 (28.1 mg, 0.11 mmol) was added, and the reaction mixture was heated to reflux for 30 minutes. Then H2O (0.5 mL) was added and then the reaction mixture was dried over Na2SO4 and filtered. 2,2′-Dipyridyl disulfide (21.4 mg, 0.11 mmol) in CHCl3 (1 mL) was added by dropwise to this solution and the resulting reaction mixture was stirred at room temperature for 30 min before it was concentrated. The residue was purified by column chromatography over silica gel (eluent: CH2Cl2/MeOH 5/1) to give the title compound (23.4 mg, 58%) as a white foam with identical characteristics to the sample described above.
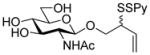
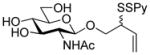
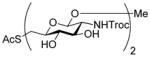
2-(Phenyloxycarbonylthioxy)-3-butenyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranoside (21)
To a stirred solution of the oxazolinone28 19 (2.5 g, 7.6 mmol) and 2-(phenoxycarbonylthioxy)-3-butenol25 20 (425 mg, 1.9 mmol) in CHCl3 (13 mL) was added, at room temperature, copper chloride (1.5 g, 11.4 mmol). The resulting mixture was heated at reflux for 14 h then allowed to cool to room temperature before saturated aqueous NaHCO3 was added. The resulting mixture was filtered through Celite® and washed twice with saturated aqueous NaHCO3. The combined organic layers were dried, filtered and concentrated. The glycoside was isolated chromatographically, eluting with hexanes/CH2Cl2/EtOAc (1:1:2) as a white foam as an approximately 1:1 mixture of diastereomers (473 mg, 45%): 1H NMR (400 MHz) δ: 7.35 (t, J = 8.0 Hz, 2 × 2H), 7.22 (t, J = 7.6 Hz, 2 × 1H), 7.12 (d, J = 6.4 Hz, 2 × 2H), 5.74–5.98 (m, 2 ×2H), 5.36 (dd, J = 3.2, 16.8 Hz, 2 × 1H), 5.28 (dd, J = 10.4, 11.6 Hz, 2 × 1H), 5.21 (dd, J = 4.4, 10.8 Hz, 2 × 1H), 5.04 (t, J = 9.6 Hz, 2 × 1H), 4.74 (t, J = 8.0 Hz, 2 × 1H), 4.02–4.28 (m, 2 × 4H), 3.86 (q, J = 8.8, 17.8 Hz, 2 × 1H), 3.82-3.66 (m, 2 × 2H), 2.04 (s, 2 × 3H), 2.00 (s, 2 × 6H), 1.92 (s, 2 × 3H); 13C NMR (100 MHz) δ: 171.1, 171.0, 170.6, 169.6, 151.2, 134.0, 133.8, 129.8, 126.5, 121.5, 119.1, 119.0, 101.4,100.7, 72.4, 72.1, 71.6, 70.4, 68.8, 62.2, 54.8, 54.6, 48.9, 48.2, 23.6, 23.5, 20,9, 20.8; ESIHRMS calcd for C25H31NO11SNa [M+Na]+: 576.1516, found 576.1516.
2-(2-Pyridyldithio)-3-butenyl 2-acetamido-2-deoxy-β-D-glucopyranoside (22)
Prepared according to the general procedure 3 as a white foam eluting from silica gel with CH2Cl2/MeOH (25:1) as an approximately 1:1 mixture of diastereomers in 60% yield over two steps: 1H NMR (400 MHz, CD3OD) δ: 8.36 (s, 2 × 1H), 7.94-7.78 (m, 2 × 2H), 7.21 (t, J = 5.6 Hz, 2 × 1H), 5.87-5.67 (m, 2× 1H), 5.19 (dd, J = 4.8, 16.8 Hz, 2 × 1H), 5.11 (d, J = 10.4 Hz, 2 × 1H), 4.47-4.39 (m, 2 × 1H), 4.16–4.50 (m, 2 × 1H), 3.86 (dd, J = 5.6, 9.6, Hz, 2 × 1H), 3.82-3.75 (m, 2 × 1H), 3.74-3.63 (m, 2 × 4H), 3.50-3.42 (m, 2 × 1H), 3.36-3.23 (m, 2 × 4H), 1.98 (s, 2 × 3H); 13C NMR (100 MHz, CD3OD) δ: 172.5, 160.5, 148.9, 148.8, 138.1, 138.0, 134.2, 133,8, 121.1, 121,0, 120.2, 121.1, 118.4, 118.3, 116.9, 102.1, 101.7, 76.9, 76.7, 74.8, 74.7, 74.6, 71.4, 70.9, 69.9, 69.8, 61.6, 61.5, 56.1, 56.0, 54.4, 54.1, 22.1, 22.0; ESIHRMS calcd for C17H24N2O6S2Na [M+Na]+: 439.0974, found 439.0978.
4-(2-Naphthylmethyloxy)-2Z-butene-1-ol (23)
A stirred solution of cis-1,4-but-2-ene-diol (2.39 g, 27.1 mmol) in THF (10 mL) was treated under an atmosphere of N2 with sodium hydride (0.38 g, 9.5 mmol) at 0 °C and stirred at room temperature for 1 h. 2-(Bromomethyl)naphthalene (2.0 g, 9.0 mmol) was added and stirring was continued at room temperature for 2 h before the reaction mixture was heated to reflux for 6 h, then cooled to room temperature, diluted with saturated aqueous NH4Cl (30 mL) and ethyl acetate (20 mL). The organic portion was separated, dried, and evaporated to dryness. The crude product was purified over silica gel using EtOAc/hexanes as eluent to give the title compound 23 as thick oil (1.76 g, 85%). 1H NMR δ: 7.87 – 7.85 (m, 3H), 7.80 (s, 1H), 7.51–7.48 (m, 3H), 5.83 (dt, J = 11.0, 6.0 Hz, 1H), 5.78 (dt, J = 11.0, 6.0 Hz, 1H), 4.69 (s, 2H), 4.17 (d, J = 6.0 Hz, 2H), 4.13 (d, J = 6.5 Hz, 2H), 2.39 (br s, 1H); 13C NMR δ: 135.6, 133.5, 133.3, 132.8, 128.5, 128.3, 128.1, 127.9, 126.9, 126.4, 126.2, 126.1, 72.8, 65.9, 58.8; ESIHRMS calcd for C15H16O2Na [M+Na]+: 251.1048, found: 251.1055.
1,2:5,6-Di-O-isopropylidene-3-O-4-(2-naphthylmethyloxy)-2Z-butenyl-α-D-glucofuranose (24)
To a stirred solution of 4-(2-naphthylmethyloxy)-2Z-butene-1-ol (23) (1.76 g, 7.7 mmol) in CH2Cl2 (20 mL) under an atmosphere of N2 was added Et3N (1.60 mL, 11.6 mmol) followed by DMAP (94 mg, 0.77 mmol) at 0 °C. A solution of methanesulfonyl chloride (0.75 mL, 9.64 mmol) in CH2Cl2 (2 mL) then was added dropwise and the reaction mixture was stirred for 2 h and then diluted with saturated NaCl solution (20 mL). The organic portion was separated and the aqueous part was again washed with CH2Cl2 (15 mL). The combined organic part was dried and evaporated to dryness. The crude mesylate (2.36 g, ~ 100%) so obtained in DMF (50 mL)was added at 0 °C to a solution of diacetone-D-glucose (2.0 g, 7.7 mmol) and sodium hydride (338 mg, 8.5 mmol) in DMF (10.0 mL) that had been stirred for 1 h. The reaction mixture was heated to 60 °C and stirred for 12 h before it was cooled and diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and washed with brine (100 mL), dried and evaporated to dryness. The crude product was purified by column chromatography using 60% EtOAc/hexanes as eluent to give the title compound as thick gum (2.65 g, 75%). [α]23D −2.0 (c 0.85); 1H NMR δ: 7.86–7.84 (m, 3H), 7.80 (s, 1H), 7.51–7.47 (m, 3H), 5.87 (d, J = 4.0 Hz, 1H), 5.85–5.83 (m, 1H), 5.79–5.74 (m, 1H), 4.69 (s, 2H), 4.52 (d, J = 4.0 Hz, 1H), 4.32–4.28 (m, 1H), 4.26–4.22 (dd, J = 13.0, 6.5 Hz, 1H), 4.19 (d, J = 6.0 Hz, 1H), 4.17–4.15 (m, 2H), 4.13–4.11(m, 1H), 4.09–4.07 (m, 1H), 4.03–3.99 (m, 1H), 3.92 (d, J = 3.0 Hz, 1H) 1.51 (s, 3H), 1.42 (s, 3H), 1.34 (s, 3H), 1.30 (s, 3H); 13C NMR δ: 135.8, 133.5, 133.3, 130.0, 129.3, 128.5, 128.1, 127.9, 126.7, 126.4, 126.2, 125.9, 112.0, 109.2, 105.5, 83.0, 81.8, 81.4, 72.7, 72.6, 67.6, 66.5, 66.0, 27.1, 27.0, 26.5, 25.6; ESIHRMS calcd for C27H34O7Na [M+Na]+: 493.2202, found: 493.2216.
1,2,4,6-Tetra-O-acetyl-3-O-[4-(2-naphthylmethyloxy)-2Z-butenyl]-D-α,β-glucopyranose (25)
Following the general procedure 10, and eluting with 50% EtOAc/hexanes the title compound was obtained as an approximately 1.4:1 mixture of stereoisomers in 89% yield. 1H NMR δ: 7.84-7.83 (m, 3H), 7.78 (s, 1H) 7.50–7.46 (m, 3H), 6.29 (d, J = 3.5 Hz, 1H), 5.81–5.77 (m, 1H), 5.67–5.58 (m, 2H), 5.09–5.03 (m, 2H), 4.99–4.97 (m, 1H), 4.67 (s, 2H, major), 4.66 (s, 2H, minor), 4.25–4.13 (m,3H), 4.12–4.04 (m, 3H), 4.00–3.97 (m, 1H), 3.81 (t, J = 9.5 Hz, 1H), 3.64–3.62 (m, 1H), 3.58–3.54 (m, 1H), 2.12–1.97 (s, 12H major + 12H minor); 13C NMR δ: 170.9, 169.7, 169.4, 169.2, 168.9, 135.7, 133.5, 133.2, 129.6, 129.4, 129.3, 129.2, 128.5, 128.5, 128.1, 128.0, 127.9, 126.7, 126.7, 126.4, 126.2, 125.9, 125.9, 92.1, 89.6, 79.7, 76.6, 73.2, 72.7, 71.6, 71.5, 70.4, 69.3, 69.1, 68.4, 67.8, 65.9, 65.8, 62.0, 61.9, 21.0, 20.9, 20.8, 20.7; ESIHRMS: calcd for C29H34O11Na [M+Na]+: 581.1999, found: 581.1998.
2,4,6-Tri-O-acetyl-3-O-[4-(2-naphthylmethyloxy)-2Z-butenyl]-α-D-glucopyranosyl trichloroacetimidate (26)
Following the general procedure 11, and eluting with 40% EtOAc/hexanes the title compound was obtained in 76% yield. [α]23D 63.8 (c 1); 1H NMR δ: 8.67 (s, 1H), 7.85–7.82 (m, 3H), 7.78 (s, 1H), 7.50–7.46 (m, 3H), 6.5 (d, J = 3.5 Hz, 1H), 5.83–5.78 (m, 1H), 5.68–5.64 (m, 1H), 5.12 (t, J = 10.0 Hz, 1H), 5.01 (dd, J = 10.0, 4.0 Hz, 1H), 4.68 (d, J = 4.5 Hz, 2H), 4.27–4.18 (m, 3H), 4.13–4.07 (m, 4H), 3.92 (t, J = 10.0 Hz, 1H), 2.07 (s, 3H), 2.02 (s, 3H), 1.94 (s, 3H); 13C NMR δ: 170.9, 169.9, 169.5, 160.8, 135.7, 133.5, 133.2, 129.6, 129.3, 128.5, 128.0, 127.9, 126.7, 126.4, 126.2, 125.9, 93.5, 91.1, 76.5, 72.7, 72.1, 70.7, 69.1, 68.5, 65.8, 61.9, 20.9, 20.9, 20.7; ESIHRMS calcd for C29H32Cl3NO10Na [M+Na]+: 682.0989, found: 682.0999.
Methyl 2,4,6-tri-O-acetyl-3-O-[4-(2-napthylmethyloxy)-2Z-butenyl]-β-D-glucopyranoside (27)
Following the general procedure 12, and eluting with 60% EtOAc/hexanes the title compound was obtained in 75% yield. [α]23D −14.5 (c 1); 1H NMR δ 7.85-7.83 (m, 3H), 7.79 (s, 1H), 7.49–7.46 (m, 3H), 5.81–5.76 (m, 1H), 5.64–5.58 (m, 1H) 5.03 (t, J = 10.0 Hz, 1H), 4.94 (dd, J = 9.5 Hz, J = 8.0 Hz, 1H), 4.68 (s, 3H), 4.27 (d, J = 8.0 Hz, 1H), 4.20 (dd, J = 12.0, 5.0 Hz, 1H), 4.14 (d, J = 6.0 Hz, 1H), 4.11 (d, J = 3.0 Hz, 1H), 4.09-4.08 (m, 2H), 3.55–3.50 (m, 2H), 3.46 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H), 2.00 (s, 3H); 13C NMR δ: 171.0, 169.5, 169.4, 135.7, 133.5, 133.2, 129.4, 128.4, 128.1, 127.9, 126.7, 126.4, 126.2, 125.9, 101.9, 79.8, 72.7 (2C), 72.5, 72.2, 69.7, 67.3, 65.8, 62.5, 56.9, 21.1, 20.9 (2C); ESIHRMS calc. for C28H34O10Na [M+Na]+: 553.2050, found: 553.2064.
Methyl 2,4,6-tri-O-acetyl-3-O-[4-hydroxy-2Z-butenyl]-β-D-glucopyranoside (28)
Following the general procedure 13, and eluting with 40% EtOAc/hexanes the title compound was obtained in 88% yield. [α]23D −25.4 (c 1); 1H NMR δ: 5.77–5.72 (m, 1H), 5.55–5.51 (m, 1H), 5.06 (t, J = 9.50 Hz, 1H), 4.98–4.95 (m, 1H), 4.34 (d, J = 8.0 Hz, 1H), 4.23 (dd, J = 12.0, 5.0 Hz, 1H), 4.15–4.11 (m, 5H), 3.62–3.58 (m, 2H), 3.48 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H); 13C NMR δ: 171.1, 169.8 (2C), 132.4, 127.9, 101.9, 79.8, 72.2 (2C), 69.3, 66.3, 62.5, 58.6, 57.1, 21.2, 21.1, 21.0; ESIHRMS calcd for C17H26O10Na [M+Na]+: 413.1424, found: 413.1425.
Methyl 2,4,6-tri-O-acetyl-3-O-[4-(phenyloxythionocarbonyloxy)-2Z-butenyl]-β-D-glucopyranoside (29)
Following the general procedure 1, and eluting with 45% EtOAc/hexanes the title compound was obtained in 90% yield. [α]23D −11.5 (c 1); 1H NMR δ: 7.45–7.42 (m, 2H), 7.32–7.29 (m, 1H), 7.12–7.10 (m, 2H), 5.84–5.79 (m, 1H), 5.76–5.72 (m, 1H), 5.10-5.06 (m, 3H), 5.01–4.97 (m, 1H), 4.35 (d, J = 7.50 Hz, 1H), 4.26–4.24 (m, 2H), 4.22 (d, J = 5.0 Hz, 1H), 4.13 (dd, J = 12.5, 2.5 Hz, 1H), 3.65–3.59 (m, 2H), 3.48 (s. 3H), 2.13 (s, 3H), 2.11 (s, 3H), 2.08 (s, 3H); 13C NMR δ: 195.1, 171.0, 169.5, 169.5, 153.7, 131.9, 129.8, 126.9, 125.1, 122.1, 101.9, 80.0, 72.4, 72.2, 69.6, 69.5 (2C), 67.2 (2C), 62.4, 57.0, 21.2, 21.1, 21.0; ESIHRMS calcd for C24H30O11SNa [M+Na]+: 549.1407, found: 549.1398.
Methyl 2,4,6-tri-O-acetyl-3-O-[(2-phenyloxycarbonylthioxy)-3-butenyl]-β-D-glucopyranoside (30)
Following the general procedure 2, and eluting with 45% EtOAc/hexanes the title compound was obtained in 90% yield as an approximately 1.25:1 mixture of stereoisomers. 1H NMR δ: 7.39–7.36 (m, 2H), 7.26–7.23 (m, 1H), 7.15–7.14 (m, 2H), 5.93–5.85 (m, 1H), 5.33 (d, J = 17.0 Hz, 1H), 5.19 (d, J = 10.5 Hz, 1H), 5.10 (t, J = 10.0 Hz, 1H), 5.03–4.99 (m, 1H), 4.33 (dd, J = 8.0, 3.0 Hz, 1H), 4.25–4.21 (m, 1H), 4.15–4.11 (m, 1H), 4.09–4.05 (m, 1H), 3.86–3.80 (m, 2H), 3.62–3.58 (m, 2H), 3.48 (s, 3H, major + minor), 2.12 (s, 3H), 2.10 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H X 3 minor); 13C NMR δ: 171.0, 169.6, 169.5, 169.4 (2 C), 151.3, 134.1, 134.0, 129.7, 126.5, 121.5, 118.7 (2 C), 102.0, 81.0, 73.9, 73.8, 72.2 (2 C), 69.6, 69.5, 62.4, 57.0, 48.9 (2 C), 21.3, 21.2 (2 C), 21.1, 21.0; ESIHRMS calcd for C24H30O11SNa [M+Na]+: 549.1407, found: 549.1385.
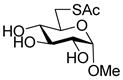
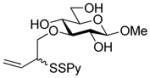
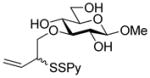
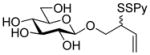
Methyl 3-O-[(2-pyridyldithio)-3-butenyl]-β-D-glucopyranoside (31)
Following the general procedure 3, and eluting with 5% MeOH/CH2Cl2 the title compound was obtained in 76% yield as an approximately 1.5:1 mixture of stereoisomers. 1H NMR (CD3OD) δ: 8.37–8.36 (m, 1H), 7.93–7.91 (m, 1H), 7.81–7.78 (m, 1H), 7.22–7.19 (m, 1H), 5.87–5.79 (m, 1H), 5.24–5. 21 (m, 1H), 5.12 (d, J = 10.0 Hz, 1H), 4.17 (dd, J = 7.5, 1.5 Hz, 1H), 4.13–4.09 (m, 1H), 4.07–4.06 (m, 1H), 4.04–4.01 (m, 1H), 3.88-3.85 (m, 1H), 3.81–3.77 (m, 1H), 3.69–3.65 (m, 1H), 3.53 (s, 3H + 3H, two isomers), 3.32 (m, 5H), 3.28–3.19 (m, 3H); 13C NMR (CD3OD) δ: 160.7, 148.8, 137.8, 134.6, 134.5, 121.0, 120.3, 118.1, 104.2, 85.9, 76.7, 73.8, 73.7 (2C), 70.1, 70.0, 61.4, 56.2, 54.9; ESIHRMS calcd for C15H21NO6S2Na [M+Na]+: 398.0708, found: 398.0700.
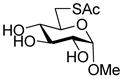
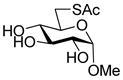
Methyl 6-acetylthio-α-D-glucopyranoside (32) was prepared by a literature method30 and had data consistent with the literature.30
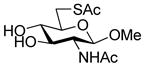
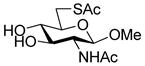
Methyl 2-acetamido-6-acetylthio-2-deoxy-β-D-glucopyranoside (33)
Diisopropyl azodicarboxylate (0.25 mL, 1.22 mmol) was added dropwise at 0 °C to a stirred solution of triphenylphosphine (326 mg, 1.24 mmol) in DMF (2 mL). The mixture was stirred at 0 °C for 1 h and gave a light yellow precipitate. A solution of methyl 2-acetamido-2-deoxy-β-D-glucopyranoside41 (240 mg, 1.02 mmol) and thioacetic acid (0.09 mL, 1.22 mmol) in DMF (1.7 mL) then was added dropwise at 0 °C and the resulting mixture was stirred for 20 h at room temperature, resulting in a clear yellow solution. The mixture was concentrated in vacuo and the residue was purified by silica gel chromatography (eluting with CH2Cl2/MeOH 15:1) to give the title compound as a white solid (176 mg, 59%) mp 195 °C; [α]RTD +36.8 (c 1, MeOH); 1H NMR (300 MHz, CD3OD) δ: 4.26 (d, J = 8.7 Hz, 1H), 3.66-3.55 (m, 2H), 3.48-3.38 (m, 4H), 3.38-3.26 (m, 4H), 3.20 (t, J = 9 Hz, 1H), 2.80 (dd, J = 8.1, 8.7 Hz, 1H), 2.32 (s, 3H), 1.94 (s, 3H); 13C NMR (75 MHz, CD3OD) δ: 195.8, 172.6, 102.2, 75.0, 74.6, 73.9, 56.0, 55.7, 30.9, 29.2, 21.8; ESIHRMS calcd for C11H19NO6S Na [M + Na]+: 316.0831, found: 316.0849.
2,4,6-Tri-O-acetyl-3-acetylthio-α,β-D-glucopyranose (35)
A stirred solution of 3432 (406 mg, 1.0 mmol) in a mixture of EtOAc (20 mL) and CH2Cl2 (10.0 mL) was treated with TiBr4 (908 mg, 2.5 mmol) and stirred at room temperature for 96 h before it was and diluted with CH2Cl2 (30.0 mL) and filtered through a pad of Celite®. The filtrate was washed with saturated aqueous NaHCO3 (50.0 mL) and the organic portion was dried and evaporated to dryness. The so-obtained crude bromide was dissolved in an acetone/water mixture (20.0 mL, 2:1), treated with Ag2CO3 (411 mg, 1.5 mmol), and stirred at room temperature for 12 h. The reaction mixture was diluted with EtOAc (30.0 mL) and filtered through a short Celite® pad and washed with saturated aqueous NaHCO3 (50.0 mL). The organic portion was dried, concentrated, and purified by column chromatography using 50% EtOAc/hexanes to give the title product in 86% yield as an approximately 2:1 mixture of stereoisomers in the form of an oil. 1H NMR δ: 5.39 (t, J = 4.0 Hz, 1H, major), 5.09–5.07 (m, 1H, major), 5.06–5.04 (m, 1H, minor), 4.95–4.92 (m, 1H, major), 4.84 (dd, J = 11.5, 8.0 Hz, 1H, minor), 4.73 (dd, J = 8.5, 8.0 Hz, 1H, minor), 4.28–4.25 (m, 1H, major), 4.23–4.15 (m, 2H, major), 4.12–4.11 (m, 1H, minor), 3.97–3.95 (m, 1H, minor), 3.85–3.83 (m, 1H, minor), 3.81–3.80 (m, 1H, major), 3.79–3.76 (m, 1H, minor), 2.32 (s, 3H, minor), 2.32 (s, 3H, major), 2.07 (s, 3H X 2), 2.06 (s, 3H X 2), 2.02 (s, 3H, major), 2.01 (s, 3H, minor); 13C NMR δ: 193.7 (2C), 171.2, 171.1 (minor), 170.8 (minor), 170.3, 169.7, 169.6 (minor), 97.1 (minor), 89.9, 75.0, 72.4 (minor), 70.3, 68.7 (minor), 67.6, 62.6 (minor), 47.7 (minor), 44.4, 30.9, 30.9 (minor), 21.1 (2C), 20.9, 20.8 (2C), 20.7 (2C), 20.7; ESIHRMS calcd for C14H20O9SNa [M+Na]+: 387.0726, found: 387.0724.
2,4,6-Tri-O-acetyl-3-acetylthio-α-D-glucopyranosyl trichloroacetimidate (36)
Following the general procedure 11, and eluting with 45% EtOAc/hexanes the title compound was obtained in 80% yield. [α]23D 52.0° (c 1); 1H NMR δ: 8.66 (s, 1H), 6.48 (d, J = 3.0 Hz, 1H), 5.21–5.13 (m, 2H), 4.23–4.16 (m, 3H), 4.09–4.06 (m, 1H), 2.31 (s, 3H), 2.04 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H); 13C NMR δ: 193.2, 170.7, 169.8, 169.5, 161.0, 92.8, 91.0, 71.5, 68.9, 66.6, 61.9, 44.7, 30.9, 20.9, 20.7, 20.6; ESIHRMS calcd for C16H20Cl3NO9SNa [M+Na]+: 529.9822, found: 529.9799.
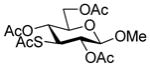
Methyl 2,4,6-tri-O-acetyl-3-acetylthio-β-D-glucopyranoside (37)
Following the general procedure 12, and eluting with 60% EtOAc/hexanes the title compound was obtained in 76% yield. [α]23D −8.7° (c 1); 1H NMR δ: 5.06 (dd, J = 11.0, 9.5 Hz, 1H), 4.95 (dd, J = 11.0, 7.5 Hz, 1H), 4.45 (d, J = 7.5 Hz, 1H), 4.26 (dd, J = 12.0, 4.5 Hz, 1H), 4.12 (dd, J = 12.0, 3.0 Hz, 1H), 3.85 (t, J = 11.0 Hz, 1H), 3.76–3.73 (m, 1H), 3.50 (s, 3H), 2.33 (s, 3H), 2.09 (s, 3H), 2.05 (s, 3H), 2.02 (s, 3H); 13C NMR δ: 193.7, 170.9, 169.5 (2C), 103.2, 74.7, 70.3, 67.8, 62.5, 57.1, 47.9, 30.8, 20.9, 20.9, 20.8; ESIHRMS calcd for C15H22O9SNa [M+Na]+: 401.0882, found: 401.0878.
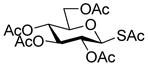
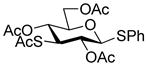
Phenyl 2,4,6-tri-O-acetyl-3-acetylthio-1-thio-β-D-glucopyranoside (38) was obtained by the literature procedure from 101 in 60% yield and had physical characteristics consistent with the literature.32
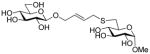
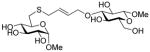
Methyl 6-[4-(β-D-glucopyranosyloxy)-2E-butenyl]thio-α-D-glucopyranoside (40)
The general coupling procedure 14 gave the title compound (17.4 mg, 57%) as a white foam after purification by column chromatography over silica gel (eluent: CH2Cl2/MeOH 1/1). (When using phosphate buffer solution as solvent, the yield is 93%). [α]D +31.3° (c 1.3, CH3OH); 1H NMR (CD3OD) δ: 5.68–5.79 (m, 2H), 4.63 (d, J = 4.0 Hz, 1H), 4.35 (dd, J = 12.0, 5.0 Hz, 1H), 4.31 (d, J = 8.0 Hz, 1H), 4.13–4.18 (m, 1H), 3.87 (d, J = 12.0 Hz, 1H), 3.60–3.68 (m, 2H), 3.57 (t, J = 9.5 Hz, 1H), 3.42 (s, 3H), 3.39 (dd, J = 10.0, 4.0 Hz, 1H), 3.33–3.36 (m, 2H), 3.30–3.31 (m, 6H), 3.17–3.28 (m, 6H), 2.91 (dd, J = 14.0, 2.0 Hz, 1H), 2.56 (dd, J = 14.0, 9.0 Hz, 1H); 13C NMR δ:130.1, 128.9, 101.9, 99.9, 76.9, 76.8, 73.9, 73.8, 73.5, 72.5, 72.4, 70.5, 68.8, 61.6, 54.3, 33.9, 32.1; ESIHRMS calcd for C17H30O11SNa [M+Na]+: 465.1407, found: 465.1407.
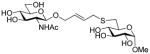
Methyl 6-[4-(2-acetamido-2-deoxy-β-D-glucopyranosyloxy)-2E-butenyl]thio-2-deoxy-β-D-glucopyranoside (41)
Prepared according to the general protocol 14 in 80% yield or in 57 % yield according to protocol 15 as a white foam eluting from silica gel in CH2Cl2/MeOH (20:1): [α]RTD +74.0° (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ: 5.76-5.58 (m, 2H), 4.63 (d, J = 4.4 Hz, 1H), 4.44 (d, J = 8.0 Hz, 1H), 4.31 (dd, J = 4, 4.8 Hz, 1H), 4.09 (dd, J = 5.6, 6.0 Hz, 1H), 3.88 (d, J = 12.0 Hz, 1H), 3.74-3.54 (m, 4H), 3.47-3.16 (m, 11H), 2.90 (dd, J = 2.4, 14.0 Hz, 1H), 2.56 (dd, J = 8.8, 13.6 Hz, 1H), 1.98 (s, 3H); 13C NMR (100 MHz, CD3OD) δ: 172.6, 129.6, 128.7, 100.5, 99.9, 76.8, 74.9, 73.8, 73.6, 72.5, 72.4, 70.9, 68.5, 61.6, 56.2, 54.3, 34.0, 32.2, 21.9; ESIHRMS calcd for C19H33NO11SNa [M + Na]+: 506.1672, found: 506.1655.
Methyl 6-[4-(2-acetamido-2-deoxy-β-D-glucopyranosyloxy)-2E-butenyl]thio-2-acetamido-2-deoxy-β-D-glucopyranoside (42)
Prepared according to the general protocol 14 in 82% yield or in 68 % yield according to protocol 15 as a white foam eluting from silica gel with CH2Cl2/MeOH (20:1): [α]RTD +41.0° (c 0.5, MeOH); 1H NMR (400 MHz, CD3OD) δ: 5.80-.60 (m, 2H), 4.40 (d, J = 8.0 Hz, 1H), 4.32 (d, J = 8.8 Hz, 1H), 4.27 (d, J = 8.8 Hz, 1H), 4.09 (dd, J = 5.2, 12.8 Hz, 1H), 3.88 (d, J = 12.0 Hz, 1H), 3.72-3.61 (m, 4H), 3.48-3.20 (m, 12H), 2.94 (d, J = 13.6 Hz, 1H), 2.62 (dd, J = 8.0, 14.4 Hz, 1H), 1.98 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz, CD3OD) δ: 172.6, 129.6, 128.8, 102.4, 102.3, 100.6, 77.0, 76.8, 74.9, 74.8, 73.6, 70.9, 68.7, 61.6, 56.1, 56.0, 55.9, 34.0, 32.1, 21.9, 21.8; ESIHRMS calcd for C21H36N2O11SNa [M+Na]+: 547.1938, found: 547.1942.
Methyl 3-deoxy-3-[4-(β-D-glucopyranosyloxy)but-2E-enylthio]-β-D-glucopyranoside (43)
Following the general procedure 15, and eluting with 12% MeOH/CH2Cl2 the title compound was obtained in 65% yield. [α]23D −2.0° (c 0.85, MeOH); 1H NMR (CD3OD) δ: 5.85 (dt, J = 15.0, 7.5 Hz, 1H), 5.75 (dt, J = 15.5, 6.5 Hz, 1H), 4.36 (d, J = 8.0 Hz, 1H), 4.32 (dd, J = 12.5, 5.0 Hz, 1H), 4.20 (d, J = 8.0 Hz, 1H), 4.17 (dd, J = 12.5, 6.5 Hz, 1H), 3.89–3.86 (m, 2H), 3.69 (dd, J = 12.0, 5.0 Hz, 1H), 3.66 (dd, J = 12.5, 5.5 Hz, 1H), 3.54 (s, 3H), 3.46–3.40 (m, 1H), 3.28–3.27 (m, 6H), 3.25–3.17 (m, 2H), 2.54 (t, J = 10.0 Hz, 1H); 13C NMR (CD3OD) δ: 130.8, 128.7, 105.3, 101.5, 79.2, 76.8, 76.6, 73.9, 73.1, 70.5, 68.7, 68.6, 61.7, 61.6, 56.0, 54.6, 33.2; ESIHRMS calcd for C17H30O11SNa [M+Na]+: 465.1401, found: 465.1407.
Methyl 3-O-[4-(1-thio-β-D-glucopyranosyl)-2E-butenyl]-β-D-glucopyranoside (44)
Following the general procedure 15, and eluting with 12% MeOH/CH2Cl2 the title compound was obtained in 70% yield. [α]23D −45.1° (c 1, MeOH); 1H NMR (CD3OD) δ: 5.79–5.77 (m, 2H), 4.38 (d, J = 9.5 Hz, 1H), 4.35–4.34 (m, 2H), 4.19–4.18 (m, 1H), 3.89–3.86 (m, 2H), 3.70–3.63 (m, 2H), 3.53 (s, 3H), 3.50–3.46 (m, 1H), 3.32–3.31 (m, 3H), 3.29–3.27 (m, 2H), 3.26–3.22 (m, 4H); 13C NMR (CD3OD) δ: 130.3, 128.9, 104.2, 84.3, 83.9, 80.5, 78.5, 76.6, 73.9, 73.2, 72.7, 70.5, 70.1, 61.8, 61.5, 56.1, 30.9; ESIHRMS calcd for C17H30O11SNa [M+Na]+: 465.1401, found: 465.1407.
Methyl 3-O-[4-(methyl α-D-glucopyranosid-6-thio)but-2E-enyl]-β-D-glucopyranoside (45)
Following the general procedure 15, and eluting with 10% MeOH/CH2Cl2 the title compound was obtained in 70% yield. [α]23D −26.0° (c 0.75, MeOH); 1H NMR (CD3OD) δ: 5.78–5.69 (m, 2H), 4.65 (d, J = 3.5 Hz, 1H), 4.36–4.35 (m, 1H), 4.19–4.18 (m, 1H), 3.86 (dd, J = 12.0, 2.5 Hz, 1H), 3.69–3.54 (m, 3H), 3.44 (s, 3H), 3.43 (s, 3H), 3.44–3.37 (m, 1H), 3.32–3.31 (m, 2H), 3.29–3.19 (m, 6H), 2.94 (dd, J = 14.0, 2.0 Hz, 1H), 2.59 (dd, J = 14.6, 8.0 Hz, 1H); 13C NMR (CD3OD) δ: 130.2, 129.3, 104.2, 99.8, 84.5, 76.6, 73.9, 73.8, 73.5, 72.8, 72.4, 70.1, 61.5, 56.2, 54.4, 34.1, 32.1; ESIHRMS calcd for C18H32O11SNa [M+Na]+: 479.1563, found: 479.1550.
Phenyl 3-deoxy-3-[4-(β-D-glucopyranosyloxy)but-2E-enylthio]-1-thio-β-D-glucopyranoside (46)
Following the general procedure 15, phenyl 2,4,6-tri-O-acetyl-3-acetylthio-1-thio-β-D-glucopyranoside (38) was coupled with sulfenyl donor 13. Chromatographic purification eluting with 8% MeOH/CH2Cl2 gave the title compound in 70% yield. [α]23D −36.0° (c 0.75, MeOH); 1H NMR (400 MHz, CD3OD) δ: 7.57–7.54 (m, 2H), 7.32–7.23 (m, 3H), 5.86–5.79 (m, 1H), 5.74–5.67 (m, 1H), 4.62 (d, J = 12.0 Hz, 1H), 4.34 (d, J = 10.0 Hz, 1H), 4.30 (dd, J = 16.5, 7.0 Hz, 1H), 4.14 (dd, J = 16.0, 9.0 Hz, 1H), 3.87 (dd, J = 15.0, 2.0 Hz, 2H), 3.69–3.62 (m, 2H), 3.46–3.26 (m, 8H), 3.18 (t, J = 11.0 Hz, 1H), 2.58 (t, J = 11.0 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ: 133.9, 131.6, 130.8, 128.7, 127.1, 101.5, 89.6, 82.7, 76.8, 76.6, 73.9, 72.4, 70.5, 68.6, 68.5, 61.8, 61.6, 56.7, 33.6; ESIHRMS calcd for C22H32O10S2Na [M+Na]+: 543.1335, found: 543.1346.
Phenyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-β-D-glucopyranoside (48)
A white solid prepared from 7142 by the general procedure 4 and eluted from silica gel with hexanes/EtOAc (2:1) in 83% yield: mp 110 °C; [α]RTD +47.3 (c 1.0); 1H NMR (400 MHz) δ: 7.55 (dd, J = 2.4, 5.6 Hz, 2H), 7.37-7.30 (m, 3H), 4.40 (d, J = 9.6 Hz, 1H), 3.92-3.83 (m, 1H), 3.77-3.68 (m, 2H), 3.63 (t, J = 9.6 Hz, 1H), 3.54-3.49 (m, 1H), 3.39 (t, J = 9.6 Hz, 1H), 3.33 (s, 3H), 3.28 (d, J = 5.6 Hz, 1H), 3.23 (s, 3H), 1.94 (dd, J = 5.6, 7.2 Hz, 1H), 1.33 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz) δ: 134.1, 129.4, 128.9, 100.4, 99.9, 86.3, 78.3, 73.2, 65.8, 61.7, 61.5, 48.3, 48.3, 17.8, 17.7; ESIHRMS calcd for C18H25N3O6SNa [M + Na]+: 434.1362, found: 434.1354.
Methyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-β-D-glucopyranoside (55)
A white foam prepared from 7242b,43 by the general procedure 4 and eluted from silica gel with hexanes/EtOAc (2:1) in 81% yield: [α]RTD = +70.3° (c 1.0); 1H NMR (400 MHz) δ: 4.2 (d, J = 8 Hz, 1H), 3.88-3.80 (m, 1H), 3.76-3.66 (m, 2H), 3.60 (dd, J = 9.6, 10.8 Hz, 1H), 3.53 (s, 3H), 3.48-3.42 (m, 1H), 3.39 (dd, J = 7.2, 8.4 Hz, 1H), 3.28 (s, 3H), 3.23 (s, 3H), 2.50 (dd, J = 5.6, 8.0 Hz, 1H), 1.31 (s, 3H), 1.26 (s, 3H); 13C NMR (100 MHz) δ: 103.6, 100.3, 99.9, 76.9, 74.2, 71.0, 66.0, 62.9, 61.2, 57.6, 48.3, 17.8, 17.7; ESIHRMS calcd for C13H23N3O7Na [M + Na]+: 365.1434, found: 365.1432.
Phenyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-6-O-p-toluenesulfonyl-β-D-glucopyranoside (73)
Prepared by the general procedure 5 as a white solid eluted from silica gel with hexanes/EtOAc (3:1) in a quantitative yield: mp 141 °C; [α]RT D +12.3° (c 1.0); 1H NMR (400 MHz) δ: 7.80 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 7.34-7.25 (m, 3H), 4.29 (t, J = 10.4 Hz, 2H), 4.21 (dd, J = 4.0, 10.8 Hz, 1H), 3.67-3.53 (m, 3H), 3.31 (d, J = 9.6 Hz, 1H), 3.28 (s, 3H), 3.23 (d, J = 7.6 Hz, 1H), 3.20 (s, 3H), 2.67 (s, 3H), 1.29 (s, 3H), 1.25 (s, 3H); 13C NMR (100 MHz) δ: 145.2, 134.4, 133.0, 130.3, 130.1, 129.3, 129.0, 128.2, 100.5, 100.1, 86.1, 77.6, 75.4, 73.0, 67.3, 65.3, 61.1, 48.6, 48.4, 21.9, 17.8, 17.7; ESIHRMS calcd for C25H31N3O8S2Na [M+Na]+: 588.1450, found: 588.1440.
Phenyl 2-azido-2-deoxy-6-acetylthio-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-β-D-glucopyranoside (74)
Prepared by the general procedure 6 and isolated as a light yellow foam eluted from silica gel with hexanes/EtOAc (6:1) in 78% yield: [α]RTD +132.1° (c 1.0), 1H NMR (400 MHz) δ: 7.57 (dd, J = 6.4, 7.2 Hz, 2H), 7.35-7.30 (m, 3H), 4.35 (d, J = 9.6 Hz, 1H), 3.67 (t, J = 9.6 Hz, 1H), 3.62-3.55 (m 1H), 3.51-3.45 (m, 2H), 3.44-3.30 (m, 2H), 3.30 (s, 3H), 3.22 (s, 3H), 3.07 (q, J = 7.2, 14.0 Hz, 1H), 2.35 (s, 3H), 1.31 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz) δ: 194.9, 134.5, 133.2, 130.7, 129.2, 129.1, 128.9, 100.4, 100.13, 86.2, 76.7, 73.0, 68.6, 61.5, 48.4, 48.3, 30.7, 30.1, 17.8, 17.7; ESIHRMS calcd for C20H27N3O6S2Na [M+Na]+: 492.1239, found: 492.1245.
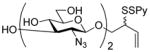
Phenyl 6-acetylthio-2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-β-D-glucopyranoside S-Oxide (54)
Prepared by the general procedure 7 and isolated as a white solid eluting from silica gel in hexanes/EtOAc (4:1) in 70% yield: mp 125 °C; [α]RTD −12.3° (c 1.0); 1H NMR (400 MHz) δ: 7.62-7.58 (m, 2H), 7.55-7.51 (m, 3H), 4.01 (dd, J = 9.6, 10.8 Hz, 1H), 3.83 (t, J = 9.6 Hz, 1H), 3.72 (d, J = 9.6 Hz, 1H), 3.54 (d, J = 9.6 Hz), 3.43-3.31 (m, 5H), 3.20 (s, 3H), 2.83 (dd, J = 8.0, 14.0 Hz, 1H), 2.19 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz) δ: 194.7, 139.1, 131.5, 129.2, 125.6, 100.5, 100.2, 91.9, 77.8, 73.3, 68.8, 57.6, 48.4, 48.3, 30.6, 29.8, 17.8, 17.7; ESIHRMS calcd for C20H27N3O7S2Na [M+Na]+: 508.1188, found: 508.1183.
Phenyl 2-azido-2-deoxy-3,4,6-tri-O-(p-methoxybenzyl)-1-thio-β-D-glucopyranoside (75)
To a stirred solution of 7142 (800 mg, 2.7 mmol) in DMF (6.4 mL) was added portionwise at 0 °C sodium hydride (542 mg, 16.1 mmol). The resulting mixture was stirred for 0.5 h at 0 °C before p-methoxybenzyl chloride (2.2 mL, 16.1 mmol) was added dropwise. The resulting mixture was allowed to warm to room temperature and stirred for 2 h. The reaction was quenched by addition of MeOH, concentrated and purified by column chromatography eluting with 0.5% Et3N in hexanes/EtOAc (4:1) to give the title compound (1.6 g, 92%) as a light yellow solid: mp 72 °C, [α]RTD −45.8° (c 1.0), 1H NMR (400 MHz) δ: 7.60 (dd, J = 7.2, 8.0 Hz, 2H), 7.32-7.24 (m, 7H), 7.12 (d, J = 8.8 Hz, 2H), 6.92-6.82 (m, 6H), 4.78 (s, 2H), 4.72 (d, J = 10.4 Hz, 1H), 4.56 (d, J = 11.2 Hz, 1H), 4.52-4.46 (m, 2H), 4.40 (d, J = 9.6 Hz, 1H), 3.80 (s, 9H), 3.76-3.66 (m, 2H), 3.58-3.42 (m, 3H), 3.32 (t, J = 9.6 Hz, 1H); 13C NMR (100 MHz) δ: 159.7, 159.6, 159.4, 133.8, 131.5, 130.5, 130.3, 130.1, 130.0, 129.8, 129.6, 129.2, 128.5, 114.2, 114.1, 114.0, 86.1, 85.0, 79.6, 76.9, 75.7, 74.9, 73.3, 65.6, 65.3, 55.5; ESIHRMS calcd for C36H39N3O7SNa [M+Na]+: 680.2406, found: 680.2404.
Phenyl 2-azido-2-deoxy-3,4,6-tri-O-(p-methoxybenzyl)-1-thio-β-D-glucopyranoside S-Oxide (47)
A white solid prepared by the general procedure 7 and eluted from silica gel with 0.5% Et3N in hexanes/EtOAc (4:1) as a mixture of diastereomers, in 92% yield: 1H NMR (400 MHz) δ: 7.71-7.63 (m, 2H), 7.51 (d, J = 4.8 Hz, 2H), 7.45 (dd, J = 2.4, 3.2 Hz, 2H), 7.32 (d, J = 8.4 Hz, 1H), 7.25 (t, J = 8.0 Hz, 3H), 7.18-6.79 (m, 3H), 6.92-6.81 (m, 7H), 4.83 (d, J = 1.6 Hz, 1H), 4.89-4.65 (m, 3H), 4.48 (dd, J = 7.2, 8.0 Hz, 3H), 4.31 (d, J = 11.6 Hz, 1H), 4.17 (dd, J = 9.6, 12.0 Hz, 1H), 3.86-3.3.77 (m, 13H), 3.74-3.67 (m, 2H), 3.66-3.57 (m, 1H), 3.56-3.45 (m, 4H); 13C NMR (100 MHz) δ: 159.7, 159.6, 140.3, 139.4, 131.6, 131.5, 130.1, 130.0, 129.9, 129.7, 129.6, 129.3, 129.2, 125.6, 124.9, 114.2, 114.1, 114.0, 113.9, 94.5, 91.8, 84.8, 84.7, 80.9, 80.4, 75.8, 75.7, 74.9, 73.5, 73.4, 68.4, 68.3, 61.0, 60.2, 55.5; ESIHRMS calcd for C36H39N3O8SNa [M+Na]+: 696.2356, found: 696.2369.
Phenyl 3,4,6-tri-O-acetyl-2-deoxy-1-thio-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside (77)
To a stirred solution of 1,3,4,6-tetra-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxycarbonylamino)-α,β-D-glucopyranose44 (76) (10 g, 19.2 mmol) in CH2Cl2 (100 mL) was added at room temperature, trimethylsilyl trifluoromethanesulfonate (4.2 mL, 23 mmol) and thiophenol (2.4 mL, 23 mmol). The resulting mixture was stirred for 4 h then, neutralized by addition of triethylamine and concentrated. Chromatographic purification (hexanes/EtOAc 3:1) afforded the title compound (9.2 g, 84%) as a light yellow solid: mp 87 °C; [α]RT D +18.5° (c 1.0); 1H NMR (400 MHz) δ: 7.49 (dd, J = 3.2, 4.0 Hz, 2H), 7.29 (dd, J = 2.0, 3.2 Hz, 3H), 4.95 (d, J = 9.6 Hz, 1H), 5.27 (t, J = 9.6 Hz, 1H), 5.01 (t, J = 9.6 Hz, 1H), 4.85 (d, J = 10.8 Hz, 1H), 4.74 (q, J = 12.0 Hz, 2H), 4.24–4.412 (m, 2H), 3.76-3.66 (m, 2H), 2.06 (s, 3H), 1.99 (s, 3H), 1.97 (s, 3H); 13C NMR (100 MHz) δ: 170.9, 169.7, 154.2, 133.1, 132.3, 129.2, 128.5, 119.2, 86.8, 77.6, 75.9, 74.7, 73.4, 68.8, 62.6, 55.2, 21.0, 20.9, 20.8; ESIHRMS calcd for C21H24Cl3NO9SNa [M+Na]+: 594.0135, found: 594.0112.
Methyl 3,4,6-tri-O-acetyl-2-(2,2,2-trichloroethoxycarbonylamino)-2-deoxy-β-D-glucopyranoside (79)
A white solid prepared by the general NIS/TfOH protocol and eluted from silica gel with hexanes/EtOAc (2:1) in 94% yield: mp 115 °C; lit.45 mp 119–124 °C; [α]RTD +12.5° (c 1.0); 1H NMR (400 MHz) δ: 5.30 (dd, J = 9.2, 10.4 Hz, 1H), 5.20 (br s, 1H), 5.07 (dd, J = 8.8, 10.0 Hz, 1H), 4.80 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 12 Hz, 1H), 4.54 (d, J = 7.6 Hz, 1H), 4.28 (dd, J = 4.0, 4.8 Hz, 1H), 4.15 (d, J = 10.0 Hz, 1H), 3.72 (d, J = 7.2 Hz, 1H), 3.64 (dd, J = 8.8, 17,6 Hz, 1H), 3.52 (s, 3H), 2.03 (s, 6H), 2.09 (s, 3H); 13C NMR (100 MHz) δ: 170.9, 169.7, 154.3, 119.2, 102.0, 74.7, 72.0, 68.9, 62.2, 57.4, 56.4, 55.1, 21.0, 20.8; ESIHRMS calcd for C16H22Cl3NO10Na [M+Na]+: 516.0207, found: 516.0211.
Methyl 3,4-O-(2,3-dimethoxybutane-2,3-diyl)-2-(2,2,2-trichloroethoxycarbonylamino)-2-deoxy-β-D-glucopyranoside (81)
Compound 79 (600 mg, 1.66 mmol) was dissolved in MeOH (8.3 mL) at room temperature and a catalytic amount of 25% NaOMe in MeOH (0.16 mmol) was added. The resulting mixture was stirred for 1 h before the pH of the solution was adjusted to 7 by addition of dry Amberlyst IR 120 resin. The resulting mixture was filtered and concentrated to give methyl 2-(2,2,2-trichloroethoxycarbonylamino)-2-deoxy-β-D-glucopyranoside (82), which was immediately subjected to the general procedure 4 and eluted from silica gel with hexanes/EtOAc (1:1) in 72% yield: white solid, mp 105 °C; [α]RT αD +123.0° (c 1.0); 1H NMR (400 MHz) δ: 5.20 (s, 1H), 4.72 (m 3H), 4.10 (dd, J = 7.2, 9.2 Hz, 1H), 3.92-3.84 (m, 1H), 3.80-3.74 (m,1H), 3.69 (t, J = 10.0 Hz, 1H), 3.58-3.53 (m, 1H), 3.50 (s, 3H), 3.40-3.28 (m, 1H), 3.25 (s, 3H), 3.21 (s, 3H), 2.01 (dd, J = 4.8, 8.0 Hz, 1H), 1.28 (s, 6H); 13C NMR (100 MHz) δ: 119.2, 119.1, 102.2, 100.2, 99.8, 74.6, 74.1, 68.3, 67.2, 61.4, 57.2, 56.0, 48.2, 48.1, 17.9, 17.8; ESIHRMS calcd for C16H26Cl3NO9Na [M+Na]+: 504.0571, found: 504.0572.
Phenyl 3,4,6-O-acetyl-2-deoxy-1-thio-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside S-Oxide (78)
A white foam prepared by the general procedure 7 and eluted from silica gel in hexanes/EtOAc (2:1) in 88% yield: mp 134 °C; [α]RTD +31.0° (c 1.0); 1H NMR (400 MHz) δ: 7.71 (dd, J = 2.5, 3.2 Hz, 2H), 7.52 (dd, J = 2.4, 3.2 Hz, 3H), 5.68 (d, J = 8.8 Hz, 1H), 5.44 (t, J = 9.6 Hz, 1H), 4.92 (t, J = 9.6 Hz, 1H), 4.80 (d, J = 10.4 Hz, 1H), 4.62 (d, J = 12.4 Hz, 1H), 4.44 (d, J = 12.0 Hz, 1H), 4.16 (m, 2H), 3.86 (dd, J = 9.6, 10.8 Hz, 1H), 3.78 (m, 1H), 2.00 (s, 9H); 13C NMR (100 MHz) δ: 170.7, 170.6, 169.6, 153.9, 138.8, 131.6, 129.1, 125.9, 95.3, 93.3, 76.4, 74.6, 72.6, 68.1, 61.7, 51.7, 20.9, 20.8; ESIHRMS calcd for C21H24Cl3NO10SNa [M+Na]:610.0084, found: 610.0067.
Phenyl 2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside (83)
To a stirred solution of phenyl 3,4,6-tri-O-acetyl-2-deoxy-1-thio-2-(2′,2′,2′-trichloroethoxycarbonylamino)-β-D-glucopyranoside46 (77) (1 g, 1.75 mmol) in MeOH (8.8 mL) was added a catalytic amount of 25% sodium hydroxide in MeOH (0.18 mmol) at room temperature. The resulting mixture was stirred for 1 h, neutralized by addition of Amberlyst IR 120 resin, filtered and concentrated to give phenyl 2-(2,2,2-trichloroethoxycarbonylamino)-1-thio-β-D-glucopyranoside (82),47 which was subjected to general protocol 4 directly, giving the title compound as a white foam, eluted from silica gel with hexanes/EtOAc (1.5:1) in 72% yield: [α]RTD +54.5° (c 1.0); 1H NMR (400 MHz) δ: 7.48 (dd, J = 2.4, 4.0 Hz, 2H), 7.31 (dd, J = 2.4, 4.4 Hz, 3H), 5.10 (d, J = 8.8 Hz, 2H), 4.75 (s, 2H) 4.10 (dd, J = 7.2, 9.6 Hz, 1H), 3.93-3.85 (m, 1H), 3.77-3.70 (m, 1H), 3.66 (t, J = 9.6 Hz, 1H), 3.62-3.56 (m, 1H), 3.41 (br s, 1H), 3.10 (dd, J = 4.4, 5.6 Hz, 1H), 3.24 (s, 3H), 3.21 (s, 3H), 1.91 (dd, J = 5.6, 7.6 Hz, 2H), 1.27 (s, 6H); 13C NMR (100 MHz) δ: 153.8, 133.0, 132.0, 129.4, 129.3, 128.4, 100.3, 99.8, 86.1, 78.1, 77.4, 74.7, 70.1, 67.0, 61.7, 54.7, 48.2, 48.1, 17.8, 17.8; ESIHRMS calcd for C21H28Cl3NO8SNa [M+Na]+: 582.0449, found: 582.0494.
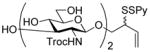
Phenyl 2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-6-O-p-toluenesulfonyl-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranoside (57)
A white solid prepared by the general procedure 5 and eluted from silica gel with hexanes/EtOAc (3:1) in a quantitative yield: mp 144 °C; [α]RTD +84.2° (c 1.0); 1H NMR (400 MHz) δ: 7.81 (d, J = 7.2 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 7.34-7.21 (m, 5H), 5.03 (dd, J = 6.4, 10.0 Hz, 2H), 4.73 (s, 2H), 4.30 (d, J = 10.8 Hz, 1H), 4.23 (dd, J = 4.0 Hz, 10.8 Hz, 1H), 4.07 (dd, J = 10.0, 11.2 Hz, 1H), 3.67 (dd, J = 2.4, 3.2 Hz, 1H), 3.59 (t, J = 9.6 Hz, 1H), 3.31-3.23 (m, 1H), 3.29 (s, 3H), 3.18 (s, 3H), 2.40 (s, 3H), 1.25 (s, 6H); 13C NMR (100 MHz) δ: 145.1, 133.3, 130.1, 129.2, 128.5, 128.2, 100.4, 100.0, 85.6, 77.6, 75.4, 74.6, 69.7, 67.7, 66.5, 54.3, 48.5, 48.2, 21.9, 17.9, 17.7; ESIHRMS calcd for C28H34Cl3NO10Na [M+Na]+: 736.0587, found: 736.0594.
Phenyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-2′-azido-2′-deoxy-3′,4′,6′-triO-(p-methoxybenzyl)]-β-D-gentiobioside (49)
Glycosyl sulfoxide 47 (1.2 g, 1.78 mmol) was premixed with acceptor 48 (880 mg, 2.14 mmol), 2,4,6-tri-tert-butylpyrimidine (886 mg, 3.57 mmol) and activated 4Å powdered molecular sieves in CH2Cl2/acetonitrile (4:3) (3.6 mL) and the resulting mixture was stirred at room temperature for 0.5 h then cooled to−80 °C before trifluoromethanesulfonic anhydride (0.33 mL, 1.96 mmol) was added dropwise. The reaction mixture was stirred at −80 °C for 1.5 h, quenched by addition of aqueous saturated NaHCO3 and then allowed to warm to room temperature. The aqueous phase was extracted twice with CH2Cl2. The organic layers were combined, dried, filtered and concentrated in vacuo. Chromatographic purification eluting with 0.5% Et3N in hexanes/EtOAc (4:1 to 2.5:1) gave the title compound (1.05 g, 62%) as a white foam: [α]RTD +29.6° (c 1.0); 1H NMR (400 MHz) δ: 7.61-7.53 (m, 2H), 7.36-7.27 (m, 5H), 7.27-7.22 (m, 3H), 7.07 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.83 (dd, J = 5.6, 8.8 Hz, 3H), 4.83-4.69 (m, 3H), 4.58-4.37 (m, 4H), 4.14 (d, J = 10.8 Hz, 1H), 3.90-3.46 (m, 16H), 3.44-3.19 (m, 11H), 1.32 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz) δ: 159.6, 159.5, 134.1, 133.8, 130.4, 130.0, 129.8, 129.3, 128.6, 114.1, 114.0, 102.4, 100.4, 100.0, 86.4, 83.1, 78.2, 75.9, 75.8, 75.5, 75.2, 74.9, 74.7, 73.4, 72.2, 71.2, 68.4, 68.1, 66.6, 66.3, 65.6, 61.7, 60.2, 55.5, 55.4, 48.3, 17.8, 17.7; ESIHRMS calcd for C48H58N6O13SNa [M+Na]+: 981.3680, found: 981.3682.
Phenyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-1-thio-[2′-azido-2′-deoxy-3′,4′,6′-tri-O-(p-methoxybenzyl)]-β-D-gentiobioside S-Oxide (50)
A white foam prepared by the general procedure 7 and eluted from silica gel with 0.5% Et3N in hexanes/EtOAc (3:1) in 88% yield, in an approximately 1:1 mixture of diastereomers: 1H NMR (400 MHz) δ: 7.68-7.60 (m, 2 × 2H), 7.55-7.45 (m, 2 × 3H), 7.33-7.20 (m, 2 × 4H), 7.12-7.02 (m, 2 × 2H), 6.91-6.79 (m, 2 × 6H), 4.80-4.66 (m, 2 × 3H), 4.54 (dd, J = 9.6, 10.8 Hz, 1 × 1H), 4.48-4.38 (m, 2 × 2H), 4.21 (dd, J = 5.6, 9.6 Hz, 1 × 1H), 4.02-3.84 (m, 2 × 3H), 3.83-3.73 (m, 2 × 10H), 3.73-3.44 (m, 2 × 6H), 3.43-3.14 (m, 2 × 9H), 1.34 (s, 2 × 3H), 1.26 (s, 2 × 3H); 13C NMR (100 MHz) δ: 159.6, 139.1, 131.7, 131.4, 130.3, 130.0, 129.9, 129.8, 129.7, 129.6, 129.4, 129.3, 125.4, 124.7, 114.2, 114.1, 114.0, 102.9, 102.1, 100.5, 100.1, 95.2, 91.7, 91.4, 82.7, 79.8, 78.8, 77.5, 75.4, 75.3, 75.0, 74.9, 74.8, 73.5, 73.4, 73.1, 68.5, 68.2, 67.7, 66.4, 66.1, 65.8, 57.7, 55.5, 55.4, 48.4, 48.3, 17.8, 17.7; ESIHRMS calcd for C48H58N6O14SNa [M+Na]+: 997.3629, found: 997.3614.
2-(Phenyloxycarbonylthioxy-but-3-enyl 2-azido-2-deoxy-3,4-O-(2,3-dimethoxybutane-2,3-diyl)-[2′-azido-2′ deoxy-3′,4′,6′-tri-O-(p-methoxybenzyl)]-β-D-gentiobioside (51)
The glycosyl sulfoxide 50 (560 mg, 0.57 mmol) was mixed with 2022b (266 mg, 1.20 mmol), 2,4,6-tri-tert-butylpyrimidine (314 mg, 1.26 mmol) and activated 4Å powdered molecular sieves in CH2Cl2/acetonitrile (4:3) (2 mL) and the resulting mixture was stirred at room temperature for 0.5 h then cooled to −60 °C before trifluoromethanesulfonic anhydride (0.15 mL, 0.86 mmol) was added dropwise. The reaction mixture was stirred at −60 °C for 8 h and was then cooled to −80 °C, quenched by addition of aqueous saturated NaHCO3, and then allowed to warm to room temperature. The aqueous phase was extracted twice with CH2Cl2. The organic layers were combined, dried, filtered and concentrated in vacuo. Chromatographic purification eluting with 0.5% Et3N in hexanes/EtOAc (6:1 to 3:1) gave the title compound (390 mg, 63%) as a white foam as an approximately 1:1 mixture of diastereomers: 1H NMR (400 MHz) δ 7.41-7.11 (m, 2 ×10H), 7.17-7.10 (m, 2 ×2H), 7.45 (d, J = 7.2 Hz, 2 × 2H), 6.90-6.79 (m, 2 ×7H), 6.03-5.92 (m, 2 ×1H) 5.43 (d, J = 16.8 Hz, 2 × 1H), 5.25 (d, J = 9.6 Hz, 2 × 1H), 4.80-4.67 (m, 2 ×4H), 4.55 (dd, J = 7.2, 11.6 Hz, 2 × 1H), 4.47-4.35 (m, 2 ×4H), 4.29-4.17 (m, 2 ×3H), 3.92-3.81 (m, 2 ×1H), 3.79 (s, 2 × 6H), 3.78 (s, 2 × 3H), 3.73–4.49 (m, 2 ×7H), 3.49-3.19 (m, 2 ×10H), 1.33 (s, 2 × 3H), 1.27 (s, 2 × 3H); 13C NMR (100 MHz) δ: 169.3, 169.2, 169.0, 159.6, 159.5, 159.4, 156.6, 151.4, 134.2, 134.1, 130.3, 130.2, 130.0, 130.0, 129.9, 129.8, 129.7, 126.4, 126.3, 124.7, 121.7, 121.6, 121.5, 120.3, 118.9, 115.7, 114.2, 114.1, 114.0, 103.0, 102.9, 102.8, 102.7, 100.2, 100.0, 83.2, 83.0, 75.4, 75.2, 74.9, 74.6, 73.4, 73.3, 71.4, 71.2, 70.8, 70.1, 70.0, 68.4, 67.1, 66.5, 63.2, 55.5, 55.4, 48.7, 48.3, 45.9, 18.0, 17.9, 17.8, 17.7; ESIHRMS calcd for C53H64N6O16SNa [M+Na]+: 1095.3997, found: 1095.4004.
2-(Phenyloxycarbonylthioxy)but-3-enyl 2,2′-diazido-2,2′-dideoxy-β-D-gentiobioside (52)
A white foam obtained by treatment of 51 with TFA according to the general procedure 9 eluted from silica gel with CH2Cl2/MeOH (20:1) as an approximately 1:1 mixture of diastereomers in 67% yield NMR (400 MHz) δ: 7.40 (t, J = 7.3 Hz, 2 × 2H), 7.26 (t, J = 7.2 Hz, 2 × 1H), 7.15 (d, J = 9.2 Hz, 2 × 2H), 6.06-5.95 (m, 2 × 1H), 5.41 (d, J = 15.2 Hz, 2 × 1H), 5.23 (d, J = 10.8 Hz, 2 × 1H), 4.48 (dd, J = 7.3, 8.8 Hz, 2 × 1H), 4.41 (d, J = 8.0 Hz, 2 × 1H), 4.28-4.18 (m, 2 × 3H), 3.93-3.60 (m, 2 × 7H), 3.47 (dd, J = 6.4, 8.0 Hz, 2 × 1H), 3.34-3.06 (m, 2 × 9H); 13C NMR (100 MHz) δ: 164.0, 134.9, 129.5, 126.2, 121.3, 117.7, 117.6, 102.7, 102.3, 102.1, 101.9, 94.7, 76.8, 76.2, 75.1, 75.0, 70.7, 70.6, 70.3, 69.0, 67.1, 61.3, 48.6; ESIHRMS calcd for C23H30N6O11SNa [M+Na]+: 621.1591, found: 621.1593.
2-(Pyridyldithio)but-3-enyl 2,2′-diazido-2,2′-dideoxy-β-D-gentiobioside (53)
A white foam prepared by the general procedure 3 and eluting from silica gel in CH2Cl2/MeOH (20:1) as an approximately 1:1 mixture of diastereomers in 65% yield over two steps: 1H NMR (400 MHz, CD3OD) δ: 8.37 (d, J = 4.0 Hz, 2 × 1H), 8.75 (dd, J = 4.0, 8.4 Hz, 2 × 1H), 7.83-7.77 (m, 2 × 1H), 7.21 (dd, J = 4.8, 7.2 Hz, 2 × 1H), 5.90-5.78 (m, 2 × 1H), 5.30-5.21 (m, 2 × 1H), 5.15 (d, J = 10.8 Hz, 2 × 1H), 4.96-4.85 (m, 2 × 1H), 4.47-4.32 (m, 2 × 2H), 4.24-3.98 (m, 2 × 3H), 3.91-3.64 (m, 2 × 7H), 4.48-3.05 (m, 2 × 9H); 13C NMR (100 MHz, CD3OD) δ: 148.9, 137.9, 133.9, 121.1, 120.3, 120.2, 118.4, 102.7, 101.8, 76.8, 76.2, 75.1, 74.9, 70.6, 70.4, 69.8, 69.0, 67.0, 61.4, 53.8; ESIHRMS calcd for C21H29N7O9S2Na [M+Na]+: 610.1366, found: 610.1379.
Methyl 2,2′-diazido-2,2′-dideoxy-6′-acetylthio-β-D-gentiobioside (56)
Glycosyl sulfoxide (54) (340 mg, 0.70 mmol) was mixed with 2,4,6-tri-tert-butylpyrimidine (244 mg, 0.98 mmol) and activated 4Å powdered molecular sieves in CH2Cl2/acetonitrile (3:1) (2.6 mL). The resulting mixture was stirred at room temperature for 0.5 h then cooled to −65 °C before trifluoromethanesulfonic anhydride (0.14 mL, 0.84 mmol) was added dropwise. The glycoside sulfoxide was preactivated for 10 min and a solution of 55 (466 mg, 1.4 mmol) in CH2Cl2 (0.8 mL) was added. The resulting mixture was stirred for 8 h at −65 °C quenched by addition of aqueous saturated NaHCO3 and then allowed to warm to room temperature. The aqueous phase was extracted twice with CH2Cl2. The organic layers were combined, dried, filtered and concentrated in vacuo. The crude protected disaccharide was subjected directly to the standard acid hydrolysis step after which the title compound was obtained as a white foam eluted from silica gel with CH2Cl2/MeOH (20:1) in 26% overall yield: [α]RTD +18.5° (c 1.0, MeOH); NMR (400 MHz) δ: 4.42 (d, J = 7.8 Hz, 1H), 4.24 (d, J = 7.8 Hz, 1H), 4.16 (dd, J = 1.8, 11.4 Hz, 1H), 3.70 (dd, J = 6.9 11.7 Hz, 1H), 3.60-3.51 (m, 5H), 3.46 (dd, J = 7.2, 8.1 Hz, 1H), 3.35 (s, 1H), 3.32-3.24 (m 8H), 3.23-3.08 ( m, 4H), 2.98 (dd, J = 8.1, 14.1 Hz, 1H), 2.33 (s, 3H); 13C NMR (100 MHz) δ: 195.7, 102.9, 102.6, 75.9, 75.2, 75.0, 74.7, 73.4, 70.6, 69.1, 67.0, 48.7, 47.0, 30.6, 29.2; ESIHRMS calcd for C15H24N6O9SNa [M+Na]+: 487.1223, found: 487.1232.
Phenyl 3′,4′,6′-tri-O-acetyl-2,2′-dideoxy-3,4-O-(2,3-dimethoxybutan-2,3-diyl)-1-thio-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobioside (84)
Glycosyl sulfoxide 78 (310 mg, 0.53 mmol) was stirred in CH2Cl2 (1.6 mL) with activated 4Å powdered molecular sieves at room temperature for 0.5 h. The resulting mixture was cooled to −65 °C before trifluoromethanesulfonic anhydride (0.10 mL, 0.58 mmol) was added dropwise. After stirring for 10 min a solution of 83 (350 mg, 0.62 mmol) in CH2Cl2 (0.4 mL) was added. The resulting mixture was stirred for 7 h at −65 °C, quenched by addition of aqueous saturated NaHCO3, and then allowed to warm to room temperature. The aqueous phase was extracted twice with CH2Cl2. The organic layers were combined, dried, filtered and concentrated in vacuo. Chromatographic purification eluting with hexanes/EtOAc (5:1 to 2:1) gave the title disaccharide as a white foam (230 mg, 43%): [α]RTD +44.2° (c 1.0); 1H NMR δ: 7.52 (d, J = 7.2 Hz, 2H), 7.43-7.33 (m, 3H), 5.36-5.06 (m, 2H), 5.00 (d, J = 7.6 Hz, 2H), 4.81-4.69 (m, 3H), 4.64 (dd, J = 8.8, 12.0 Hz, 1H), 4.52 (d, J = 8.0 Hz, 1H), 4.28-4.21 (m, 2H), 4.15-4.02 (m, 3H), 3.75-3.52 (m, 4H), 3.39 (t, J = 9.6 Hz, 2H), 3.20 (s, 3H), 3.17 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H), 1.26, (s, 3H), 1.24 (s, 3H); 13C NMR δ: 169.7, 169.3, 168.4, 153.1, 152.5, 132.2, 128.5, 127.7, 100.2, 99.0, 98.7, 94.5, 84.9, 78.1, 75.7, 73.5, 71.2, 70.5, 67.7, 67.3, 66.6, 66.3, 60.9, 54.9, 53.4, 47.0, 46.9, 19.7, 19.6, 16.6, 16.5; ESIHRMS calcd for C36H46Cl6N2O17SNa [M+Na]+: 1043.0549, found: 1043.0546.
2-(Phenyloxycarbonylthioxy)but-3-enyl 3′,4′,6′-tri-O-acetyl-2,2′-dideoxy-3,4-O-(2,3-dimethoxybutan-2,3-diyl)-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobioside (85)
The thioglycoside 84 was converted to the corresponding glycosyl sulfoxide according to general procedure 7 and 280 mg of this sulfoxide (0.27 mmol) was mixed with 2022b (120 mg, 0.54 mmol) and activated 4Å powdered molecular sieves in CH2Cl2 (1.2 mL) and stirred at room temperature for 0.5 h then cooled to −60 °C before trifluoromethanesulfonic anhydride (0.07 mL, 0.40 mmol) was added dropwise. The reaction mixture was stirred at −60 °C for 12 h, then was cooled to −75 °C, quenched by addition of aqueous saturated NaHCO3 and allowed to warm to room temperature. The aqueous phase was extracted twice with CH2Cl2. The organic layers were combined, dried, filtered and concentrated in vacuo. Purification by column chromatography eluting with hexanes/EtOAc (4:1 to 2:1) gave the title compound as a white foam as an approximately 1:1 mixture of diastereomers (202 mg, 40% over two steps): 1H NMR (400 MHz) δ: 7.36 (t, J = 7.2 Hz, 2 × 2H), 7.25-7.13 (m, 2 × 3H), 6.1-5.85 (m, 2 × 1H), 5.40 (d, J = 16.8 Hz, 2 × 1H), 5.35-5.20 (m, 2 × 3H), 5.05 (dd, J = 8.8, 9.6 Hz, 2 × 2H), 4.85-4.65 (m, 2 × 6H), 4.30-3.95 (m, 2 × 6H), 3.90-3.80 (m, 2 × 1H), 3.75-3.50 (m, 2 × 5H), 3.45 (dd, J = 8.0, 10.0 Hz, 2 × 1H), 3.25 (s, 2 × 3H), 3.15 (s, 2 × 3H), 2.08 (s, 2 × 3H), 1.98 (s, 2 × 6H), 1.28 (s, 2 × 3H), 1.24 (s, 2 × 3H); 13C NMR (100 MHz) δ: 170.9, 170.6, 169.7, 154.2, 151.3, 134.1, 133.8, 129.7, 126.4, 121.6, 121.5, 119.2, 119.1, 101.1, 100.1, 99.9, 95.7, 76.9, 74.7, 72.2, 71.9, 71.6, 70.2, 68.8, 68.6, 68.3, 67.7, 62.2, 62.1, 56.5. 55.7, 55.4, 49.0, 48.6, 48.2, 48.1, 29.9, 21.0, 20.8, 17.9, 17.8; ESIHRMS calcd for C41H52Cl6N2O20SNa [M+Na]+: 1157.0885, found: 1157.0890.
2-(Phenyloxycarbonylthioxy)but-3-enyl 3′,4′,6′-tri-O-acetyl-2,2′-dideoxy-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobioside (86)
Cleavage of the bis(acetal) groups from 85 according to the general procedure 9 gave the title compound as a white foam that eluted from silica gel with hexanes/EtOAc (1:1 to 1:3) as an approximately 1:1 mixture of diastereomers in 68% yield NMR (400 MHz) δ: 7.38 (dd, J = 7.2, 8.4 Hz, 2 × 2H), 7.28-7.21 (m, 2 × 1H), 7.16 (dd, J = 4.0, 8.4 Hz, 2 × 2H), 6.00–6.57 (m, 2 × 1H), 5.55-5.45 (d, J = 16.8 Hz, 2 × 1H), 5.40 (d, J = 16.8 Hz, 2 × 1H), 5.25 (d, J = 10.4 Hz, 2 × 1H), 5.25-5.20 (m, 2 × 1H), 5.05 (t, J = 9.6 Hz, 2 × 1H), 4.80-4.65 (m, 2 × 4H), 4.55 (br s, 2 × 1H), 4.30-4.05 (m, 2 × 5H), 3.90-3.60 (m, 2 × 6H), 3.60-3.30 (m, 2 × 4H), 2.10 (s, 2 × 3H), 2.03 (s, 2 × 6H); 13C NMR (100 MHz) δ: 171.1, 171.0, 169.7, 155.3, 154.6, 151.3, 134.1, 133.8, 129.8, 126.5, 121.6, 121.5, 119.4, 119.3, 101.5, 95.5, 77.0, 75.1, 75.0, 74.8, 74.7, 74.3, 72.2, 71.5, 69.3, 68.7, 62.1, 58.3, 58.1, 56.4, 48.8, 48.4, 29.9, 21.1, 20.8; ESIHRMS calcd for C35H42Cl6N2O18SNa [M+Na]+: 1043.0182, found: 1043.0209.
2-(2-Pyridyldithio)but-3-enyl 2,2′-dideoxy-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobioside (62)
Installation of the disulfide moiety on 86 by the standard procedure 3 gave the title compound as a white foam eluting from silica gel with CH2Cl2/MeOH (20:1) as an approximately 1:1 mixture of diastereomers in 62% yield over two steps: 1H NMR (400 MHz, CD3OD) δ: 8.36 (d, J = 4.8 Hz, 2 × 1H), 7.91 (dd, J = 7.2, 8.4 Hz, 2 × 1H), 7.87-7.79 (m, 2 × 1H), 7.20 dd, J = 4.8, 7.2 Hz, 2 × 1H), 5.90-5.74 (m, 2 × 1H), 5.15 (dd, J = 9.6, 16.8 Hz, 2 × 2H), 4.85-4.64 (m, 2 × 10H), 4.53-4.33 (m, 2 × 2H), 4.21-4.00 (m, 2 × 2H), 3.88 (d, J = 12.0 Hz, 2 × 1H), 3.82-3.59 (m, 2 × 4H), 3.50-3.21 (m, 2 × 9H); 13C NMR (100 MHz, CD3OD) δ: 155.8, 149.1, 148.8, 138.1, 134.1, 133.8, 122.2, 121.0, 120.3, 118.6, 118.3, 102.1, 101.8, 101.7, 76.8, 75.8, 74.6, 74.5, 74.4, 71.1, 70.9, 70.0, 69.8, 69.0, 61.6, 58.0, 57.8, 54.4, 54.3, 29.6; ESIHRMS calcd for C27H35Cl6N3O13S2Na [M+Na]+: 905.9637, found: 905.9640.
Methyl 2,2′-dideoxy-3,4;3′,4′-di-O-(2,3-dimethoxybutan-2,3-diyl)-2,2′-bis(2,22-trichloroethoxycarbonylamino)-6′-O-p-toluenesulfonyl-β-D-gentiobioside (59)
A white foam prepared by the general procedure 8 from 57 and 58 and eluted from silica gel with hexanes/EtOAc (5:1 to 3:1) in 89% yield: [α]23D = +72.7° (c 1.0); 1H NMR (400 MHz) δ: 7.82 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 5.10 (br s, 2H), 4.88 (br s, 1H), 4.80-4.62 (m, 5H), 4.31 (d, J = 10.8 Hz, 1H), 4.16 (dd, J = 5.6, 10.8 Hz, 1H), 4.12-3.97 (m, 3H), 3.68 (dd, J = 6.4, 8.8 Hz, 3H), 3.59-3.45 (m, 5H), 3.40-3.12 (m, 14H), 2.45 (s, 3H), 1.28 (s, 6H), 1.25 (s, 6H); 13C NMR (100 MHz) δ: 154.6, 154.1, 145.2, 132.9, 130.1, 128.4, 128.2, 101.9, 101.6, 100.2, 99.8, 95.8, 95.7, 74.8, 74.5, 73.8, 73.6, 72.3, 71.7, 69.7, 69.2, 68.5, 67.7, 67.0, 57.3, 57.2, 56.5, 55.7, 55.6, 48.5, 48.4, 48.2, 48.1, 29.9, 21.9, 18.9, 17.8; ESIHRMS calcd for C38H54Cl6N2O19SNa [M+Na]+: 1107.1070, found: 1107.1086.
Methyl 6′-acetylthio-2,2′-dideoxy-3,4;3′,4′-di-O-(2,3-dimethoxybutan-2,3-diyl)-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobioside (60)
A light yellow foam prepared by displacement of the tosylate from 59 according to the general procedure 6 and eluted from silica gel with hexanes/EtOAc (6:1) in 71% yield: [α]RTD +82.3° (c 1.0), 1H NMR (400 MHz) δ: 5.16-5.0 (m, 1H), 4.92-4.83 (m, 1H), 4.78-4.63 (m, 5H), 4.12-3.99 (m, 3H), 3.74-3.65 (m, 2H), 3.64-3.56 (m, 1H), 3.53-3.42 (m, 6H), 3.33 (dd, 5.2, J = 7.2 Hz, 3H), 3.23 (s, 3H), 3.21 (s, 3H), 3.20 (s, 3H), 3.18 (s, 3H), 3.04 (q, J = 10.0 Hz, J = 18.8 Hz, 1H), 2.33 (s, 3H), 1.29 (s, 3H), 1.27 (s, 3H); 13C NMR (100 MHz) δ: 195.1, 154.6 154.0, 129.2, 128.8, 101.8, 101.6, 100.1, 100.0, 99.8, 95.8, 77.6, 75.9, 74.8, 74.6, 74.2, 74.1, 72.7, 72.5, 70.1, 68.4, 67.8, 57.6, 57.3, 55.9, 48.5, 48.3, 48.1, 30.7, 30.2, 19.0, 17.9, 17.8; ESIHRMS calcd for C33H50Cl6N2O17SNa [M+Na]+: 1011.0859, found: 1011.0869.
Methyl 2,2′-dideoxy-2,2′-bis(2,2,2-trichloroethoxycarbonylamino)-6′-acetylthio-β-D-gentiobioside (61)
Removal of the bisacetal groups from 60 according to the general procedure 9 gave the title compound as a white foam eluted from silica gel with CH2Cl2/MeOH (20:1) in 68% yield: [α]RTD −6.0° (c 1.0); NMR (400 MHz) δ: 4.84-4.69 (m, 6H), 4.60 (s, 1H), 4.45 (d, J = 7.6 Hz, 1H), 4.29 (d, J = 8.8 Hz, 1H), 4.11 (d, J = 10.4 Hz, 1H), 3.72-3.56 (m, 2H), 3.45-3.29 (m, 7H), 3.21 (dd, J = 8.8, 9.4 Hz, 3H), 2.90 (dd, J = 7.2, 13.6 Hz, 1H), 2.34 (s, 3H); 13C NMR (100 MHz) δ: 196.0, 155.9, 102.4, 102.1, 91.5, 75.6, 75.0, 74.6, 74.4, 74.2, 74.1, 74.0, 72.1, 71.4, 69.5, 48.5, 47.4, 30.8, 29.3, 25.6, 19.8; ESIHRMS calcd for C21H30Cl6N2O13SNa [M+Na]+:782.9497, found: 782.9501.
3-O-{2,4,6-Tri-O-acetyl-3-O-[4-(2-naphthylmethyloxy)but-2Z-enyl]-β-D-glycopyranosyl}-1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (87)
Following the general procedure 12, diacetone-D-glucose was glycosylated with trichloroacetimidate 26 and after eluting with 70% EtOAc/hexanes from silica gel the title compound was obtained in 60% yield. [α]23D −15.7° (c 1); 1H NMR δ: 7.86–7.83 (m, 3H), 7.79 (s, 1H), 7.51–7.46 (m, 3H), 5.84 (d, J = 3.5 Hz, 1H), 5.81–5.76 (m, 1H), 5.62–5.57 (m, 1H), 5.03 (t, J = 10.0 Hz, 1H), 4.92 (dd, J = 9.5, 7.5 Hz, 1H), 4.68 (s, 2H), 4.50 (d, J = 8.0 Hz, 1H), 4.42 (d, J = 4.0 Hz, 1H), 4.35 (dd, J = 12.0, 6.0 Hz, 1H), 4.27 (dd, J = 5.5, 3.0 Hz, 1H), 4.25–4.24 (m, 1H), 4.18–4.11 (m, 3H), 4.08–4.03 (m, 3H), 3.97 (dd, J = 9.0, 6.0 Hz, 1H), 3.53–3.49 (m, 2H), 2.08 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.50 (s, 3H), 1.43 (s, 3H), 1.35 (s, 3H), 1.32 (s, 3H); 13C NMR δ: 170.9, 169.4, 168.9, 135.7, 133.5, 133.2, 129.5, 129.3, 128.5, 128.1, 127.9, 126.7, 126.5, 126.2, 125.9, 112.3, 108.8, 105.3, 99.6, 83.0, 81.2, 80.8, 79.7, 73.3, 72.7, 72.6, 69.5, 67.3, 66.4, 65.8, 62.4, 27.1, 26.8, 26.6, 25.5, 20.9; ESIHRMS calcd for C35H47NO17Na [M+Na]+: 776.2742, found: 776.2740.
2,2′,4,4′,6,6′-Hexa-O-acetyl-3′-O-[4-(2-naphthylmethyloxy)but-2Z-enyl]-α-D-laminaribiosyl trichloroacetimidate (89)
Following the general procedure 10, the acetonide groups were cleaved and the acetate groups were reinstalled to give the peracetate 88 in 60% yield as a mixture of stereoisomers that was applied directly to the general procedure 11, after which elution from silica gel with 70% EtOAc/hexanes gave the title compound in 68% yield as yellow oil. [α]23D +28.0° (c 1); 1H NMR δ: 8.70 (s, 1H), 7.85 – 7.82 (m, 3H), 7.77 (s, 1H), 7.50–7.45 (m, 3H), 6.46 (d, J = 4.0 Hz, 1H), 5.79–5.74 (m, 1H), 5.59–5.54 (m, 1H), 5.12–5.06 (m, 2H), 4.99 (t, J = 9.5 Hz, 1H), 4.89–4.85 (m, 1H), 4.66 (s, 2H), 4.54 (d, J = 8.5 Hz, 1H), 4.23 (dd, J = 12.0, 5.0 Hz, 1H), 4.19 (dd, J = 12.5, 4.5 Hz, 1H), 4.16–4.03 (m, 8H), 3.55–3.49 (m, 2H), 2.08 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 1.98 (s, 3H), 1.97 (s, 3H); 13C NMR δ: 170.9, 170.8, 169.7, 169.4, 169.3, 169.1, 160.8, 135.7, 133.5, 133.2, 129.5, 129.2, 128.5, 128.1, 127.9, 126.7, 126.5, 125.9, 101.4, 93.4, 79.8, 76.3, 72.7, 72.3, 72.1, 72.0, 70.5, 69.4, 67.5, 66.8, 65.9, 62.3, 61.8, 20.9, 20.9, 20.9, 20.9, 20.8, 20.8, 20.7; ESIHRMS calcd for C41H48Cl3NO18Na [M+Na]+: 970.1835, found: 970.1845.
Methyl 2,2′,4,4′,6,6′-hexa-O-acetyl-3′-O-[4-(2-naphthylmethyloxy)but-2Z-enyl]-β-D-laminaribioside (90)
Following the general procedure 12, and eluting with 75% EtOAc/hexanes the title compound was obtained in 60% yield. [α]23D −17.0° (c 1); 1H NMR δ: 7.85-7.83 (m, 3H), 7.78 (s, 1H), 7.49–7.45 (m, 3H), 5.79–5.74 (m, 1H), 5.59–5.55 (m, 1H), 5.03–4.91 (m, 3H), 4.86 (t, J = 9.0 Hz, 1H), 4.66 (s, 2H), 4.46 (d, J = 8.5 Hz, 1H), 4.30–4.25 (m, 2H), 4.19–4.18 (m, 2H), 4.11 (d, J = 6.5 Hz, 2H), 4.07 (d, J = 6.0 Hz, 2H), 4.00 (dd, J = 12.0, 2.5 Hz, 1H), 3.85 (t, J = 9.0 Hz, 1H), 3.69–3.67 (m, 1H), 3.51–3.49 (m, 2H), 3.47 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H), 2.02 (s, 3H), 2.02 (s, 3H), 1.98 (s, 3H); 13C NMR δ: 171.1, 170.9, 169.5, 169.4, 169.4, 169.0, 135.7, 133.5, 133.2, 129.5, 129.2, 128.5, 128.1, 127.9, 126.7, 126.4, 126.2, 125.9, 101.7, 101.4, 79.8, 78.8, 72.9, 72.7, 72.1, 72.1, 72.0, 69.3, 68.7, 66.8, 65.9, 62.4, 62.3, 56.8, 21.2, 21.0, 20.9, 20.9, 20.8, 20.7; ESIHRMS calcd for C40H50O18Na [M+Na]+: 841.2895, found 841.2870.
Methyl 2,2′,4,4′,6,6′-hexa-O-acetyl-3′-O-[4-hydroxybut-2Z-enyl]-β-D-laminaribioside (91)
Following the general procedure 13, and eluting with 90% EtOAc/hexanes the title product was obtained in 85% yield. [α]23D −37.0° (c 1); 1H NMR δ: 5.75–5.69 (m, 1H), 5.50–5.45 (m, 1H), 5.02 (t, J = 9.5 Hz, 1H), 4.97–4.90 (m, 2H), 4.86 (dd, J = 9.5, 8.5 Hz, 1H), 4.49 (d, J = 8.0 Hz, 1H), 4.31–4.28 (m, 2H), 4.20–4.16 (m, 2H), 4.14–4.09 (m, 4H), 4.02 (dd, J = 12.0, 2.5 Hz, 1H), 3.85 (t, J = 9.5 Hz, 1H), 3.67–3.64 (m, 1H), 3.59–3.52 (m, 2H), 3.45 (s, 3H), 2.13 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H); 13C NMR δ: 171.0, 170.8, 169.7, 169.6, 169.5, 169.1, 132.3, 127.7, 101.7, 101.4, 79.8, 78.9, 72.9, 72.1, 72.0, 71.7, 68.8, 68.6, 65.9, 62.4, 62.3, 58.5, 56.8, 21.2, 21.0, 21.0, 20.9, 20.9, 20.7; ESIHRMS calcd for C29H42O18Na [M+Na]+: 701.2269, found: 701.2289.
Methyl 2,2′,4,4′,6,6′-hexa-O-acetyl-3′-O-[4-(phenyloxythionocarbonyloxy)but-2Z-enyl]-β-D-laminaribioside (92)
Following the general procedure 1, and eluting with 70% EtOAc/hexanes the title product was obtained in 88% yield. [α]23D −32.0° (c 1); 1H NMR δ 7.43–7.40 (m, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.09–7.07 (m, 2H), 5.80–5.75 (m, 1H), 5.71–5.66 (m, 1H), 5.05–5.02 (m, 3H), 4.97–4.87 (m, 3H), 4.48 (d, J = 8.0 Hz, 1H), 4.31–4.28 (m, 2H), 4.21–4.17 (m, 4H), 4.02 (dd, J = 12.5, 2.5 Hz, 1H), 3.86 (t, J = 9.5 Hz, 1H), 3.68–3.65 (m,1H), 2.12 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.02 (s, 3H); 13C NMR δ: 195.1, 171.0, 170.8, 169.5, 169.4, 169.1, 153.6, 131.8, 129.8 (2C), 126.9, 125.1, 122.1 (2C), 101.7, 101.4, 80.4, 78.8, 77.0, 72.9, 72.1, 72.0, 71.9, 69.5, 69.2, 68.6, 66.8, 62.4, 62.2, 56.8, 21.1, 21.1, 21.0, 20.9 (2C), 20.7; ESIHRMS calcd for C36H46O19SNa [M+Na]+: 837.2252, found: 837.2239.
Methyl 2,2′,4,4′,6,6′-hexa-O-acetyl-3′-O-[2-phenyloxycarbonylthioxy]but-3-enyl]-β-D-laminaribioside (93)
Following the general procedure 2, and eluting with 70% EtOAc/hexanes the title product was obtained as an approximately 1:1 mixture of stereoisomers in 90% yield. 1H NMR δ: 7.39–7.36 (m, 2H), 7.24 (t, J = 7.5 Hz, 1H), 7.16–7.15 (m, 1H), 7.14–7.13 (m, 1H), 5.19 (dd, J = 10.0, 3.5 Hz, 1H), 4.10–5.05 (m, 1H), 4.99–4.91 (m, 3H), 4.49 (d, J = 8.5 Hz, 1H), 4.33–4.29 (m, 2H), 4.19–4.18 (m, 2H), 4.05–4.01 (m, 2H), 3.86 (t, J = 9.5 Hz, 1H), 3.82–3.77 (m, 2H), 3.47 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H), 2.09 (s, 3H × 3), 2.08 (s, 3H × 2), 2.08 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H); 13C NMR δ: 171.0, 171.9, 169.5, 169.4 (2C), 169.1, 151.3, 133.9, 133.8, 129.7, 126.5, 121.5, 118.8, 118.7, 101.7, 101.4, 81.0, 80.8, 78.8, 73.5, 73.4, 72.9, 72.1, 72.1, 71.9, 71.7, 69.1, 68.9, 68.6, 62.4, 62.2, 56.8, 48.9, 48.7, 21.2, 21.1, 21.0 (2C), 20.9, 20.9, 20.9 (2C), 20.7 (2C); ESIHRMS calcd for C36H46O19SNa [M+Na]+: 837.2252, found: 837.2224.
Methyl 3′-O-(2-pyridin-2-yldithio)but-3-enyl-β-D-laminaribioside (63)
Following the general procedure 3, and eluting with 10% MeOH/CH2Cl2 the title product was obtained in 68% yield as an approximately 1.1:1 mixture of stereoisomers. 1H NMR δ: 8.37–8.36 (m, 1H), 7.94–7.90 (m, 2H), 7.82–7.78 (m, 1H), 7.22–7.19 (m, 1H), 5.87–5.79 (m, 1H), 5.25–5.21 (m, 1H), 5.14–5.12 (m, 1H), 4.55 (dd, J = 7.5, 4.5 Hz, 1H), 4.23 (dd, J = 7.5, 1.5 Hz, 1H), 4.12 (dd, J = 10.5, 6.0 Hz, 1H, minor), 4.08 – 4.07 (m, 1H), 4.03 (dd, J = 10.0, 6.0 Hz, 1H, minor), 3.90–3.86 (m, 2H), 3.80 (dd, J = 15.0, 6.5 Hz, 1H), 3.70 (dd, J = 11.5, 5.5 Hz, 1H), 3.65 – 3.61 (m, 1H), 3.54 (s, 3H major + 3H minor), 3.43–3.33 (m, 7H), 3.23 (t, J = 8.5 Hz, 1H); 13C NMR δ: 160.7, 148.8, 137.8, 134.6, 134.5, 121.0, 120.3, 118.1, 104.1, 103.7, 86.9, 85.7, 85.6, 76.8, 76.4, 74.4, 74.3, 73.7, 73.2, 69.9, 69.8, 68.8, 61.4, 61.3, 56.2, 54.9, 54.8; ESIHRMS calcd for C22H33NO11S2Na [M+Na]+: 574.1393, found: 574.1385.
1-Thio-hepta-O-acetyl-β-D-laminaribiose (65)
A stirred solution of peracetyl laminaribiosyl bromide38 (698 mg, 1.0 mmol) in acetone/water (5.0 mL) was treated with thiourea (114 mg, 1.5 mmol) and heated to reflux for 4–6 h. After cooling to room temperature the solvents were evaporated and a stirred solution of the residue in CH2Cl2/water (10.0 mL) was treated with sodium metabisulfite (2.0 mmol) and heated to reflux under an atmosphere of N2 for 2–4 h. The reaction mixture was diluted with CH2Cl2 (10.0 mL) and washed with saturated aqueous NaHCO3 (10.0 mL). The combined organic portion was dried over Na2SO4 and evaporated to dryness. The crude product was purified by column chromatography over silica gel 70% (EtOAc/hexanes) to give the title product as colorless oil in 65% yield. [α]23D −18.2° (c 1); 1H NMR δ: 5.13 (t, J = 9.5 Hz, 1H), 5.06 (t, J = 9.5 Hz, 1H), 4.99–4.94 (m, 2H), 4.91–4.88 (m, 1H), 4.60 (d, J = 7.5 Hz, 1H), 4.42–4.36 (m, 2H), 4.19-4.12 ( m, 2H), 4.03 (dd, J = 10.5, 2.0 Hz, 1H), 3.85 (t, J = 9.5 Hz, 1H), 3.70–3.66 (m, 2H), 2.29 (d, J = 10.5 Hz, 1H), 2.17 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 2.01(s, 3H), 2.00 (s, 3H), 1.97 (s, 3H); 13C NMR δ: 170.9, 170.7, 170.6, 169.6, 169.5, 169.5, 169.4, 101.1, 80.1, 79.0, 76.6, 75.3, 73.2, 71.9, 71.3, 68.2, 68.1, 62.5, 61.8, 21.3, 21.0, 20.8, 20.7, 20.7, 20.6, 20.5; ESIHRMS calcd for C26H36O17SNa [M+Na]+: 675.1571, found: 675.1541.
Benzyl 2-O-benzoyl–4,6-O-benzylidene-2′,4′,6′-tri-O-acetyl-3′-acetylthio-β-D-laminaribioside (95)
Following the general procedure 12, benzyl 2-O-benzoyl–4,6-O-benzylidene-β-D-glucopyranoside (94) was glycosylated with the trichloroacetimidate 36. Purification was achieved by eluting from silica gel with 70% EtOAc/hexanes. The title product was obtained in 25% yield. [α]23D −47.7° (c 1); 1H NMR (400 MHz) δ: 7.99–7.97 (m, 2H), 7.64–7.60 (m, 1H), 7.51–7.45 (m, 4H), 7.35–7.33 (m, 3H), 7.21–7.16 (m, 1H), 7.14–7.12 (m, 4H), 5.57 (s, 1H), 5.35 (t, J = 10.0 Hz, 1H), 4.97 (t, J = 9.6 Hz, 1H), 4.90 (dd, J = 11.2, 7.2 Hz, 1H), 4.84 (d, J = 8.4 Hz, 1H), 4.64–4.58 (m, 3H), 4.38 (dd, J = 10.8, 4.8 Hz, 1H), 4.11–4.04 (m, 2H), 3.91–3.79 (m, 3H), 3.59 (t, J = 11.2 Hz, 1H), 3.52–3.46 (m, 2H), 2.21 (s, 3H), 1.94 (s, 3H), 1.92 (s, 3H), 1.68 (s, 3H); 13C NMR (100 MHz) δ: 193.7, 169.4, 165.6, 137.4, 136.8, 133.5, 130.2, 130.0, 129.9, 129.3, 128.7, 128.6, 128.4, 128.1, 128.0, 126.3, 119.2, 102.1, 101.5, 99.8, 79.3, 79.1, 77.6, 77.3, 76.9, 74.4, 73.6, 70.6, 69.9, 68.8, 67.8, 66.8, 62.3, 47.9, 30.7, 20.9, 20.7, 20.2; ESIHRMS calcd for C41H44O15SNa [M+Na]+: 831.2299, found: 831.2259.
Benzyl 2-O-benzoyl–2′,4′,6′-tri-O-acetyl-3′-acetylthio-β-D-laminaribioside (64)
Compound 95 (41 mg, 0.05 mmol) was stirred in 50% aqeous AcOH (1.0 mL) at 50 °C for 4–6 h. After the solvents were evaporated the residue was purified by column chromatography over silica gel eluting with 90% EtOAc/hexanes to afford the title product in 90% yield. [α]23D −38.7° (c 1); 1H NMR (400 MHz) δ: 7.99–7.97 (m, 2H), 7.64–7.61 (m, 1H), 7.51–7.47 (m, 2H), 7.19–7.14 (m, 1H), 7.12–7.11 (m, 4H), 5.25 (t, J = 11.0 Hz, 1H), 4.99–4.92 (m, 2H), 4.82–4.79 (m, 1H), 4.63–4.59 (m, 1H), 4.53 (d, J = 10.5 Hz, 2H), 4.17–4.08 (m, 2H), 3.99–3.94 (m, 1H), 3.83–3.75 (m, 4H), 3.73–3.71 (m, 1H), 3.67–3.62 (m, 2H), 3.41–3.36 (m, 1H), 2.23 (s, 3H), 2.07 (s, 3H X 2), 1.97 (s, 3H); 13C NMR δ: 193.4, 170.7, 169.4, 169.3, 165.1, 137.0, 133.6, 130.0, 129.8, 128.7, 128.5, 128.0, 127.9, 102.7, 99.7, 85.7, 75.8, 74.8, 72.5, 70.7, 69.9, 69.5, 67.8, 63.1, 62.3, 47.9, 30.7, 20.8, 20.6, 20.1; ESIHRMS calcd for C34H40O15SNa [M+Na]+: 743.1986, found: 743.1971.
Methyl 6-[4-O-(2,2′-azido-2,2′-dideoxy-β-D-gentiobiosyloxy)but-2E-enyl]thio-2,2′-azido-2,2′-dideoxy-α-D-gentiobioside (66)
Thiol precursor 56 (40 mg, 0.09 mmol) was dissolved in DMF (0.8 mL) at room temperature and hydrazine acetate (12 mg, 0.13 mmol) was added. The resulting mixture was stirred for 0.75 h and then directly transferred to a stirred solution of sulfenyl donor 53 (56 mg, 0.1 mmol) in MeOH (1 mL) at room temperature. The resulting mixture was stirred until TLC showed complete consumption of the thiol (18 h). Silver nitrate (26 mg, 0.20 mmol) was then added and the resulting mixture was stirred at room temperature, with exclusion of light, for an additional 36 h. The resulting mixture was filtered through a pad of Celite® and silica gel and concentrated. Chromatographic purification eluting with CH2Cl2/MeOH (15:1) afforded the title compound as a white foam in 52% yield: [α]RTD +65.8° (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ: 5.84-5.67 (m, 2H), 4.62 (s, 2H), 5.52-4.36 (m, 5H), 4.23 (dd, J = 9.5, 12.0 Hz, 5H), 3.87 (d, J = 12.0 Hz, 2H), 3.83-3.62 (m, 5H), 3.56 (s, 3H), 3.54-3.43 (m, 5H), 3.42-3.05 (m, 15H), 2.95 (d, J = 15.0 Hz, 1H), 2.63 (q, J = 7.0 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ: 130.4, 128.6, 102.9, 102.6, 100.6, 77.2, 76.8, 76.1, 75.9, 75.2, 75.1, 74.8, 72.9, 70.6, 70.5, 70.3, 69.1, 69.0, 68.9, 67.0, 66.9, 61.3, 56.2, 33.9, 31.4; ESIHRMS calcd for C29H46N12O17SNa [M+Na]+: 889.2722, found: 889.2736.
Methyl 6-[4-O-(2,2′-dideoxy-2,2′-(2,2,2-trichloroethoxycarbonylamino)-β-D-gentiobiosyloxy)but-2E-enyl]thio-2,2′-dideoxy-(2,2,2-trichloroethoxycarbonylamino-α-D-gentiobioside (67)
Thiol precursor 61 (30 mg, 0.04 mmol) was dissolved in DMF (0.5 mL) at room temperature and hydrazine acetate (12 mg, 0.13 mmol) was added. The resulting mixture was stirred for 1.25 h and directly transferred to a stirred solution of sulfenyl donor 62 (42 mg, 0.05 mmol) in MeOH (0.8 mL). The resulting mixture was stirred until TLC showed complete consumption of the thiol compound (20 h). Triphenylphosphine (13 mg, 0.05 mmol) was then added and the resulting mixture was stirred at room temperature for an additional 40 h before it was concentrated. Purification by silica gel chromatography CH2Cl2/MeOH (15:1) afforded the title compound as a white foam in 54% yield: [α]RTD +72.6° (c 1.0, MeOH); 1H NMR (400 MHz, CD3OD) δ: 5,85-5.58 (m, 2H), 4.92 (d, J = 4.1 Hz, 3H), 4.93-4.86 (m, 12H), 4.85 (d, J = 4.0 Hz, 2H), 4.80 (d, J = 12.1 Hz, 2H), 4.73 (dd, J = 9.7, 12.2, 3Hz), 4.62 (dd, J = 8.9, 11.3, 2Hz), 4.54-4.37 (m, 2H), 4.36-4.24 (m, 2H), 4.19 (d, J = 4.0 Hz, 2H), 4.13-4.01 (m, 1H), 3.89 (d, J = 11.3, 1H), 3.77-3.59 (m, 4H), 3.53-3.18 (m, 18H), 2.96 (d, J = 13.0 Hz, 1H), 2.63 (dd, J= 5.7, 8.1 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ: 153.8, 153.6, 130.4,128.7, 102.8, 102.5, 100.7, 78.2, 77.7, 76.2, 75.9, 75.8, 75.3, 75.2, 74.9, 72.5, 72.3, 70.7, 70.6, 70.4, 69.2, 69.0, 68.6, 67.5, 67.3, 61.5, 56.3, 33.9, 32.2; ESIHRMS calcd for C41H58Cl12N4O25SNa [M+Na]+: 1480.9271, found: 1480.9316.
Benzyl 3-deoxy-3-[4-(β-D-laminaribiosyloxy)but-2E-enylthio]-β-D-laminaribioside (68)
Following the general procedure 15, the title compound was obtained in 50% yield. [α]23D −16.0° (c 0.75, MeOH); 1H NMR (CD3OD) δ: 7.44 – 7.42 (m, 2H), 7.36–7.32 (m, 2H), 7.29–7.27 (m, 1H), 5.88–5.83 (m, 1H), 5.76–5.71 (m, 1H), 4.94 (d, J = 12.0 Hz, 1H), 4.69 (d, J = 8.5 Hz, 1H), 4.58 (t, J = 8.0 Hz, 1H), 4.47–4.41 (m, 2H), 4.31 (dd, J = 13.0, 5.0 Hz, 1H), 4.20 (dd, J = 13.0, 7.5 Hz, 1H), 3.93–3.87 (m, 14H), 3.74–3.26 (m, 12H), 2.59 (t, J = 10.0 Hz, 1H); 13C NMR (CD3OD) δ: 137.7, 131.2, 128.6, 128.2, 128.1, 127.6, 105.3, 104.2, 101.6, 100.7, 86.9, 86.8, 79.4, 76.9, 76.5, 76.4, 76.2, 74.4, 73.8, 73.4, 73.3, 72.6, 70.6, 70.4, 69.0, 68.9, 68.7, 68.4, 63.2, 61.7, 61.5, 61.4, 53.9, 33.3; ESIHRMS calcd for C35H54O21SNa [M+Na]+: 865.2776, found: 865.2768.
Methyl 3-O-[4-(1-thio-β-D-laminaribiosyl)but-2E-enyl]-β-D-laminaribioside (69)
Following the general procedure 15, the title compound was obtained in 55% yield. [α]23D −15.0° (c 0.75, MeOH); 1H NMR (CD3OD):δ 5.79–5.77 (m, 2H), 4.57 (d, J = 7.5, 4.0 Hz, 2H), 4.46 (d, J = 9.5 Hz, 1H), 4.36–4.35 (m, 2H), 4.24 (d, J = 7.5 Hz, 1H), 3.89 (dd, J = 7.0, 2.0 Hz, 4H), 3.72–3.67 (m, 2H), 3.66–3.62 (m, 3H), 3.58–3.53 (m, 2H), 3.54 (s, 3H), 3.52–3.47 (m, 2H), 3.45–3.21 (m, 13H); 13C NMR (CD3OD) δ: 130.3, 129.1, 104.1, 104.0, 103.7, 88.3, 86.9, 83.6, 83.3, 80.2, 76.9, 76.8, 76.6, 76.4, 74.6, 74.4, 73.2, 72.5, 72.4, 70.4, 70.0, 69.0, 68.9, 61.7, 61.4, 56.1, 30.8; ESIHRMS calcd for C29H50O21SNa [M+Na]+: 789.2463, found: 789.2451.
4-(1-Thio-β-D-laminaribiosyl)but-2E-enyl β-D-laminaribioside (70)
Following the general procedure 15, the title compound was obtained in 55% yield. [α]23D −11.0° (c 0.75, MeOH); 1H NMR (CD3OD) δ: 5.86–5.80 (m, 1H), 5.77–5.71 (m, 1H), 4.59 (d, J = 11.0, 7.5 Hz, 2H), 4.44–4.41 (m, 2H), 4.33 (dd, J = 12.5, 4.5 Hz, 1H), 4.20 (dd, J = 12.0, 7.5 Hz, 1H), 3.91–3.89 (m, 4H), 3.75–3.62 (m, 5H), 3.59–3.55 (m, 2H), 3.53–3.18 (m, 15H); 13C NMR (CD3OD) δ: 130.2, 128.8, 104.1 (2C), 103.9, 100.9, 88.6, 87.3, 83.1, 80.2, 76.9, 76.8, 76.5, 76.4, 74.4, 74.4, 73.2, 72.4, 70.4, 69.0, 68.9, 66.5, 61.7, 61.5, 30.6; ESIHRMS calcd for C28H48O21SNa [M+Na]+: 775.2306, found: 775.2328.
Supplementary Material
Scheme 10.

Preparation of 2-Deoxy-2-(trichloroethoxycarbonylamino)glucose-Based Monosaccharyl Units
Scheme 11.

Preparation of the N-Troc Glucosamine–Based Disaccharyl Sulfenyl Donor 62
Scheme 12.

Preparation of the Laminaribiosyl 3′-Sulfenyl Donor 63
Scheme 13.

Preparation of the 1-Deoxy-mercaptolaminaribiose Precursor 65
Acknowledgments
We thank the NIH (GM62160) for support of this work.
Footnotes
Supporting Information Available. Full experimental details and copies of the 1H and 13C NMR spectra of all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Kent SBH. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 2.(a) Brown GD, Gordon S. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]; (b) Palma AS, Feizi T, Zhang Y, Stoll MS, Lawson AM, Díaz-Rodríguez E, Campanero-Rhodes MA, Costa J, Gordon S, Brown GD, Chai W. J Biol Chem. 2006;281:5771–5779. doi: 10.1074/jbc.M511461200. [DOI] [PubMed] [Google Scholar]; (c) Adams EL, Rice PJ, Graves B, Ensley HE, Yu H, Brown GD, Gordon S, Monteiro MA, Papp-Szabo E, Lowman DW, Power TD, Wempe MF, Williams DL. J Pharmacol Expt Ther. 2008;325:115–123. doi: 10.1124/jpet.107.133124. [DOI] [PubMed] [Google Scholar]
- 3.Jamois F, Ferrières V, Guégan JP, Yvin JC, Plusquellec D, Vetvicka V. Glycobiology. 2005;15:393–407. doi: 10.1093/glycob/cwi020. [DOI] [PubMed] [Google Scholar]
- 4.Descroix K, Ferrieres V, Jamois F, Yvin JC, Plusquellec D. Minirev Med Chem. 2006;6:1341–1349. doi: 10.2174/138955706778993058. [DOI] [PubMed] [Google Scholar]
- 5.(a) Zeng Y, Ning J, Kong F. Tetrahedron Lett. 2002;43:3729–3733. [Google Scholar]; (b) Zeng Y, Ning J, Kong F. Carbohydr Res. 2003;338:307–311. doi: 10.1016/s0008-6215(02)00455-x. [DOI] [PubMed] [Google Scholar]
- 6.Mo KF, Li H, Mague JT, Ensley HE. Carbohydr Res. 2009;344:439–447. doi: 10.1016/j.carres.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 7.Sylla B, Descroix K, Pain C, Gervaise C, Jamois F, Yvin JC, Legentil L, Nugier-Chauvin C, Daniellou R, Ferrieres V. Carbohydr Res. 2010;345:1366–1370. doi: 10.1016/j.carres.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 8.(a) Humbel BM, Konomi M, Takagi T, Kamasawa N, Ishijima SA, Osumi M. Yeast. 2001;18:433–444. doi: 10.1002/yea.694. [DOI] [PubMed] [Google Scholar]; (b) Kollar R, Reinhold B, Petrakova E, Yeh HJ, Ashwell G, Drgonova J, Kapteyn JC, Klis FM, Cabib E. J Biol Chem. 1997;272:17762–17775. doi: 10.1074/jbc.272.28.17762. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cheon HS, Lian Y, Kishi Y. Org Lett. 2007;9:3323–3326. doi: 10.1021/ol0713335. [DOI] [PubMed] [Google Scholar]; (b) Meppen M, Wang Y, Cheon HS, Kishi Y. J Org Chem. 2007;72:1941–1950. doi: 10.1021/jo061990v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hsu MC, Lee J, Kishi Y. J Org Chem. 2007;72:1931–1940. doi: 10.1021/jo061991n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itoh Y, Rice JD, Goller C, Pannuri A, Taylor J, Meisner J, Beveridge TJ, Preston JF, Romeo T. J Bacteriology. 2008;190:3670–3680. doi: 10.1128/JB.01920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerca N, Maira-Litran T, Jefferson KK, Grout M, Goldmann DA, Pier GB. Proc Natl Acad Sci USA. 2007;104:7528–7533. doi: 10.1073/pnas.0700630104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Gening ML, Tsvetkov YE, Pierb GB, Nifantieva NE. Carbohydr Res. 2007;342:567–575. doi: 10.1016/j.carres.2006.08.010. [DOI] [PubMed] [Google Scholar]; (b) Melean LG, Love KR, Seeberger PH. Carbohydr Res. 2002;337:1893–1916. doi: 10.1016/s0008-6215(02)00299-9. [DOI] [PubMed] [Google Scholar]; (c) Fridman M, Solomon D, Yogev S, Baasov T. Org Lett. 2002;4:281–283. doi: 10.1021/ol017054d. [DOI] [PubMed] [Google Scholar]; (d) Yang F, Du Y. Carbohydr Res. 2003;338:495–502. doi: 10.1016/s0008-6215(02)00494-9. [DOI] [PubMed] [Google Scholar]; (e) Yang F, He H, Du Y. Tetrahedron Lett. 2002;43:7561–7563. [Google Scholar]
- 13.Petitou M, van Boeckel CAA. Angew Chem Int Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 14.Boons GJ. Tetrahedron. 1996;52:1095–1121. [Google Scholar]
- 15.(a) Fraser-Reid B, Lu J, Jayaprakash KN, Lopez JC. Tetrahedron: Asymmetry. 2006;17:2449–2463. [Google Scholar]; (b) Joe M, Bai Y, Nacario RC, Lowary TL. J Am Chem Soc. 2007;129:9885–9901. doi: 10.1021/ja072892+. [DOI] [PubMed] [Google Scholar]
- 16.(a) Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; (c) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (d) Meldal M, Tornøe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 17.(a) Dedola S, ANS, Field RA. Org Biomol Chem. 2007:1006–1017. doi: 10.1039/b618048p. [DOI] [PubMed] [Google Scholar]; (b) Dondoni A. Chem Asian J. 2007;2:700–708. doi: 10.1002/asia.200700015. [DOI] [PubMed] [Google Scholar]; (c) Nicotra F, Cipolla L, Peri F, La Ferta B, Redaelli C. Adv Carbohydr Chem Biochem. 2007;61:353–398. doi: 10.1016/S0065-2318(07)61007-5. [DOI] [PubMed] [Google Scholar]; (d) Gyorgydeak Z, Thiem J. Adv Carbohydr Chem Biochem. 2006;60:103–182. doi: 10.1016/S0065-2318(06)60004-8. [DOI] [PubMed] [Google Scholar]; (e) Pieters RJ, Rijkers DTS, Liskamp RMJ. QSAR Comb Sci. 2007;26:1181–1190. [Google Scholar]; (f) Santoyo-Gonzalez F, Hernandez-Mateo F. Top Heterocycl Chem. 2007;7:133–177. [Google Scholar]; (g) Hanson SR, Greenberg WA, Wong CH. QSAR Comb Sci. 2007;26:1243–1252. [Google Scholar]; (h) Wilkinson BL, Bornaghi LF, Houston TA, Poulsen S-A. In: Drug Des Res Perspectives. Kaplan SP, editor. Nova Science Publishers; Hauppauge: 2007. pp. 57–102. [Google Scholar]
- 18.(a) Dondoni A. Angew Chem Int Ed. 2008;47:8995–8997. doi: 10.1002/anie.200802516. [DOI] [PubMed] [Google Scholar]; (b) van Dijk M, Rijkers DTS, Liskamp RMJ, van Nostrum CF, Hennink WE. Bioconjugate Chem. 2009;20:2001–2016. doi: 10.1021/bc900087a. [DOI] [PubMed] [Google Scholar]; (c) Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 19.(a) Shiu HY, Chan TC, Ho CM, Liu Y, Wong MK, Che CM. Chem Eur J. 2009;15:3839–3850. doi: 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]; (b) Floyd N, Vijayakrishnan B, Koeppe JR, Davis BG. Angew Chem Int Ed. 2009:48. doi: 10.1002/anie.200903135. [DOI] [PubMed] [Google Scholar]; (c) Lo Conte M, Pacifico S, Chambery A, Marra A, Dondoni A. J Org Chem. 2010;75:4644–4647. doi: 10.1021/jo1008178. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (b) Appendino G, Bacchiega S, Minassi A, Cascio MG, De Petrocellis L, Di Marzo V. Angew Chem Int Ed. 2007;46:9312–9315. doi: 10.1002/anie.200703590. [DOI] [PubMed] [Google Scholar]; (c) Bock VD, Hiemstra H, van Maarseveen JH. Eur J Org Chem. 2006:51–68. [Google Scholar]
- 21.(a) Crich D, Krishnamurthy V, Hutton TK. J Am Chem Soc. 2006;128:2544–2545. doi: 10.1021/ja057521c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crich D, Brebion F, Krishnamurthy V. Org Lett. 2006;8:3593–3596. doi: 10.1021/ol061381+. [DOI] [PubMed] [Google Scholar]; (c) Crich D, Krishnamurthy V, Brebion F, Karatholuvhu M, Subramanian V, Hutton TK. J Am Chem Soc. 2007;129:10282–10294. doi: 10.1021/ja072969u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang D, Devarie-Baez NO, Pan J, Wang H, Xian M. Org Lett. 2010;12:5674–5676. doi: 10.1021/ol102491n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li Z, Wang C, Fu Y, Guo QX, Liu L. J Org Chem. 2008;73:6127–6136. doi: 10.1021/jo800747g. [DOI] [PubMed] [Google Scholar]
- 22.(a) Crich D, Zou Y, Brebion F. J Org Chem. 2006;71:9172–9177. doi: 10.1021/jo061439y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crich D, Yang F. J Org Chem. 2008;73:7017–7027. doi: 10.1021/jo8015314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalker JM, Lin YA, Boutureira O, Davis BG. Chem Commun. 2009:3714–3716. doi: 10.1039/b908004j. [DOI] [PubMed] [Google Scholar]
- 24.(a) Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1996. [Google Scholar]; (b) Lundblad RL. Chemical Reagents for Protein Modification. 3. CRC Press; Boca Raton: 2005. [Google Scholar]
- 25.Crich D, Subramanaian V, Karatholuvhu M. J Org Chem. 2009;74:9422–9427. doi: 10.1021/jo902012m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Overman LE, Roberts SW, Sneddon HF. Org Lett. 2008;10:1485–1488. doi: 10.1021/ol8002942. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gais HJ, Böhme A. J Org Chem. 2002;67:1153–1161. doi: 10.1021/jo010668b. [DOI] [PubMed] [Google Scholar]
- 27.Thompson C, Ge M, Kahne D. J Am Chem Soc. 1999;121:1237–1244. [Google Scholar]
- 28.Nakabayashi S, Warren CD, Jeanloz RN. Carbohydr Res. 1986;150:C7–C10. doi: 10.1016/0008-6215(86)80028-3. [DOI] [PubMed] [Google Scholar]
- 29.(a) Schmidt RR. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Dekker; New York: 1997. pp. 283–312. [Google Scholar]; (b) Schmidt RR, Jung K-H. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 5–59. [Google Scholar]
- 30.Trimnell D, Stout EI, Doane WM, Russell CR, Beringer V, Saul M, Van Gessel G. J Org Chem. 1975;40:1337–1339. [Google Scholar]
- 31.Volante RP. Tetrahedron Lett. 1981;22:3119–3122. [Google Scholar]
- 32.Stick RV, Stubbs KA. Tetrahedron: Asymmetry. 2005;16:321–335. [Google Scholar]
- 33.Cumpstey I, Ramstadius C, Akhtar T, Goldstein IJ, Winter HC. Eur J Org Chem. 2010:1951–1970. [Google Scholar]
- 34.(a) Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]; (b) Tsuda T, Nakamura S, Hashimoto S. Tetrahedron. 2004;60:10711–10737. [Google Scholar]
- 35.(a) Kahne D, Walker S, Cheng Y, Engen DV. J Am Chem Soc. 1989;111:6881–6882. [Google Scholar]; (b) Crich D, Lim LBL. Org React. 2004;64:115–251. [Google Scholar]; (c) Aversa MC, Barattucci A, Bonaccorsi P. Tetrahedron. 2008;64:7659–7683. [Google Scholar]
- 36.Sliedregt LAJM, van der Marel GA, van Boom JH. Tetrahedron Lett. 1994;35:4015–4018. [Google Scholar]
- 37.(a) Hense A, Ley SV, Osborn H, Owen DR, Poisson J-F, Warriner SL, Wesson KE. J Chem Soc, Perkin Trans. 1997;1:2023–2031. [Google Scholar]; (b) Montchamp JL, Tian F, Hart ME, Frost JW. J Org Chem. 1996;61:3897–3900. doi: 10.1021/jo960170n. [DOI] [PubMed] [Google Scholar]
- 38.Takeo K. Carbohydr Res. 1979;77:245–251. [Google Scholar]
- 39.Nicolaou KC, Mitchell HJ, Rodriguez RM, Fylaktakidou KC, Suzuki H, Conley SR. Chem Eur J. 2000;6:3149–3165. doi: 10.1002/1521-3765(20000901)6:17<3149::aid-chem3149>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 40.Demko ZP, Sharpless KB. Org Lett. 2001;3:4091–4094. doi: 10.1021/ol010220x. [DOI] [PubMed] [Google Scholar]
- 41.Whitworth GE, Macauley MS, Stubbs KA, Dennis RJ, Taylor EJ, Davies GJ, Greig IR, Vocadlo DJ. J Am Chem Soc. 2007;129:635–644. doi: 10.1021/ja065697o. [DOI] [PubMed] [Google Scholar]
- 42.(a) Buskasa T, Garegg PJ, Konradsson P, Maloisel JL. Tetrahedron: Asymmetry. 1994;5:2187–2194. [Google Scholar]; (b) Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 43.Boulineau FP, Wei A. Carbohydr Res. 2001;334:271–279. doi: 10.1016/s0008-6215(01)00203-8. [DOI] [PubMed] [Google Scholar]
- 44.Dullenkopf W, Castro-Palomino JC, Manzoni L, Schmidt RR. Carbohydr Res. 1996;296:135–147. doi: 10.1016/s0008-6215(96)00237-6. [DOI] [PubMed] [Google Scholar]
- 45.Higashi K, Nakayama K, Uoto K, Shioya E, Kusama T. Chem Pharm Bull. 1991;39:590–592. doi: 10.1248/cpb.39.3244. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Magnusson G. Carbohydr Res. 1996;295:41–55. doi: 10.1016/s0008-6215(96)90118-4. [DOI] [PubMed] [Google Scholar]
- 47.Yan F, Mehta S, Eichler E, Wakarchuk WW, Gilbert M, Schur MJ, Whitfield DM. J Org Chem. 2003;68:2426–2431. doi: 10.1021/jo026569v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.