

Abstract
Cushing’s syndrome (CS) is uncommon in childhood. CS may be either dependent or independent of adrenocorticotrophic hormone (ACTH). ACTH independent micronodular adrenocortical (MAD) disease may present in the second to third decade of life or between ages 2–3years. It may occur in isolation, or as a part of the Carney complex and it represents an elusive entity to diagnose. We present a 3 year 7 month old boy with isolated MAD (iMAD). Abdominal CT revealed prominent mildly lobulated anteromedial margin of adrenals with nodular appearance. Cardiac echo, thyroid and testicular ultrasounds performed as a work up for Carney complex were normal. Bilateral adrenalectomy confirmed MAD as the cause of CS. We present the history’ and identification of a unique case of iMAD.
Keywords: Cushing’s syndrome, micronodular adrenocortical disease, Carney complex, primary pigmented adrenocortical disease (PPNAD), hypercorticolism
INTRODUCTION
Cushing’s syndrome is uncommon in childhood and is most often caused by excessive glucocorticoid administration1. Estimates of the incidence rate of Cushing’s syndrome are near 2 per 1,000,000 inhabitants in both the adult and pediatric populations combined2 The manifestations of the syndrome are quite protean and may differ with respect to the age of presentation. The classic features of Cushing’s syndrome are well known to most clinicians and include weight gain with fat redistribution affecting the face (leading to ‘moon face’), trunk (leading to a buffalo hump’), neck, abdomen, wide purplish striae and poor wound healing. This constellation of findings is often seen in advanced cases especially in the adult population however, they may not be present in children. Weight gain and growth arrest are the most reliable and earliest indicators of hypercorticolism in children3. Pediatric Cushing’s syndrome may be manifested by somatic growth impairment occurring secondary to the action of glucocorticoids on long bones. The suppressive effect of glucorticoids on growth may be due to a) an indirect increase in the hypothalamic secretion of somatostatin, b) indirect suppressive effect on growth hormone receptors and IGFI production and c) direct action on epiphyseal cartilage to inhibit sulfation, mineralization and cell proliferation4.
Pediatric Cushing’s syndrome may be classified as either adrenocoticotropic hormone (ACTH) dependent or ACTH independent in accordance with the source responsible for producing the excess cortisol. ACTH independent causes may be due to exogenous glucocorticoid administration, adrenocortical tumors or primary disease, macronodular adrenocortical hyperplasia or causes due to the McCune Albright’s syndrome5. Micronodular adrenocortical disease may exist as isolated micronodular adrenocortical disease (iMAD), which may be sporadic or familial and its primary variant primary pigmented nodular adrenocortical disease (PPNAD). The presence of lipofuscin derived from the residual bodies containing the end products of oxidative damage to lipid molecules is responsible for imparting the color to the nodules6 Micronodular adrenocortical disease and its variant PPNAD may be a part of the Carney complex, which is a multiple endocrine neoplastic syndrome. Micronodular adrenocortical disease and its variant PPNAD are most often bilateral7. Patients with iMAD may present with classical Cushing’s syndrome, atypical Cushing’s syndrome (ACS) or periodic Cushing’s syndrome (PCS). ACS is characterized by asthenic body habitus, short stature and muscle wasting whereas PCS presents with periods of hypercorticolism interspersed with periods of normal cortisol production8. We present a case of an almost 4 year old child who had sudden onset and progressive weight gain. He underwent biochemical testing revealing ACTH independent Cushing’s syndrome. Radiological tests initially failed to reveal the cause of his hypercorticolism. Bilateral adrenalectomy was performed and he was found to have micronodular adrenocortical disease (iMAD) as a cause for his presentation.
PATIENT REPORT
A 3 year and 7 month old white male presented to our facility with a 3-month history of weight gain amounting to approximately 10 pounds (4.5 kilograms). He was referred by his primary care physician in light of a sudden and progressive history of weight gain. Per his parents, he suddenly became ‘chubby’ with new onset facial acne. He was described as having a ‘greasy teenager smell’ and also a voracious appetite. Dad also noted that he was always a tall child however; in addition to this he had suddenly become heavy. Labs performed at the primary care physician’s office showed a serum cortisol of 968.3 nmol/L (118.6–617.4nmol/L) [35.1 mcg/dl (4.3–22.4 mcg/dl)] at 10:25 am, random blood glucose 5.6mmol/L [100 mg/dl], thyroid stimulating hormone (TSH) 0.52μIU/ml (0.55–7.10IμIU/ml) and total thyroxine 8.0 μg/dl (4.7–11.6 μg/dl). On his initial presentation to our urgent care department, anthropometrics included a weight of 20 kgs (97th centile) and a height of 101.3 cm (75th centile), blood pressure (B.P.) 126/79 (greater than the 95th centile for age) however, B.P. was normal for age at the primary care physician’s office 6 hrs earlier.
On physical examination he had swollen cheeks imparting a moon face with no peri-orbital swelling (please see Figures 1 and 2). Other notable positive findings included facial acne, a humplike appearance to the nape of his neck and fullness to his supraclavicular region. His abdominal examination was unremarkable and his testicular volume was prepubertal. He had no pubic hair or phallic enlargement.
Figure 1.

Patient 6 months prior to presentation.
Figure 2.

Patient 1 day prior to presentation.
He was admitted to hospital in order to expedite the workup of hypercorticolism due to the severity and rapidity of his presentation. During his hospitalization, labs obtained included serum cortisol 22.3 mcg/dl (4.3–22.4 mcg/dl) [614.3 nmom/L (118.6–617.4 nmol/L)] at noon, adrenocorticotropic hormone (ACTH) 1.11 pmol/L (174.6–1780.73 pmol/L) [ <5pg/ml (7–50 pg/ml)], dehydroepiandrosterone sulphate (DHEAS) [31mcg/dl (6–35mcg/dl)], dehydroepiandrostene (DHEA) 2669.9 pmol/L (381.4–1491 pmol/L) [77 (11–43mg/dl)], testosterone by liquid chromatography mass spectrometry (LCMS) 970.9 pmom/L (normal <173.37 pmol/L [28ng/dl (normal <5 ng/dl)], androstenedione 9008.4pmol/L (174.58–1780.73pmol/L) [258ng/m1 (5–51 ng/ml)], prolactin 11mcg/L (I-16mcg/L) [11 ng/ml (1–16 ng/ml)]. Magnetic Resonance Imaging (MRI) of the abdomen and head were performed while awaiting ACTH results in order to determine the etiology for hypercortisolism however, both were normal. It was the view of the local radiology department that MRI was quite sensitive in ruling out adrenal pathology hence no other imaging modality was deemed likely to reveal the existence of any pathology.
After a one night admission the patient was discharged and a midnight salivary cortisol test arranged. The results were 20.14nmoml/L [0.730mcg/dl] and 17.38 nmoml/L [0.630mcg/dl] (0.01–0.090 mcg/dl). He was readmitted after 17 days due to a concern by his primary care physician for malignant hypertension. Systolic blood pressures ranged between 130–144 and diastolic blood pressures 76–82. Amlodipine therapy was initiated after a consultation had occurred with the nephrology service. A midnight serum cortisol level was also obtained on this occasion. It was 865.64nmomIL [31.4 mcg/dl] which represents an elevation. This added further support for the assessment of hypercorticolism. ACTH level was 1.11 pmoml/L [<5pg/ml] and testosterone level was 1213.6pmomI/L [35 ng/dl]. An inhouse 24 hour urine collection for free cortisol was unsuccessful.
Prior to our referral to the National Institutes of Health, Bethesda, MD (NIH) synthetic urine and serum glucocorticoid screens were performed at the Mayo Laboratories in order to rule out an exogenous source for hypercortisolism. The synthetic serum glucocorticoid screen was negative; however; the synthetic urine glucorticoid screen was positive for prednisolone 0.95mcg/dl (cutoff 0.05mcg/dl) and prednisone 0.08mcg/dl (cutoff 0.05 mcg/dl). A sequential low dose dexamethasone (20mcg/kg/day divided Q 6hrly) and high dose dexamethasone (80 mcg/kg/day divided Q 6hrly) suppression test with 24 hour urine collection for free cortisol and 17 hydroxy-corticosteroids were performed. The serum cortisol post low and high dose dexamethasone failed to suppress and were 896 nmol/L [32.5 mcg/dl] and 838.6 nmol/L [30.4 mcg/dl] respectively with ACTH <1.11 pmol/L [<5pg/ml] and 1.33 pmol/L [6 pg/ml] respectively (reference range 1.56–11.1 pmol/L [(reference range 7–50 mg/dl)]. 24 hour urine collection done simultaneously with the suppression tests was unreliable due to relative dehydration secondary to a viral illness which resulted in a decreased urinary volume. In slightly over a 2 week period from his initial presentation the patient had become easily fatigued with a diminution of his energy level. His facial acne was also worsening. A referral was made to the NIH for further evaluation and definitive management.
CLINICAL TESTS
Biochemical
The patient was transferred to the NIH Warren Magnuson’s Clinical Center post parental consent. Repeated tests confirmed the presence of hypercorticolism with a loss of the diurnal variation for cortisol secretion 841.4–940.7 nmol/L[(midnight cortisol 30.5 and 34.1mcg/dl)] and a concomitantly suppressed ACTH level<1.11pmol/L [(<5pg/ml)]. Corticotropin Releasing Hormone (CRH) stimulation testing failed to show a rise in ACTH levels and his serum cortisol level remained elevated and unresponsive to CRH (please see Table 1). Cortisol levels pre and post an 8mg overnight dexamethasone suppression test were 1015.2 nmol/L[36.8mcg/dl] and 1048.3 nmol/L [38.0 mcg/dl] respectively and this corroborated with the previous results which showed a failure to suppress cortisol production by dexamethasone. A random urine cortisol showed an increase in cortisol/creatinine ratio 1310.8nmol/mmol [4,200mcg/g] creatinine 0.69–27.8 nmol/mmol [(2.2–89mcg/g)].
Table 1.
shows a lack of increase in cortisol production after ACTH and suppressed ACTH levels during CRH stimulation.
| Time (mins) | -5 | 0 | 15 | 3- | 45 |
| CRH Test (cortisol in mcg/dl) | 33.7 | 36 | 32.3 | 30.7 | 32.6 |
| CRH test (ACTH in PT/ml) | <5 | <5 | <5 | <5 | <5 |
Radiological
Thyroid ultrasound revealed two small echodense lesions measuring 3mm in diameter located in the right thyroid gland and were thought to be non-specific. Testicular/scrotal ultrasound showed no evidence of calcifying masses in the testes. However, there was a bilateral increase in echogenic material noted in the area of the head of the epididymis which had been interpreted as being developmental. Bone age evaluation done at a chronological age of 3 years and 7 months old closely corresponded to that of a 2 year old male with standard deviation of 5.4 months per Greulich and Pyle method. Lastly, adrenal Computed Tomography (CT) scan revealed mildly lobulated prominence of the adrenal glands along the anteromedial margins bilaterally which may contain small nodules with no bulky lymphadenopathy (please see Figure 3). Cardiac echocardiogram performed prior to the patient’s departure for the NIH was negative for the existence of atrial myxoma.
Figure 3.
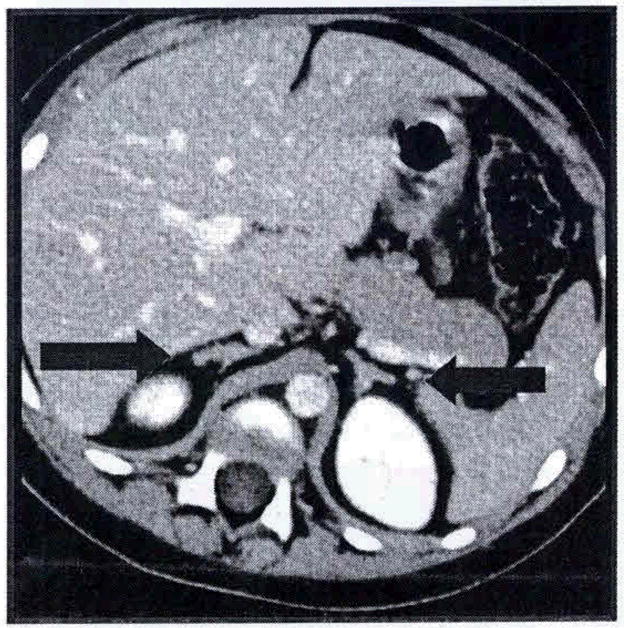
Arrows demonstrate nodularity to the adrenal gland, characteristics of nodular disease.
Tissue Analysis
Tissue for genetic analysis was obtained at the time of bilateral adrenalectomy; DNA was tested for the coding sequence of the PRKAR1A, PDE11A and PDE8B genes.
RESULTS
Clinical evaluation
The patient was evaluated over a seven week period. He displayed a loss of the diurnal variation of cortisol secretion and suppression of his ACTH levels. He failed to suppress his cortisol secretion on both the low dose and high dose dexamethasone suppression tests and a Liddle’s test which involves sequential low and high dose dexamethasone suppression testing. It was not possible to demonstrate the expected greater than 50% increase in both urinary free cortisol and 17 hydroxy-corticosteroids due to the relative dehydration of the patient during an intercurrent viral illness.
Histopathology and DNA studies
The left adrenal gland measured 4.5 X 3.0 X1.6cm and weighed 13.39 grams whereas the right adrenal gland measured 4.3 X 3.0 X 1.5cm and weighed 6.68 grams. Both glands had a tan yellow nodular cut surface with the left gland having a dominant nodule measuring 0.2 cm grossly (Figures 4 and 5). Microscopically, both glands showed multiple nodules of adrenocortical hyperplasia with oncocytic changes and occasional hyperplastic nodules outside the gland’s capsule. There was no accumulation of pigment in either gland and this was consistent with micronodular adrenocortical disease (MAD).
Figure 4.
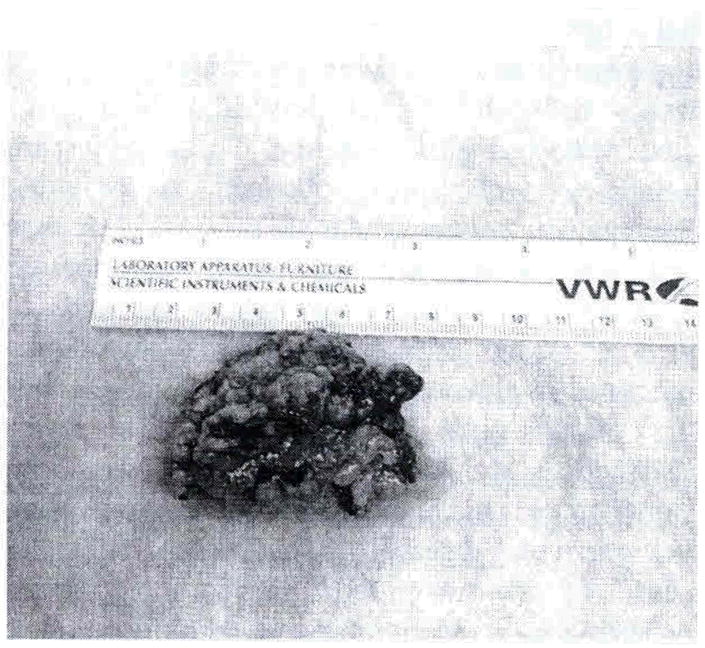
Gross anatomy of right adrenal gland displaying nodularity.
Figure 5.
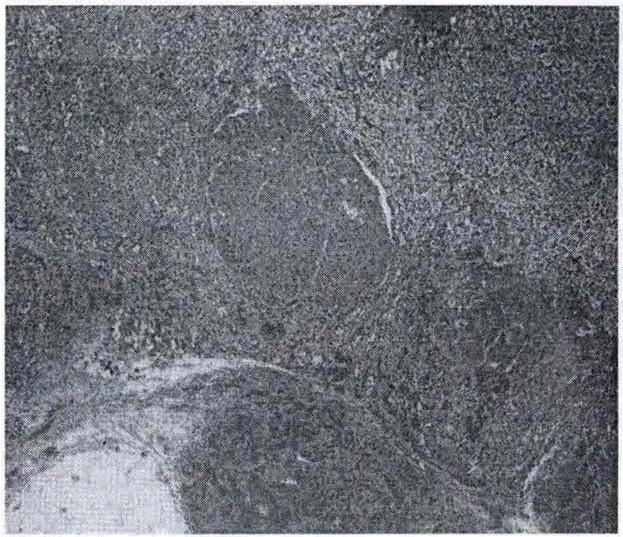
Histology of right adrenal gland displaying nodularity.
Clinical Follow Up
Six months after the adrenalectomy the patient continues to do well while taking hydrocortisone (10mg/m2/day) and fludrocortisone 100μg/day) replacements. He has no signs of Cushing’s syndrome and his acne has totally resolved (Figure 6). His energy level while taking supplementation is back to his premorbid state.
Figure 6.

Patient 6 months post bilateral adrenalectomy.
DISCUSSION
Cushing’s syndrome is rare in infancy. Adrenocortical tumors (ACT) are an important cause. Southern Brazil has a relatively high incidence of ACT and the incidence there is 3.4–4.2 per 1,000,000 children per year which is estimated to be 10–15 times higher than that existing in other geographic locations. In this setting, Cushing’s syndrome occurs as a result of tumors which usually are secondary to a germline p53 mutation in young children. Such mutations are not as common in the adult9,10 population.
In terms of etiology, Cushing’s syndrome may either be dependent or independent of ACTH. Micronodular adrenocortical (MAD) disease is considered ACTH independent and it may be divided into three groups. These are: isolated micronodular adrenocortical disease (iMAD), primary pigmented nodular adrenocortical disease (PPNAD) as a part of the Carney complex, which is a syndrome of multiple endocrine neoplasia and finally, isolated PPNAD. PPNAD has 2 peak ages of presentation, occurring as a part of the Carney complex usually in the second to third decade of life or less frequently between the ages of 2–3 years 11,12,13.
There may be difficulty in diagnosing Cushing’s syndrome in children as presentation may not involve the typical features as outlined prior. Hence, a high index of suspicion should exist for making the diagnosis. In our patient insidious weight gain in addition to a relatively stable linear height percentile was demonstrated and this is often deemed as a presenting feature in the pediatric population4.
Micronodular adrenocortical disease (MAD) and its variant PPNAD are often difficult to diagnose and one must first demonstrate the existence of hypercorticolism. Hypercorticolism is however difficult to demonstrate in the pediatric population as the 24 hour urine collection which is taken as the gold standard for proving its existence, poses a challenge to obtain14. Night time salivary cortisol is able to screen for hypercorticolism and has a diagnostic accuracy of 93% which is similar to that of urinary free cortisol15. Our patient had an elevation of his midnight salivary cortisol near 7–8 times the upper limits of normal in addition to a serum cortisol of 865.01 nmol/L [31.4mcg/dl] at midnight.
Since our patient’s presentation was quite sudden and progressive, the possibility of exposure to exogenous glucocorticoids was ruled out by performing serum and urine synthetic glucocorticoid screens16,17. While the serum synthetic glucocorticoid screen was negative for all steroids, the synthetic urine glucocorticoid screen was positive for all steroids with the exception of prednisone and prednisolone. This was likely due to cross reactivity from high serum cortisol levels.
Post demonstration of hypercorticolism an etiology of excess cortisol production was searched for. A lack of suppression of cortisol production involving low dose and high dose dexamethasone suppression tests and repeatedly suppressed adrenocorticotrophic hormone (ACTH) levels strongly pointed towards the existence of an adrenal pathology. A corroboration of biochemical evidence was sought pointing in the direction of micronodular adrenocorrtical disease (MAD) by the performance of a Liddle test. This test involves obtaining baseline cortisol levels as well as a 24 hr urine collection for free cortisol (UFC) and 17 hydroxycorticosterone (17OHCS) levels. Sequential low and high dose dexamethasone suppression testing is done with UFC l7OHCS levels determined post low and high dose dexamethasone administration. These values are compared with those obtained at baseline. Patients with micronodular adrenocortical disease and its variant primary pigmented nodular adrenocortical disease (PPNAD) respond to dexamethasone suppression with a paradoxical increase in glucocorticoid secretion on the last day of this test. Urinary free cortisol levels obtained on the last day of the test have been shown to bear the highest accuracy for diagnosing micronodualr disease18. The exact molecular cause of the increased glucocorticoid levels occurring after dexamethasone administration is presently unknown, however, increased glucocorticoid receptor present in the nodules may play a role in the response to dexamethasone19. The Liddle test performed on our patient was non-diagnostic possibly due to him having an intercurrent viral illness which resulted in him having an inadequate urine output, thus precluding an accurate assessment of his urine volume during his illness.
Ovine corticotrophin releasing hormone (CRH) stimulation testing by failing to produce elevated levels of ACTH and cortisol further supported the evidence for the existence of an adrenal pathology in our patient. A lack of response from the pituitary gland after stimulation is postulated to occur as a result of suppression of the hypothalamo-pituitary unit by the autonomous secretion of glucocorticoid20,21. His random urine cortisol/creatinine ratio was grossly elevated at 1310.8 nmol/mmol [4,200 mcg/g] creatinine and this provided further confirmation of hypercorticolism22,23.
Thyroid, testicular/scrotal ultrasounds, cardiac echocardiography performed in our patient to rule out features of the Carney complex were all negative. The latter is an autosomal dominant syndrome characterized by multiple neoplasias which include myxomas, endocrine tumors and lentiginosis24.
A bone age evaluation performed at a chronologic age of 3 years and 7 months was more than two standard deviations delayed. This was unexpected and may be argued as being due to the relatively short duration of testosterone increase precluding an untoward effect on bone maturity.
Radiological evidence supporting the existence of micronodular adrenocortical disease (MAD) may be operator dependent. Computer tomography (CT) and magnetic resonance imaging (MRI) have been shown to demonstrate subtle features of nodularity as had been seen in our patient25. Although not universal in its occurrence, radiological findings have been described as a “bead on a string appearance” by some authors and this is due to hyperplastic nodules with intervening atrophic adrenal glandular tissue.
In our patient, histologically, both adrenal glands displayed nodularity with an absence of prominent pigmentation. This resulted in an assessment of isolated micronodular adrenocortical disease (iMAD) and not primary pigmented nodular adrenocortical disease (PPNAD) which would have been characterized by prominent pigmentation secondary to the presence of lipofuscin.
Molecular analysis thus far has revealed an innocent polymorphism involving the PPARKAR1A gene and analysis is ongoing for PDE11A and PDE8B gene mutations26,27.
MAD and its variant PPNAD, apart from being rare, are also difficult to diagnose. A high index of suspicion should exist for these conditions as the cause of CS in the pediatric population especially when an ACTH independent source of hypercortisolism is demonstrated. Sequential and extensive biochemical work up is often necessary to prove these diagnoses after confirmation of hypercorticolism. Hence, despite negative imaging, one needs to continue the search for an etiology.
References
- 1.Aron D, Findling J, Tyrrell JB. Greenspan’s Basic and Clinical Endocrinology. 8. 2007. Glucocorticoids and adrenal androgens; p. 378. [Google Scholar]
- 2.Findling JW, Ralf H. Screening and Diagnosis of Cushing’s syndrome. Endocrinol Metab CLinic N Am. 2005;34:385–402. doi: 10.1016/j.ecl.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Devoe DJ, Miller WL, Conte FA, Kaplan SL, Grumbach MM, Rosenthal SM, et al. Long term outcome of children and adolescents following transphenoidal surgery for Cushings disease. J Clinic Endocrinol Metab. 1997;82:3196–3202. doi: 10.1210/jcem.82.10.4290. [DOI] [PubMed] [Google Scholar]
- 4.Miller WL, Achermann JC, Fluck CE. Sperling Pediaric Endocrinology, 3‴ Edition. 2008. The Adrenal Cortex and its Disorders; pp. 444–511. [Google Scholar]
- 5.Savage MO, Chan LF, Grossman AB, Storr HL. Work up and management of Pediatric Cushing’s syndrome. Current Opinion in Endocrinology, Diabetes and Obesity. 2008;15:346–351. doi: 10.1097/MED.0b013e328305082f. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki Y, Park MN, Mori T, Kwashima S. The difference in autofluorescence features of lipofuscin between brain and adrenal. Zoolog Sci. 1995;12:283–288. doi: 10.2108/zsj.12.283. [DOI] [PubMed] [Google Scholar]
- 7.Stratakis CA, Kirschner LS. Clinical and genetic analysis of primary bilateral adrenal diseases (micro and macronodular disease) leading to Cushing’s syndrome. Horm Metab Res. 1998;30:456–463. doi: 10.1055/s-2007-978914. [DOI] [PubMed] [Google Scholar]
- 8.Mellinger RC, Smith RW., Jr Studies of the adrenal hyperfunction in 2 patients with atypical Cushing’s syndrome. J Clin Endocrinol Metab. 1956;16:350–366. doi: 10.1210/jcem-16-3-350. table. [DOI] [PubMed] [Google Scholar]
- 9.Michalkiewicz E, Sandrini R, Figuerredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–845. doi: 10.1200/JCO.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 10.Wagner J, Portwine C, Robin K, et al. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst. 1994;86:1701–1710. doi: 10.1093/jnci/86.22.1707. [DOI] [PubMed] [Google Scholar]
- 11.Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. 2007;3:748–757. doi: 10.1038/ncpendmet0648. [DOI] [PubMed] [Google Scholar]
- 12.Gunther DF, Bourdeau I, Matyakhina L, et al. Cushing’s syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab. 2004;89:3173–3182. doi: 10.1210/jc.2003-032247. [DOI] [PubMed] [Google Scholar]
- 13.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 14.Niemann LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, et al. The diagnosis of Cushing’s syndrome: AN Endocrine Society Clinical Practice Guideline. J Clinical Endocrinol Metab. 2008;93(5):1526–1540. doi: 10.1210/jc.2008-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaffing RI, Papanicolaou DA, Niemann LK. Nighttime salivary cortisol measurement as a simple noninvasive outpatient screening test for Cushing’s syndrome in children and adolescents. Journal of Pediatrics. 2000;137(1):30–35. doi: 10.1067/mpd.2000.106226. [DOI] [PubMed] [Google Scholar]
- 16.Lin CL, Wu TJ, Machacek DA, Jiang NS, et al. Urinary free cortisol and cortisone determined by high performance liquid chromatography in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 1997;82:151–155. doi: 10.1210/jcem.82.1.3687. [DOI] [PubMed] [Google Scholar]
- 17.Villanueva RB, Brett E, Gabrilove JL. A cluster of cases of factitious Cushing’s syndrome. Endocrine Practice. 2000;6920:143–7. doi: 10.4158/EP.6.2.143. [DOI] [PubMed] [Google Scholar]
- 18.Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Neimann LK, Chrousos GP, Papanicolaou OA. Paradoxical response to dexamethasone in the diagnosis of PPNAD. Ann Intern Med. 199;131:585–591. doi: 10.7326/0003-4819-131-8-199910190-00006. [DOI] [PubMed] [Google Scholar]
- 19.Bordeau I, Lacroix A, Schurch W, Caron P, Antakly T, Stratakis CA. PPNAD: Pararadoxical responses of cortisol secretion to dexamethasone OCCUI in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88:3931–3937. doi: 10.1210/jc.2002-022001. [DOI] [PubMed] [Google Scholar]
- 20.Levine RJ, Schulte HM, Gallucci WT, Cutler GB, Loriaux OJ, Chrousos GP. Ovine cortitropin-releasing hormone stimulation test in normal children. J Clin Endocrinol Metab. 1986;62:390. doi: 10.1210/jcem-62-2-390. [DOI] [PubMed] [Google Scholar]
- 21.Gomez Muguniza MT, Chrousos GP. Periodic Cushing syndrome in a short boy: usefulness of ovine cortitropin releasing hormone test. J Pediatr. 1989;115:270–273. doi: 10.1016/s0022-3476(89)80081-2. [DOI] [PubMed] [Google Scholar]
- 22.Legro RS, Lin HM, Demers LM, Lloyd T. Urinary free cortisol increases in adolescent Caucasian females during perimenarche. J Clinic Endocrinol Metab. 2003;88(1):215–219. doi: 10.1210/jc.2002-020256. [DOI] [PubMed] [Google Scholar]
- 23.Gomez MT, Malozowski S, Winterer J, Vamvakopoulos NC, Chrousos GP. Urinary free cortisol values in normal children and adolescents. J Pediatr. 1991 Feb;118(2):256–8. doi: 10.1016/s0022-3476(05)80496-2. [DOI] [PubMed] [Google Scholar]
- 24.Stratakis CA, Carney JA, Jing-Ping Lin, Papanicolaou DA, Karl M, Kastner DL, Pras E, Chrousos GP. Carney complex, a Familial Multiple Neoplasia and Lentiginosis Syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doppman JL, Travis WD, BIeman L, Miller DL, Chrousos GP, Gomez MT, et al. Cushing syndrome due to primary pigmented nodular adrenocortical disease: fiundings at CT and MR imaging. Radiology. 1989;172:415–420. doi: 10.1148/radiology.172.2.2748822. [DOI] [PubMed] [Google Scholar]
- 26.Horvath A, Boikos S, Giatzakis C, et al. A genome wide scan identifies mutations in the gene encoding phosphodiesterase 11 A (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet. 2006;38:794–800. doi: 10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 27.Groussin L, Horvath A, Jullian E, et al. PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab. 2006;91:1943–1949. doi: 10.1210/jc.2005-2708. [DOI] [PubMed] [Google Scholar]