

Summary
The transition of ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) is a critical step in breast cancer progression. We introduce a simple microfluidic 3D compartmentalized system in which mammary epithelial cells (MCF-DCIS) are co-cultured with human mammary fibroblasts (HMFs), which promotes a transition from DCIS to IDC in vitro. The model enables control of both spatial (distance-dependence) and temporal (transition from larger clusters) aspects within the microenvironment, allowing recapitulation of the in vivo environment in ways not practical with existing experimental models. When HMFs were cultured some distance (0.5-1.5 mm) from the MCF-DCIS cells, we observed an initial morphological change, suggesting soluble factors can begin the transition. However, cell-cell contact with HMFs allowed the MCF-DCIS cells to complete the transition to invasion. Uniquely, the compartmentalized platform enables the analysis of the intrinsic second harmonic generation signal of collagen, providing a label-free quantitative analysis of DCIS-associated collagen remodeling. The arrayed microchannel-based model is compatible with existing infrastructure and, for the first time, provides a cost effective approach to test for inhibitors of pathways involved in DCIS progression to IDC allowing a screening approach to the identification of potential therapeutic targets. Importantly, the model can be easily adapted and generalized to a variety of cell-cell signaling studies.
Introduction
Cancer kills when the primary tumor becomes invasive and metastasis ensues. While Paget first hypothesized that the appropriate microenvironment is essential for metastatic spread to distant sites over 100 years ago, surprisingly little is known about the molecular mechanisms involved in tumor progression1. A critical step in cancer progression is from non-invasive to invasive – in breast cancer, this is known as the transition from Ductal Carcinoma In Situ (DCIS) to Invasive Ductal Carcinoma (IDC)2-5. DCIS is observed clinically and has been recapitulated in mouse models, yet we lack an in vitro model that would allow screening for inhibitors and further our ability to elucidate the underlying molecular mechanisms that govern the progression to IDC. Prior xenograft models have shown that MCFDCIS.com (MCF-DCIS) cells form lesions that resemble comedo-type DCIS and which eventually become invasive. This invasion is accelerated, when various stromal fibroblasts are co-inoculated2, 3, 6, 7. As well, there are a myriad of molecules (e.g. growth factors, hormones, cytokines, extracellular matrix (ECM) molecules, and matrix metalloproteinases) which may play essential roles in DCIS progression2-5, 8, 9. Thus, there is a clear need for a reliable and efficient in vitro culture model to enable higher throughput exploration of this critical step in cancer progression.
Existing 3-dimensional (3D) in vitro models, including a DCIS to IDC progression model, have employed mixed co-cultures of carcinoma cells with stromal fibroblasts in 3D gels (e.g. collagen I and matrigel)3, 10. In these models morphological change and marker analysis serve as primary readouts. These models are well-established and employed to investigate the paracrine signaling effect between two different cell types in cancer research. However, existing models require a large amount of cells and reagents (e.g., typically performed in 48 well plates) and possess limited functionality (e.g., limited manipulation of the microenvironment). Miniaturization technology, in particular microfluidics, has shown promise in overcoming these limitations. While most microfluidic cell culture has focused on 2D applications, a few microfluidic 3D culture systems have been developed. For example, in addition to a simple mixed co-culture method, compartmentalized or layered co-culture platforms have been demonstrated by various research groups in attempts to create more in vivo-like phenotypes and analyze cell migration11-14. While work to date has shown the ability of microfluidics to use smaller cell numbers, less reagent and to provide additional microenvironment control, they have not found widespread use in biology research. In part, the lack of adoption may be due to the requirement for tube connections and specialized equipment (e.g. syringe pumps) and their incompatibility with existing equipment (e.g. plate readers). Thus, the attributes of current in vitro models and current microfluidic systems present both a need and an opportunity for improved 3D co-culture models.
Here we report a simple microfluidic 3D compartmentalized co-culture system that supports the DCIS to IDC transition in vitro and enables information-rich in vitro assays. The platform uses two input and one output ports, and sample loading and fluid changes are accomplished using surface-tension driven pumping (a.k.a. passive pumping15). Passive pumping is compatible with automated liquid handlers enabling hands-free high throughput operation16, 17. Laminar flow patterning allows the loading of MCF-DCIS cells adjacent to stromal fibroblasts recapitulating the general spatial relationship observed in vivo5,6. The compartmentalization allows examination of distance-dependent effects (e.g., invasion at the interface vs. at a distance) along with paracrine signaling effects. Furthermore, the two cell types can be loaded sequentially at different time points in the same culture platform (e.g., 6 days monoculture of MCF-DCIS cells in one compartment, followed by filling another compartment with stromal fibroblasts). Temporally separated loading allows the formation of larger DCIS clusters more closely mimicking in vivo DCIS clusters in size. As primary readouts, we examined cell morphology (circularity(Circ), roundness(Round), and aspect ratio(AR)), Collagen IV localization and E-cadherin expression and compared them to in vivo data. Due to the complexity of cluster morphology, three different shape descriptors were used to quantify the observations. In addition to circularity and aspect ratio of clusters, roundness was also measured to define the degree of curvature. The roundness represents the degree of sharp corners and projections. Protrusive filamentous actin (F-actin) structure is considered to be a good indicator for invasive transition and was examined using phalloidin staining20,21. An intriguing benefit of compartmentalized coculture is the ability to visualize collagen structure (through second harmonic generation (SHG) imaging18) that is reconstructed by MCF-DCIS cells only, not by fibroblasts, as each compartment can be imaged separately. This imaging process provided us a unique quantitative readout for DCIS transition to IDC because invasive clusters altered collagen structure considerably compared to non-invasive clusters. In cancer and many other diseases, paracrine signaling is implicated in disease pathogenesis, yet our ability to study these effects in vitro are limited (e.g. transwells, conditioned media) typically requiring large samples and are not amendable to automation particularly in 3D culture. The in vitro system presented can serve as a platform to efficiently elucidate such pathways. In this example, we focus on the progression from DCIS to IDC in breast cancer, but the method can be easily adapted and generalized to a variety of signaling studies.
Results
Design of Channel and Matrix Composition
We developed a compartmentalized, 3D microscale culture system to enable the recapitulation of aspects of the in vivo environment and to provide a more information rich assay for investigating the cancer progression in vitro. Microscale systems provide practical benefits, including greatly reduced number of cells required and automated arrayed operation. Of equal or greater importance, compartmentalization enabled the seeding of epithelial cells next to stromal fibroblasts and the investigation of distance-dependent paracrine interaction (e.g., more interaction at the interface) - functionality not available in traditional co-culture systems. Microchannels with two inputs/one output, as shown in Fig. 1, were prepared and loaded with MCF-DCIS cells (the invasiveness was confirmed by traditional invasion assay [Fig. S1]3) in one compartment and with human mammary fibroblasts (HMF) in the other compartment by pumping through two different inlet ports simultaneously. The microchannels can also be arrayed (Fig. 1b) to be used in screening experiments such as neutralizing antibodies and inhibitors.
Fig 1.

3D compartmentalization and the invasive transition of MCF-DCIS cells. (a) Passive pumping allows for the loading and compartmentalization of 3D cultures. Drops of cell containing polymer solutions are loaded onto the inlet ports. Laminar flow leads to two side-by-side 3D compartments. (b) Yellow and green food coloring solutions are pumped into Y-shaped PDMS microchannels to demonstrate compartmentalization by passive pumping. Scale bar represents 6mm. Y-shaped channels are arrayed (4×3 array). Scale bar is 2cm. (c) The invasive transition of MCF-DCIS cells in compartmentalized culture is observed at interface. Cells in control side retained noninvasive morphology. These cells were imaged after 12 days of culture. Scale bar represents 30μm. (d) Representative morphology of MCF-DCIS cells after 7 days culture with a spacer gel of specific width between MCF-DCIS and HMF compartments. Scale bar represents 150μm. Bar graph presents the values from shape descriptor analysis, showing decreasing circularity and roundness and increasing aspect ratio as gap distance decreases. Error bars represent standard error. *: p<0.005 compared with the control group. **: p<0.0005 compared with gap ‘0’ group.
For 3D cultures, the physical properties and composition of the ECM are crucial factors. Thus, determining the proper ECM composition that supports the transition to invasive MCF-DCIS cells was our first focus. Three different matrix conditions (matrigel, 1.3 mg/ml collagen I, 1:1 mixture of matrigel and collagen I with final collagen I concentration of 0.8 or 1.3 mg/ml (mixed gel)) were initially selected based on previous studies. Experiments revealed that while matrigel was well suited to maintain cohesive MCF-DCIS clusters in 3D cultures, the matrigel was not sufficient to maintain HMF cells' differentiation and survival, or to support the invasive transition (as defined via established markers of invasion described in the next section) of MCF-DCIS cells, even in co-cultures with HMF cells. Furthermore, MCF-DCIS cells in collagen I alone did not grow in 3D clusters (Fig. S2). The 1:1 mixed gel did support both cell types and the invasive transition (Fig. 1). Similarly, Seton-Rogers et al did not observe invasive activity of MCF10A cells in response to ErbB2 and TGFβ stimulation in Matrigel, suggesting that the presence of collagen I is critical for the invasive activity19. Two different final concentrations of collagen I in mixed gels were evaluated, and MCF-DCIS cells in lower concentration gel (0.8 mg/ml) showed more obvious transitions determined by longer and more extended morphology (Circ: 0.12±0.1, Round: 0.40±0.14, AR: 2.94±1.45) than the higher concentration gel (1.3 mg/ml) (slightly elongated and shorter; Circ: 0.40±0.17, Round: 0.67±0.16, AR: 1.58±0.49). These experiments were done both in macro (48 well) and micro systems with similar results. Thus, 1:1 mixed gels with a 0.8 mg/ml collagen concentration were used for all subsequent experiments.
Loading Process and Culture
In traditional mixed co-cultures, two cell types are mixed together and cultured in a single compartment, making it challenging to separate out characteristics of one cell type or another. For example, it is difficult to image one cell type or the collagen production/remodeling due to one cell type because of the presence of the other cell type. Compartmentalized co-culture systems facilitate observation of one cell type independently and also allow for new observations, such as distance-dependent effects. Each cell type was prepared in a mixed matrix and co-injected into the `Y' shaped channel to form separate but adjacent compartments and cultured for longer than 9 days. Interestingly, MCF-DCIS cells near the interface showed invasive growth (Circ: 0.21±0.11, Round: 0.54±0.22, AR: 2.29±1.23), while the cells upstream, before the two channels join (>1.5 mm from the interface), retained rounded cluster morphology (Circ: 0.47±0.17, Round: 0.79±0.13, AR: 1.3±0.29) providing a built-in monoculture control (Fig. 1c). This distance dependency was further distinguished by inserting a blank spacer gel (same ECM composition) of specific width (i.e., 0, 0.5, 0.75, and 1.5 mm) between the two cell-containing compartments (Fig. 1d). These configurations were achieved by preparing microchannels of three or four input ports (Fig. S3). The spacer gel minimized direct contact of MCF-DCIS and HMF cells, but still allowed diffusion of molecules. After 7 days of culture, morphological differences between control (mono-culture), contact co-culture (no spacer) and co-culture with spacer gel were observed.. Specifically, based on a shape descriptor (Circ, Round, and AR) analysis shown in a bar graph in Fig. 1d, a clear invasive transition was observed in contact co-culture, a short protrusive morphology was observed when a gel spacer separated the two cell types and maintenance of rounded clusters was observed in mono control (either at sufficient distance from the fibroblast compartment or in a separate channel). In order to examine any possible microfluidic system specific influence, transwell (liquid spacing) and layered gel (gel spacing) co-cultures were also performed and similar trends were observed (Fig. S4).
In addition to the co-injected culture, the MCF-DCIS and HMF cells were loaded sequentially at different time points (e.g., 6 days' culture of MCF-DCIS cells alone in one compartment, followed by filling the other compartment with HMFs). This results in larger DCIS clusters before adding HMFs, more closely mimicking the in vivo DCIS clusters (Fig 2b). After the addition of fibroblasts, the DCIS clusters again transitioned to an invasive morphology, more widely spread (Circ: 0.14±0.05, Round: 0.34±0.13, AR: 3.33±1.08) than the transition that occurred in co-injected culture (Fig. 2c, 2d). However, the clusters in channel upstream still retained rounded morphology, confirming the distance-dependent transition. Thus, the control of both the spatial and temporal parameters of the two cell compartments results in an in vitro model that more closely mimics the DCIS progression in vivo than other existing in vitro models3,6.
Fig 2.

Co-injection and Sequential injection processes. (a) MCF-DCIS cells (left) and HMF cells (right) are co-injected and compartmentalized. The inset shows MCF-DCIS cells imaged 4 hours after loading, showing the size of cells from which the transition process was initiated. (b) The aqueous compartment filled with culture media (right) is created next to the MCF-DCIS cells containing gel compartment, which is replaced by HMF cells containing gel after 6 days' mono-culture of MCF-DCIS cells. The inset shows MCF-DCIS clusters cultured for 6 days, showing the size of cell clusters from which the transition process was initiated. Scale bars in A and B represent 0.5mm, and the ones in insets are 30μm. The invasive transition of MCF-DCIS cells in co-injected culture model (c) and sequentially injected culture model (d), showing more widely spread invasive transition in the sequentially injected culture model after a total of 10 days' culture (6 days' mono-culture, followed by 4 days' co-culture). Co-injected cells were cultured together for 10 days. Scale bars in c and d represent 30μm.
In order to validate that the transition we observed was invasive and not branching morphogenesis, we examined several established markers of invasion. It has been reported that DCIS invasive progression was accompanied by a loss of the basement membrane constituent collagen IV, a change in cell clusters' shape, and a loss of epithelial cells' adhesion2, 6. Thus, we examined the localization of collagen IV and the loss of E-cadherin in addition to morphology identification for both co-injected and sequentially-injected cultures. As can be seen in Fig. 3a, the control clusters (noninvasive) showed deposition of collagen IV at the periphery of clusters and E-cadherin at cell-cell junctions reliably, while co-cultured clusters (invasive) showed the breakdown of collagen IV and the partial loss of E-cadherin. Similar to morphology differences, the invasive clusters in sequentially-injected co-culture showed more substantial decrease of both collagen IV and E-cadherin expression than the clusters in co-injected culture (loss of expression observed mainly at the leading edge of invasion). F-actin structures have served as indicators of cancer cell invasion (more protrusive in invading clusters)20, 21. Phalloidin staining of the MCF-DCIS clusters revealed a F-actin rich protrusive structure (approximately 2.9 times higher than mono culture at the leading edges) (Fig. 3b). These endpoints all suggest a transition to an invasive phenotype similar to in vivo.
Fig 3.



Invasion marker validation for in vitro and in vivo models. (a) In vitro model validation. Co-injected cells were cultured for 10 days and fixed, and Sequentially injected cells were cultured a total of 10 days (6 days mono-culture and 4 days co-culture). Cell nuclei (blue) are labeled with TO-PRO-3. Areas of dotted square shows cells losing collagen IV (green) and E-cadherin (red) expression. HMF were GFP labeled, and are shown in collagen IV image on the right side (marked with arrows). The control (Ctrl) cluster was cultured at the control side of co-injected culture and fixed after 10 days culture. Scale bar is 50 μm. (b) F-actin structure analysis.(i) F-actin structure of co-cultured MCF-DCIS cells for 8 days (6 days mono-culture and 2 days co-culture), showing highly condensed actin at the invading edge.. (ii) F-actin structure of MCFDCIS cells mono-cultured for 8 days. Scale bar represent 30 um. (c) In vivo model validation. H & E staining showed that lesions resembling high-grade, comedo-type DCIS developed approximately 2 to 3 weeks after inoculation. Partial loss of collagen IV and E-cadherin observed in both in vivo and in vitro models mark transition to invasive phenotype. Scale bar is 200 μm.
To further validate the in vitro model, we have also used the MCF-DCIS model in vivo as described by Miller6, 7 (Fig. 3c). H & E staining showed that lesions resembling high-grade, comedo-type DCIS developed approximately 2 to 3 weeks after inoculation. After 3 to 5 weeks, the epithelial-stromal interface broke down, indicating invasion. Immunofluorescent staining showed that the comedo-type DCIS lesions were surrounded with Col IV positive cell layers and positive E-cadherin staining, which broke down when lesions became invasive, consistent with the in vitro model.
Second harmonic signal quantification
Increasing evidence has shown that the fibrillar collagen network in tumor and normal tissues are different due to the reconstruction of ECM architecture during invasive progression22-24. SHG is a nonlinear optical method where the emitted light has exactly half the wavelength of the two incident photons18,25. Because collagen is one of the strongest harmonophores, SHG has been widely used to image collagen and capture intrinsic characteristics of collagen networks12, 26-28. Several research groups have begun to utilize the SHG properties of collagen in tissue samples as a potential cancer diagnostic parameter. For example, Provenzano et al defined changes in collagen orientation that accompany tumor progression29, and Han et al employed SHG intensity information in an attempt to reveal the overall morphology of tumor collagen fibrils and provide a diagnostic ability to detect malignancy (via scoring of morphology or of average intensity)24.
Here, we present the use of SHG intensity information to further define the invasive phenotype of the MCF-DCIS clusters in our co-culture model. It has been shown that cancer invasion accompanies collagen rearrangement by utilizing contractility events of cancer cells to reorganize ECM to provide contact guidance for 3D invasion/migration31. The SHG signal analysis methods used here provide a simple and straightforward means to quantify collagen rearrangement in 3D assays that can be automated and scaled for improved throughput. Both fibroblasts and cancer cells are known to change collagen structure during culture; thus, in a mixed co-culture platform where cancer cells and fibroblasts are mixed in the same compartment, it is difficult to separate cancer cell versus fibroblast collagen remodeling (Fig. S5). Uniquely, our compartmentalized culture system makes possible imaging of collagen structures altered by MCF-DCIS cells alone because each cell type is in a separate compartment. Interestingly, we observed that fibroblast migration into the epithelial compartment was inhibited in the presence of DCIS cells (Fig. S6). GFP-transfected fibroblasts used in the work helped to validate the presence (or absence) of fibroblasts. In the DCIS compartment (in the absence of fibroblasts) two types of collagen rearrangement were observed (Fig. S7). A strong local rearrangement was observed in areas of active invasion which was distinct from the more diffuse signal observed due to contractile forces between cell clusters31. Our subsequent analysis focused on the local rearrangement associated with the invasive front. Our pilot experiments showed a higher SHG intensity at the leading edge of the invasion, indicating interactions of cells with ECM components (e.g., reorganization, deposition, and degradation) were accompanied for the invasive progression of MCF-DCIS clusters. Accordingly, we introduced two SHG signal analyses: intensity profile analysis and area-based analysis to provide quantitative measures of collagen rearrangement associated with invasive transition.
Intensity profile analysis was done to measure SHG intensity around the periphery of the cell clusters (Fig. 4). Noninvasive clusters (grown in mono-culture) showed a comparatively homogeneous intensity profile, with one sharp peak at the periphery and a rapid decrease to near zero except in regions pulled by close clusters (No. 3 in Mono-A). However, once the cluster transitioned to invasive, there was significant alteration to the collagen network, presumably caused by remodeling of the collagen during the malignant process. The highest peak around invasive clusters (toward invading direction) was at least 1.5 times higher than the highest peak around noninvasive clusters, and a gradual decrease of intensity from the peak was observed.
Fig 4.

SHG signal intensity profiles around MCF-DCIS cell clusters. Two co-cultured clusters (co-A and co-B) show heterogeneous intensity profiles around clusters, while two mono-cultured clusters (mono-A and mono-B) show relatively homogeneous intensity profiles around clusters. Cluster outlines were found after F-actin staining of clusters. Collagen structures (green) were visualized by SHG signal and merged with F-actin-stained cells (red). Seven points were randomly selected around clusters, and lines (approximately 50 μm) were radially extended out to the surrounding collagen matrix from the points. Intensity profiles on the lines were measured. Scale bar represents 25 μm.
Area-based SHG signal analysis was devised to quantify the percentage of affected collagen area (PAC) by MCF-DCIS clusters and to provide a score for diagnosing the degree of invasiveness of the clusters. The PAC was determined as the number of active (or “on”) pixels in a thresholded image divided by the total number of pixels in the image; the result was multiplied by 100 (Fig. 5a). As an initial trial, we randomly selected 150 images each for both mono and co-cultures and evaluated scores from 0 to 18 to estimate the degree of invasiveness. While most noninvasive clusters (135 out of 150) fell into a range between 0 and 3, invasive clusters spread out between 2 and 17, depending on the degree of the extension of the clusters (Fig. 5b). For example, the PAC of a mono-A (noninvasive) in Fig 4 was 2.31%, while the PAC of short (co-A) and long (co-B) invasive clusters were 4.37% and 18.80% accordingly (Fig. 5a). The non-parametric Mann-Whitney test demonstrated a statistically significant difference between the two groups (P < 0.0001, two-tailed). The results suggest that area-based analysis provides a method to index the degree of invasiveness.
Fig 5.

Area-based SHG signal analysis provides quantitative estimation of the degree of invasive transition of MCF-DCIS cells within a specific area. (a) Three sequential steps involved in image processing for an area-based analysis. The original images (OG) were first thresholded with the value obtained from a blank gel, and the thresholded images (TH) were then converted to binary (BN) for pixel counting. The percentage of affected area in mono-culture (mono-A in Fig 4) is 2.32%, whereas that in co-culture (co-B in Fig 4) is 18.80%. SHG signal values from a blank gel (containing no cells) were applied for thresholding to distinguish SHG signals altered by cells. The numbers represent the percentage of affected collagen area (PAC) in an image. (b) The bar graph is generated after analyzing 150 images each (identical sizes of 1.5 mm2) for both mono- and co-cultured clusters. The bar graph indicates the number of images in a specific range, showing that most of mono-cultured clusters fall into a range between 0 and 3, whereas co-cultured clusters show a range between 1 and 17. Schematic illustration above the bar graph shows representative appearance of clusters in an area corresponding to the PAC range. Circularity and roundness decreases approximately from 1 to 0, while aspect ratio increases from 1 to 8 as PAC increases. Noninvasive clusters apart produce a value close to ‘0’, and more invasive clusters yield a value close to ‘17’.
Discussion
Our compartmentalized co-culture system has several advantages over conventional co-culture methods (summarized in Table 1). First, the channel design (i.e. arrayed, Fig. 1b) and the loading process (i.e., passive pumping) are compatible with existing high-throughput infrastructure, facilitating rapid adoption and implementation. The platform is operationally robust while allowing control of the matrix structure16, 17. Additionally, the contraction of collagen-based gels after polymerization and during cell culture provide a path for rapid media exchange, simplifying channel design for 3D cultures28. Importantly, PDMS is known to absorb hydrophobic molecules and can disrupt paracrine signaling in 2D culture30 and, thus, could be a potential source of bias or artifact. However, based on the validation experiments performed here, we postulate that secreted molecules might be largely retained in a 3D matrix, thus attenuating the absorption influence of PDMS as compared to 2D culture. Importantly, our method is not dependent on elastomeric material properties, and, thus, further increases in sensitivity may be possible by moving to other materials (e.g., polystyrene).
Table 1.
Summary of unique capabilities of the developed in vitro system.
| Schematic | Functionality/Outcome | |
|---|---|---|
| Microchannel design and Operation |
|
- Simple and adaptable channel design - Surface-tension driven pumping - Compatible with existing infrastructures (pipetting robot, laser scanner) - Create more in vivo-like mircoenvironment |
| Spatial and temporal controls |
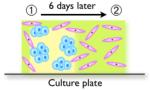
|
- Investigate distance depended behavior
(interface vs. control) - Co-injection: transition from single DCIS cells - Sequential injection: transition from larger DCIS clusters |
| Readout |
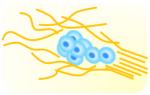
|
- Compartmentalization enables imaging of DCIS
associated collagen structure - Quantitative scoring of the degree of invasive transition of DCIS |
| Capable assays |

|
- Adaptable to various screening applications:
neutralizing antibodies, candidate drugs, siRNA - Adaptable to other cell types to create in vitro models for other diseases |
Second, the spatial control of the microenvironment allows recapitulation of aspects of the in vivo environment in vitro, and provides the ability to explore the distance-dependent effects of signaling. Compartmentalization of multiple layers in microfluidic channels is achieved by flowing various solutions simultaneously. This is in contrast to the serial loading process required to create multiple layers in 48 wells in which each layer needs to be completely polymerized (4-6 hours per layer) prior to adding another layer. Furthermore, imaging/monitoring cells through the layered gels is challenging due to the thickness of the gels. Thus, the micro scale system allows efficient study of the distance dependence of the invasive transition. The transition occurred actively around the interface where MCF-DCIS and HMF cells were close to each other and decreased as the distance between the cell types increased. At sufficient distance, no transition to invasive phenotype was observed (Circ: 0.44±0.21, Round: 0.83±0.10, AR: 1.22±0.18). Our observations that the transition to an invasive phenotype is dependent on distance from the stromal cells provides new insights into the signaling process. That is, when both soluble factors and cell-cell contact are present (zero spacing between compartments) the transition is accelerated, when only soluble factor signaling is present (spacer gel present) the transition is incomplete, and when there is minimal soluble factor signaling (distance is large enough) DCIS clusters retain their rounded morphology with no signs of a transition. It is also possible that, in addition to cell-cell contact, matrix rearrangement may have actively occurred at the interface by stromal fibroblasts, causing severe invasive transition. Even though we have not fully characterized whether the transition is dependent on either cell-cell contact or matrix remodeling, these observations illustrate how the compartmentalized system facilitates inquiry into relevant parameters. While it is possible to make similar observations in traditional co-culture systems (Fig. S4), it is not efficient (for example, observing fibroblast migration in a layered macro system would typically be performed using confocal microscopy, while in the shallow side-by-side micro scale compartments one can readily observe relative cell positions via phase contrast microscopy). The observed distance dependence effects provide insights, which help to shape our thinking about how the transition to invasion is regulated. For example, the data suggests that soluble factors may initiate the transition to invasion, but cell-cell contact (or matrix remodeling by stromal fibroblasts) may be required for invasion to continue.
Third, the temporal control providing the capability to form larger DCIS clusters prior to the introduction of the stromal cells takes an additional step closer to the in vivo organization. For this sequentially loaded culture, we have observed that HMFs encircle the pre-existing DCIS-containing gel due to the collagen contraction that occurs during initial DCIS culture. Additionally, this encasement of DCIS-containing gel may change the soluble factor gradient in the system. While this can be viewed as a step backwards in terms of imaging as the cell types now overlap one another, it is a step forward in terms of creating a structural organization more like the in vivo organization. Importantly, the adaptability of the approach allows for the control of these structural aspects without any fundamental changes to the device concept or operation. For example, a filler gel could be introduced prior to the stromal compartment introduction to maintain the side-by-side organization.
Fourth, the ability to compartmentalize by cell type facilitates readouts from one compartment without image overlap between cell types, thereby improving signal and simplifying image analysis. Here we used SHG signal intensity around the MCF-DCIS clusters to provide a unique quantitative readout of DCIS cell-associated collagen structure and a score of the degree collagen rearrangement near MCF-DCIS clusters. Both intensity profiles and area-based analyses show distinct differences between invasive and noninvasive clusters. In addition, the difference between short (less invasive) and long (more invasive) invasive clusters was detectable. While the two analyses revealed similar tendencies, each method provides unique information. In other words, the intensity profile analysis provides localized information such as heterogeneous collagen density distribution around a cluster and the invading direction of the cluster, while area-based analysis provides extensive information from a specific image (not limited to one cluster). Area-based analysis data not only simplifies the process of obtaining a quantitative measure of invasiveness, but also supports automation of analysis providing a path to high-throughput analysis. While our focus was on SHG signal intensity and collagen rearrangement in this study, the importance of collagen fiber alignment and its effect on the cell invasion process has been highlighted recently. For instance, Provenzano et al have shown that alignment of collagen perpendicular to the tumor-explant boundary promotes local invasion of mammary epithelial cells31. In our culture mode, we have, indeed, observed well-aligned collagen structures around invasive clusters generated by MCF-DCIS cells (Fig. S8). Our in vitro data is consistent with precedent observations and suggests that collagen fiber alignment and its contact guidance is one of the factors in DCIS progression to IDC, thus warranting further investigation. In addition, the platform enables efficient investigation of various ECM conditions that will facilitate further study of matrix concentration and composition - parameters that have been impractical to study using traditional assays.
Conclusion
We have presented a simple compartmentalized 3D co-culture model that supports the DCIS to IDC transition in vitro. The model was validated using accepted markers of invasion and compared to an in vivo xenograft model. The model represents a significant advance in our capability to investigate DCIS to IDC progression in breast cancer by making it possible and practical to explore both spatial and temporal effects while enabling new endpoints (e.g. SHG imaging of DCIS-associated collagen). In this study, the ability to examine distance dependence uncovered potentially new insights about the transition to invasion suggesting the possibility of a two-step process via two different progression mechanisms, which include first a soluble factor based progression and then cell-cell contact signaling involved progression. These observations were made possible by the unique functionality of the microscale model and have important implications in guiding the way we think about the transition and the development of therapeutic approaches to inhibit the transition. Importantly, the simplicity of the microfluidic system enables efficient investigation of the mechanisms involved in DCIS progression and allows screening approaches to identify pathways involved. For example, the small volumes required per endpoint open the door to the use of neutralizing antibodies or siRNA approaches. The flexibility of the system will allow it to be readily adapted to create relevant in vitro 3D models for other diseases where soluble factor signaling between different cell types is important.
Experimental
1) Microchannel design and fabrication
Devices were fabricated using soft lithography. Two layers of SU8-100 (Microchem Corp, Newton, MA) were spun and exposed individually and developed to generate a mold for 200 μm height fluidic channel and 400 μm deep fluid injection ports on a Silicon wafer. Polydimethylsiloxane (PDMS, Sylgard 184 Silicon Elastomer Kit, Dow Corning) was molded over the SU-8 master and sandwiched between transparency film and weights to allow access to the ports. The fabricated PDMS channels were sterilized in 70% ethanol and attached to polystyrene cell culture dishes (TPP AG, Switzerland). For multiphoton and confocal laser scanning microscopy, PDMS channels were attached to a glass bottom culture dish (P50G-0-30-F, MatTek corp, Ashland, MA).
For compartmentalization, a ‘Y’ shaped channel was designed. The distance between the two input ports was 8.8mm center-to-center to be compatible with a multi-pipette, and the width of main channel was 1.5mm. Three and four input channels were designed to add a spacer gel, and the width of main channels were 1.5mm and 3mm respectively. The resistance from each input to the main channel was equal to achieve laminar flow patterning.
2) Cell culture
GFP-labeled Human mammary fibroblast (HMF; originally termed RMF/EG) cells were provided by Dr. Kuperwasser34 and were cultured in DMEM (Mediatech Inc, Manassas, VA) supplemented with 10% calf serum (CS), 2mM L-glutamine, and penicillin/streptomycin. MCF-DCIS.com cells7 were purchased from Asterand (Detroit, MI), and were cultured in DMEM-F12 (50:50) supplemented with 5% horse serum (HS), 2mM L-glutamine, and penicillin/streptomycin. All cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
3) Sample preparation for in vitro 3D culture
For collagen sample preparation, the cells were trypsinized, added to culture media, counted and centrifuged (300g, 3 min). Two cells were co-cultured at a 1:2 ratio (MCFDCIS:HMF), and thus, MCF-DCIS cells were resuspended in culture media at a concentration of 3×106 cells/ml and HMF cells were resuspended at a concentration of 6×106 cells/ml. The total final cell number in a channel was approximately 750 cells (250 MCF-DCIS cells and 500 HMF cells). Collagen was prepared initially at a concentration of 2.0 mg/ml by neutralizing an acidic collagen solution (Collagen I, rat tail, BD Biosciences) with HEPES buffer (pH 7.7). Cells and culture media were added to Matrigel to make 50% Matrigel, and they were added to neutralized collagen I gel to achieve a final concentration of 1.3 mg/ml. For mixed gel condition, neutralized collagen gel and Matrigel were mixed with equal volume, and the collagen I concentration (0.8 mg/ml) was adjusted by cell suspension and culture media. To apply an additional nucleation phase of collagen polymerization before channel loading, the neutralized sample was kept at 4 °C for at least 15 min28. We have previously confirmed good cell viability in our 3D culture platform for 9 days28.
4) Device operation
The PDMS channels were pre-filled with serum-free DMEM media prior to load. Sample loading was done by passive pumping that requires two different sizes of droplets to create different inner pressure. 15 μl droplet was first dispensed at the output port, and then 8 μl droplets were dispensed at the input ports simultaneously using a multi-pipette, the latter volume always being smaller than the first. For sequential loading process, MCF-DCIS samples were pumped with non-gelling viscosity matched solution (PEG-8000,180mg/ml, Promega), which was pumped out of the channel after gelation. The created aqueous channel was filled with culture media containing 2% matrigel. After 6-days culture, the HMF samples were pumped in for co-culture.
Before and during channel loading, the prepared samples were kept in an ice chamber, and they were well-mixed to obtain uniform cell density in each channel. After the loading process was completed, the channel was placed in a water-containing plastic chamber to prevent evaporation and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 6 min to polymerize the sample. The channels were flipped every two minutes to avoid cell settlement to the bottom side before gelation. Afterwards, culture media was added to both inlet and outlet ports, and was replaced every other day.
5) Invasion assay
The invasiveness of MCF-DCIS cells was assayed by using BD BioCoat™ Growth Factor Reduced (GFR) Matrigel invasion chamber (8 μm pore size PET membrane, 6-well). MCF-DCIS cells were prelabeled by using cell tracker dye (CellBrite™ Green cytoplasmic membrane staining kit, Biotium, Inc). We resuspended 4 × 104 MCF-DCIS cells alone or mixed with 8 × 104 HMF cells in 2ml of serum-free DMEM/F12, and seeded in the upper compartment of chamber. The lower compartment was filled with 2.5ml of DMEM/F12 supplemented with 20% HS as chemoattractant. After incubation at 37 °C in a humid atmosphere for 36 hrs, filters were rinsed with PBS. Remaining cells on the upper surface were wiped away with a wet cotton swab, and those on the lower surface were fixed with 4% paraformaldehyde, and stained with Hoechst (Hoechst 33342, Molecular Probe). The number of invaded cells per microscopic view was counted and averaged.
6) Immunofluorescent staining
The gels were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature and washed 3 times with PBS. Collagen gels were then treated with 0.1M glycine in PBS at 4 °C for overnight to reduce autofluorescence followed by PBS washing (3X). The channels were blocked with 3% Fetal Bovine Serum (FBS) at least 1 hr at 4 °C and incubated with primary antibodies (1:50, Rabbit polyclonal Collagen IV antibidy, Mouse monoclonal E-cadherin, Abcam) at 4 °C overnight. After washing with PBS (4X), the secondary antibody (1:200, Alexa 488-conjugated anti-rabbit, Alexa 594-conjugated anti-mouse, Invitrogen) was added and incubated at 4 °C overnight followed by PBS washing (4X). Lastly, To-Pro-3 (1:500, Invitrogen) was added and incubated for at least 2 hrs at 4 °C, followed by PBS washing. For filamentous actin staining, phalloidin solution (1:50, alexa fluor 594 phalloidin, Invitrogen) was added after glycine treatment, incubated at 4 °C for overnight, and washed 3 times with PBS.
7) Imaging and analysis
Brightfield images were acquired on an inverted microscope (Eclipse Ti-U, Nikon) using the NIS-Element imaging system (Diagnostic Instruments, Inc.). Confocal microscopy (Biorad MRC 1024 confocal scanning laser microscopy on an inverted Nikon Eclipse TE300) was used to image immunofluorescently labeled cells. All Multiphoton laser scanning microscopy (MPLSM) and Second Harmonic Generation (SHG) imaging was done on an Optical Workstation that was constructed around a Nikon Eclipse TE300. A MaiTai Deepsee Ti:sapphire laser (Spectra Physics, Mountain View, CA) excitation source tuned to 890 nm was utilized to generate both Multiphoton excitation and SHG. The beam was focused onto the sample with a Nikon (Mehlville, NY) 20X Super Fluor air-immersion lens (numerical aperture (NA) = 1.2). All SHG imaging was detected from the back-scattered SHG signal with a H7422 GaAsP photomultiplier detector (Hamamatsu, Bridgewater, NJ), and the presence of collagen confirmed by filtering the emission signal with a 445 nm (narrow-band pass) filter (TFI Technologies, Greenfield, MA) that isolated the SHG signal. Acquisition was performed with WiscScan (http://www.loci.wisc.edu/software/wiscscan), a laser scanning software acquisition package developed at LOCI (Laboratory for Optical and Computational Instrumentation, University of Wisconsin, Madison, WI).
All SHG signal processing and analysis was performed using MATLAB (version 7.8. Natick, Massachusetts: The MathWorks Inc., 2009.). All of the SHG intensity analysis performed in the paper were done only with mixed matrix, and SHG intensity correlates to the concentration of the mixed matrix (Fig S9). To obtain the SHG area measurements, a threshold was applied to each image by subtracting the mean intensity of a blank gel of the same composition to estimate SHG signal intensity altered by cells. The images were then converted to binary images, and the area of increased SHG intensity expressed as the ratio of the total number of “on” pixels (or pixels with a value of 1) to the total number of pixels in the image. To create the SHG intensity profiles, images of the phalloidin-stained DCIS clusters were used to create a binary mask which was then applied to the corresponding SHG images. Lines were extended radially out from the DCIS cluster outlines into the surrounding matrix, and the intensity of each pixel along the line was measured. 7 different intensity profiles from one cluster were combined using Plot software (Plot 0.997, Michael Wesemann) and smoothened by Fast Fourier Transform (FFT).
The morphology analysis of MCF-DCIS clusters was done by using shape descriptor measurement of ImageJ software for circularity, roundness, and aspect ratio. Circularity was measured by applying the formula of 44π ×area/perimeter2. A value of 1.0 indicates a perfect circle, and as the value approaches 0.0, it indicates an increasingly elongated polygons. The roundness of clusters was measured by using the formula of 4 × area/(π × major_axis2). Aspect ratio was the ratio of major axis over minor axis.
8) In vivo experiment and analysis
Xenograft lesions were generated by injecting 1 × 105 MCF-DCIS cells in 0.2 ml of Matrigel subcutaneously into 6- to 8- week-old female nude mice (NCR Nude outbred, Taconic Farms, Germantown, PA). Mice were maintained for up to 5 weeks after injection before euthanized by CO2 narcosis. Xenografts were removed and fixed in 10% formalin. Formalin-fixed, paraffin-embedded xenografts were cut into 5-micron thick sections, which were deparaffinized and rehydrated through graded alcohols. The antigens were retrieved by boiling the sections in 10 mM sodium citrate buffer (pH 6.0). The sections were blocked with 5% goat serum and incubated with antibodies against human Collagen IV and E-cadherin (Abcam Inc, Cambridge, MA) at 4°C overnight. After washing with PBS, the sections were incubated with appropriate fluorescenceconjugated secondary antibodies (Invitrogen, Eugene, OR). Sections from the same formalin-fixed, paraffin-embedded xenografts were also subjected to hematoxylin-eosin (H&E) staining.
Insight, innovation, integration.
The presented model is an important innovation in our capability to investigate breast cancer progression from DCIS to IDC by making it possible and practical to explore spatial (distance-dependency) effect while enabling new endpoints (imaging of DCIS-associated collagen). The ability to examine distance dependence uncovered new insights about the transition to invasion suggesting two different mechanisms are involved - a soluble factor based progression and a cancer cell-fibroblast direct contact progression. These observations were made possible by the unique functionality of the microscale model and have important implications in guiding the way we think about the transition and the development of therapeutic approaches to inhibit the transition. Furthermore, while we focused on breast cancer progression for this paper, the flexibility of the system will allow it to be readily adapted to create relevant in vitro 3D models for other diseases where soluble factor signaling between different cell types is important.
Supplementary Material
Acknowledgements
This study was supported by NIH grants K25-CA104162, 2R01 CA107012-06 ,1R33CA137673-01, the Wisconsin Partnership Program, the DARPA Micro/nano Fluidics Fundamentals Focus Center, the Korea Research Foundation Grant (KRF-2008-220-D00133), and NLM training grant (NLM 5T15LM007359). D. J. Beebe has equity in Bellbrook Labs which has licensed technology reported in this manuscript.
Bibliographic references
- 1.Paget S. The Lancet. 1889;133:571–572. [Google Scholar]
- 2.Hu M, Yao J, Carroll D, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y. Cancer Cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu M, Peluffo G, Chen H, Gelman R, Schnitt S, Polyak K. Proc Natl Acad Sci USA. 2009;106:3372–3377. doi: 10.1073/pnas.0813306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizki A, Weaver VM, Lee S-Y, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, Mian IS, Borowsky AD, Jensen RA, Idowu MO, Chen F, Chen DJ, Petersen OW, Gray JW, Bissell MJ. Cancer Research. 2008;68:1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burstein H, Polyak K, Wong J, Lester S, Kaelin C. New England Journal of Medicine. 2004;350:1430–1441. doi: 10.1056/NEJMra031301. [DOI] [PubMed] [Google Scholar]
- 6.Tait LR, Pauley RJ, Santner SJ, Heppner GH, Heng HH, Rak JW, Miller FR. Int J Cancer. 2007;120:2127–2134. doi: 10.1002/ijc.22572. [DOI] [PubMed] [Google Scholar]
- 7.Miller FR, Santner SJ, Tait L, Dawson PJ. J Natl Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- 8.Muthuswamy S, Li D, Lelievre S, Bissell M, Brugge J. Nature cell biology. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryan B, Schnitt S, Collins L. Mod Pathol. 2006;19:617–621. doi: 10.1038/modpathol.3800570. [DOI] [PubMed] [Google Scholar]
- 10.Sadlonova A, Novak Z, Johnson M, Bowe D, Gault S, Page G, Thottassery J, Welch D, Frost A. Breast Cancer Res. 2005;7:R46–R59. doi: 10.1186/bcr949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong A, Perez-Castillejos R, Love J, Whitesides G. Biomaterials. 2008;29:1853–1861. doi: 10.1016/j.biomaterials.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang CP, Lu J, Seon H, Lee AP, Flanagan LA, Kim HY, Putnam AJ, Jeon NL. Lab Chip. 2009;9:1740–1748. doi: 10.1039/b818401a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung S, Sudo R, Mack P, Wan C, Vickerman V, Kamm R. Lab Chip. 2009;9:269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 14.Tsang V, Chen A, Cho L, Jadin K, Sah R, Delong S, West J, Bhatia S. The FASEB Journal. 2007;21:790. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 15.Walker G, Beebe D. Lab Chip. 2002;2:131–134. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- 16.Meyvantsson I, Warrick J, Hayes S, Skoien A, Beebe D. Lab Chip. 2008;8:717–724. doi: 10.1039/b715375a. [DOI] [PubMed] [Google Scholar]
- 17.Puccinelli JP, Su X, Beebe DJ. JOURNAL OF THE ASSOCIATION FOR LABORATORY AUTOMATION. 2010;15:25–32. [Google Scholar]
- 18.Williams RM, Zipfel WR, Webb WW. Biophys J. 2005;88:1377–1386. doi: 10.1529/biophysj.104.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seton-Rogers SE, Lu Y, Hines LM, Koundinya M, LaBaer J, Muthuswamy SK, Brugge JS. Proc Natl Acad Sci U S A. 2004;101:1257–1262. doi: 10.1073/pnas.0308090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson M, Sahai E. Clinical and Experimental Metastasis. 2009;26:273–287. doi: 10.1007/s10585-008-9174-2. [DOI] [PubMed] [Google Scholar]
- 21.Gaggioli C, Hooper S, Hidalgo-Carcedo C. Nature cell biology. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 22.Hompland T, Erikson A, Lindgren M, Lindmo T, de Lange Davies C. Journal of biomedical optics. 2008;13:054050. doi: 10.1117/1.2983664. [DOI] [PubMed] [Google Scholar]
- 23.Falzon G, Pearson S, Murison R. Phys Med Biol. 2008;53:6641–6652. doi: 10.1088/0031-9155/53/23/001. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Burke RM, Zettel ML, Tang P, Brown EB. Optics Express. 2008;16:1846–1859. doi: 10.1364/oe.16.001846. [DOI] [PubMed] [Google Scholar]
- 25.Provenzano PP, Eliceiri K, Keely PJ. Clinical and Experimental Metastasis. 2009;26:357–370. doi: 10.1007/s10585-008-9204-0. [DOI] [PubMed] [Google Scholar]
- 26.Alexander S, Koehl GE, Hirschberg M, Geissler EK, Friedl P. Histochem Cell Biol. 2008;130:1147–1154. doi: 10.1007/s00418-008-0529-1. [DOI] [PubMed] [Google Scholar]
- 27.Brown E, McKee T, diTomaso E, Pluen A, Seed B, Boucher Y, Jain RK. Nature medicine. 2003;9:796–800. doi: 10.1038/nm879. [DOI] [PubMed] [Google Scholar]
- 28.Sung KE, Su G, Pehlke C, Trier S, Eliceiri K, Keely PJ, Friedl A, Beebe D. Biomaterials. 2009;30:4833–4841. doi: 10.1016/j.biomaterials.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET, Beebe DJ. Lab Chip. 2009;9:2132–2139. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Biophys J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Proc Natl Acad Sci USA. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.