

Abstract
While P2rx7 has been proposed as a type 1 diabetes (T1D) susceptibility gene in NOD mice, its potential pathogenic role has not been directly determined. To test this possibility, we generated a new NOD stock deficient in P2X7 receptors. T1D development was not altered by P2X7 ablation. Previous studies found CD38 knockout (KO) NOD mice developed accelerated T1D partly due to a loss of CD4+ invariant NKT (iNKT) cells and Foxp3+ regulatory T-cells (Tregs). These immunoregulatory T-cell populations are highly sensitive to NAD induced cell death (NICD) activated by ADP-ribosyltransferase-2 (ART2)-mediated ADP-ribosylation of P2X7 receptors. Therefore, we asked if T1D acceleration was suppressed in a double knockout (DKO) NOD stock lacking both P2X7 and CD38 by rescuing CD4+ iNKT-cells and Tregs from NICD. We demonstrated P2X7 was required for T1D acceleration induced by CD38 deficiency. The CD38 KO-induced defects in homeostasis of CD4+ iNKT-cells and Tregs were corrected by co-ablation of P2X7. T1D acceleration in CD38-deficient NOD mice also requires ART2 expression. If increased ADP-ribosylation of P2X7 in CD38-deficient NOD mice underlies disease acceleration, then a comparable T1D incidence should be induced by co-ablation of both CD38 and ART2, or CD38 and P2X7. However, a previously established NOD stock deficient in both CD38 and ART2 expression is T1D resistant. The current study revealed the presence of a T1D resistance gene closely linked to the ablated Cd38 allele in the previously reported NOD stock also lacking ART2, but not in the newly generated CD38/P2X7 DKO line.
Introduction
The P2X7 receptor is an ATP-gated ion channel widely expressed in a variety of cell types including those in the immune system (1). Brief stimulation of the P2X7 receptor with ATP reversibly opens a membrane channel and rapidly triggers influx of Na+ and Ca2+, and efflux of K+, while its prolonged activation leads to formation of a nonselective pore that allow the passage of molecules up to 900 Da (2, 3). In the immune system, the function of P2X7 has been mostly studied in macrophages and T-cells. ATP-mediated P2X7 activation facilitates macrophage maturation and IL-1β secretion (4). It has also been shown that effective T-cell activation requires ATP release and the autocrine feedback through P2X7 receptors (5, 6). Recent studies have identified an alternative mechanism that indirectly activates P2X7 receptors. ADP-ribosyltransferase-2 (ART2)-mediated and NAD-dependent ADP-ribosylation of P2X7 also activates this receptor (7). Two isoforms of ART2 (ART2.1 and ART2.2) encoded by tandem genes are expressed in mice (8). Both ART2.1 and ART2.2 are expressed by T-cells in most inbred mouse strains (9-12). Expression of ART2.1 is induced in macrophages in response to several inflammatory stimuli (13). ART2.1-mediated ADP-ribosylation has been shown to lower the threshold required for P2X7 activation in macrophages in response to extracellular ATP (14). On the other hand, ART2-mediated ADP-ribosylation of P2X7 receptors on T-cells results in NAD induced cell death (NICD) (7).
Polymorphisms in the P2rx7 gene resulting in a single change at amino acid residue 451 (Pro or Leu) of the encoded protein have been described in common inbred mouse strains (15). Autoimmune type 1 diabetes (T1D) susceptible NOD mice harbor the “wild-type” allele encoding proline at position 451 that allows for high sensitivity to ATP (15). Conversely, the “mutant” allele expressed by C57BL/6 and C57BL/10 mice encodes a leucine at the same position that significantly increases the threshold required for ATP-induced P2X7 activation (15). Considering the diverse roles of P2X7 in the immune system, it has been proposed as a candidate T1D susceptibility (Idd) gene in NOD mice (16). However, this hypothesis has not been directly tested. We previously showed T-cell mediated autoimmune T1D was accelerated in CD38 knockout (KO) NOD mice (17). Disease acceleration in NOD.CD38-/- mice required the expression of ART2 molecules (17). T1D acceleration in NOD.CD38-/- mice was associated with a loss of CD4+ invariant NKT (iNKT)-cells and Foxp3+ regulatory T-cells (Treg) (17, 18). CD38 is an ectoenzyme that utilizes NAD to generate multiple calcium (Ca2+)-mobilizing metabolites, including adenosine diphosphate ribose (ADPR) and cyclic ADPR (cADPR) (19). CD38 counter-regulates ART2-dependent ADP-ribosylation levels of T-cell surface proteins including P2X7 by competing for their common NAD substrate (20). Therefore, in the absence of the NAD hydrolyzing activity of CD38, elevated concentrations of extracellular NAD allow for higher levels of ART2-mediated ADP-ribosylation of P2X7 receptors on T-cells and their subsequent loss by NICD. CD4+ iNKT-cells are extremely sensitive to NICD, most likely due to high expression levels of both ART2 and P2X7 (18, 21). Similarly, Tregs have also been reported to be highly sensitive to NICD (22).
The collective results described above suggested, but did not directly assess whether T1D acceleration in NOD.CD38-/- mice also relies on the presence of P2X7 molecules. It was also unknown if concurrent deficiency of P2X7 molecules could restore iNKT cells and Tregs in the CD38 knockout stock to the levels observed in standard NOD mice. Furthermore, it remains to be determined if independently of its role in mediating the loss of immunoregulatory populations through an NICD mechanism, whether P2X7 might also contribute to T1D development through its capacity to serve as an ATP sensor in macrophages and T-cells that respectively promotes IL-1β secretion and effector function activation (23). To directly test these possibilities, we determined if T1D development was altered in a newly generated P2X7-deficient NOD stock. We also determined if a functional interaction between CD38 and P2X7 molecules modulates T1D development.
Materials and Methods
Mice
P2rx7 KO mice on the C57BL/6J background were originally obtained from the Induced Mutant Resource at The Jackson Laboratory (JAX stock number 5576; ref (24)), and the mutation backcrossed to the NOD/LtDvs strain. Linkage markers delineating all known Idd genetic loci have been fixed to homozygosity for NOD alleles in the strain now designated NOD.P2X7-/-, currently maintained at the N9 backcross generation with a congenic interval of <49 Mb (the proximal boundary between D5Mit259 and P2rx7; the distal boundary between D5Mit138 and D5Mit98). The donor strain congenic interval of the previously reported NOD.CD38-/- mouse stock has been further shortened (ref (17) and Figure 6A). The newly developed NOD.CD38-/- stock was then used to generate NOD.CD38-/-.P2X7-/- mice. Because the Cd38 and P2rx7 genes are both on Chromosome (Chr.) 5, this required identification and breeding of a double recombinant (17). NOD.ART2-/- mice have been previously described (17). A stock of NOD mice deficient in both CD38 and ART2 molecules was also generated with the newly developed NOD.CD38-/- strain, denoted as NOD.CD38-/-<short>.ART2-/- hereafter. To distinguish the new NOD.CD38-/-<short>.ART2-/- stock from the previously described NOD.CD38-/-.ART2-/- stock with the longer congenic interval around the Cd38 locus, the latter is denoted as NOD.CD38-/-<long>.ART2-/- hereafter.
Figure 6.

Identification of a NOD T1D susceptibility genetic locus closely linked to Cd38 on Chromosome 5. (A) Genetic map showing the Cd38 and P2rx7 congenic regions in the indicated strains. The physical locations of the markers/genes are based on NCBI Build 37. NA, not available. (B) T1D incidence of female NOD mice and stocks deficient in CD38 and/or ART2 molecules. *P<0.005 compared to NOD mice (Kaplan–Meier log-rank analysis).
Flow cytometry
Red blood cell (RBC) depleted single cell suspensions were prepared from spleens and pancreatic lymph nodes. P2X7 expression by T-cells was analyzed by staining leukocytes with the unconjugated HANO43 antibody and allophycocyanin (AP)-conjugated anti-CD3ε (clone 145-2C11) followed by FITC-conjugated polyclonal goat anti-rat immunoglobulin. Regulatory T-cells (Treg) were identified by surface staining with anti-CD4 (clone RM4-5) and anti-CD25 (clone PC61), and intracellular staining with anti-Foxp3 (clone FJK-16s) according to instructions of the kit from eBioscience (San Diego, CA). For iNKT-cell staining, cells were first treated with Fc block (anti-CD16/CD32, clone 2.4G2) at room temperature for 10 min and then incubated with a cocktail containing anti-CD4, anti-TCRβ and the α-galactosylceramide (α-GalCer) analogue PBS-57 loaded CD1d-tetramer (obtained from NIH tetramer core facility) at 4°C for 30 min. Stained cells were washed and analyzed on a FACSCalibur flow cytometer (Becton Dickinson) using CellQuest software. Anti-CD4, anti-CD3ε, anti-CD25, and anti-TCRβ were purchased from BD Biosciences (San Jose, CA). Anti-P2X7 was obtained from Enzo Biosciences (Plymouth Meeting, PA). To analyze iNKT cell functional activity, mice were injected i.v. with 2 μg of α-GalCer (Enzo Life Sciences). Production of IFN-γ and IL-4 by iNKT cells were analyzed by intracellular staining at 2 hours after α-GalCer administration. Stained cells were analyzed on a LSRII flow cytometer (Becton Dickinson). Both IFN-γ and IL-4 antibodies are from eBioscience.
NICD analysis
To determine the effect of NAD on T-cell viability, RBC depleted splenocytes (5×106/ml) were incubated in previously described RPMI 1640 tissue culture medium (25) containing varying concentrations of NAD (Sigma, St. Louis, MO) at 37°C for 30 min. Cells were then washed and resuspended in Annexin V friendly FACS buffer, and stained with Annexin V (BD Biosciences) and anti-CD3ε at 4°C for 30 min as described (18). Propidium iodine was added to all samples before analyses on a FACSCalibur flow cytometer (Becton Dickinson).
T1D incidence study
T1D was assessed by weekly monitoring of glycosuria development with Ames Diastrix (Bayer, Diagnostics Division, Elkhart, NJ), and disease onset defined as two consecutive values of ≥3.
In vitro Treg assay
Treg assays were performed as previously described (26). Briefly, splenic CD4 T-cells were purified from 6-8 week old mice by depleting B220+, CD8+, and CD11b+ cells with the previously described magnetic bead system. The CD25+ fraction was further isolated by staining with biotinylated anti-CD25 (clone 7D4) followed by streptavidin conjugated microbeads (Miltenyi Biotec). CD4+CD25+ cells were then enriched by positive selection using a LS column (Miltenyi Biotec). The CD4+CD25- fraction was used as the effector population. Effector cells were labeled with CFSE as previously described. Labeled effector cells (5×104) were co-cultured in triplicate with indicated numbers of CD4+CD25+ Tregs in the presence of 2×105 NOD.scid splenocytes and 5 μg/ml anti-CD3 (clone 145-2C11, BD Bioscience) in round bottomed 96-well tissue culture plates in a final volume of 200 μl of the previously described culture medium (25). Proliferation of effector cells was determined after three days of culture by CFSE dilution.
Results
Expression levels of P2X7 receptors dictate the sensitivity of mature T-cells to NICD
We first confirmed the absence of P2X7 protein expression and function in NOD.P2X7-/- mice. P2X7 expression was assessed by flow cytometry and functional activity assayed by the sensitivity of mature T-cells to NICD. As shown in Figure 1A, expression of P2X7 on the surface of splenic T-cells was completely absent in the NOD.P2X7-/- mice. P2X7 was expressed at an intermediate level in NOD.P2X7+/- mice compared to wild-type and KO littermates (Fig. 1A). Splenic T-cells from NOD.P2X7-/- mice were completely resistant to NICD (Figure 1B). On the other hand, splenic T-cells from NOD.P2X7+/- and wild-type littermates were sensitive to NICD (Fig. 1B). Interestingly, P2X7 expression levels directly correlated with the proportions of T-cells undergoing NICD (Fig. 1B). A small but significant proportion of NOD.P2X7+/- and wild-type T-cells also underwent apoptosis in the absence of exogenous NAD. This was most likely due to intracellular NAD released from lysed RBC or other dead cells during the preparation process (27).
Figure 1.
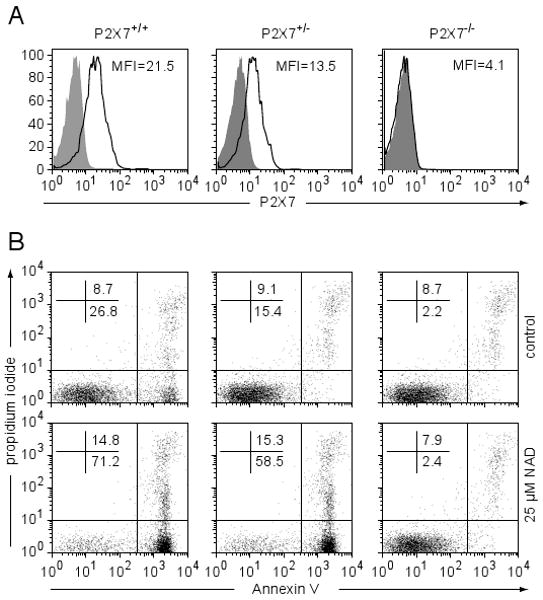
T-cells from NOD.P2X7-/- mice are resistant to NICD. (A) Flow cytometric analysis of P2X7 expression on total splenic T-cells (gated on CD3+). A representative histogram from at least 5 mice per genotype is shown. Shaded area, unstained control; open area, with anti-P2X7. (B) Splenocytes were incubated with or without 25 μM NAD at 37°C for 30 min. Apoptotic cells were determined by Annexin V and PI staining (gated on CD3+ cells). Similar results were obtained from another two experiments.
P2X7 deficiency alone does not alter T1D development in NOD mice
To determine if P2X7 plays a role in the development of T1D in NOD mice, disease onset rates were compared for littermates carrying the wild-type (+/+), heterozygous (+/-), or homozygous (-/-) P2rx7 KO alleles. In both males and females, no significant differences were found between any genotypes (Fig. 2).
Figure 2.

T1D incidence of NOD.P2X7-/- females (A) and males (B) and their wild-type and heterozygous littermate controls. No statistical significance differences were found between genotypes in both sexes.
CD38 deficiency-induced T1D acceleration in NOD mice is P2X7 dependent
Our previous studies suggested, but did not definitively demonstrate, that CD38 deficiency-induced T1D acceleration in NOD mice needs an intact NICD pathway, i.e., ART2-mediated ADP ribosylation and activation of P2X7 molecules (17). To directly test this hypothesis, we intercrossed CD38 and P2X7-deficient NOD stocks and generated NOD.CD38-/-.P2X7-/- mice. We directly compared the T1D development rates of wild-type, CD38 KO, P2X7 KO, and CD38/P2X7 double KO (DKO) NOD mice side by side. Consistent with the previously published results (17), the NOD.CD38-/- stock exhibited greatly accelerated T1D development compared to standard NOD mice (Fig. 3). Although P2X7 deficiency alone did not significantly affect disease onset (Fig 2), its absence completely abrogated accelerated T1D development in CD38-deficient DKO mice (Fig. 3; P=0.2712, NOD versus NOD.CD38-/-.P2X7-/-). Therefore, we conclude that ADP-ribosylation of P2X7 receptors and consequent triggering of NICD play an important role to promote accelerated T1D development in NOD.CD38-/- mice. We noted that the rate of T1D development is slightly greater in NOD.CD38-/-.P2X7-/- than in NOD.P2X7-/- mice. This suggests that in the absence of CD38 molecules, ART2 may promote T1D development independent of P2X7 by ADP-ribolsylation of other proteins. This possibility will be the subject of future studies.
Figure 3.

Type 1 diabetes incidence of NOD, NOD.CD38-/-, NOD.P2X7-/-, and NOD.CD38-/-.P2X7-/- females. *P<0.005 compared to all other three strains (Kaplan–Meier log-rank analysis). There is no statistical difference between NOD and NOD.P2X7-/- or NOD and NOD.CD38-/-.P2X7-/- mice. #The T1D development rate between NOD.P2X7-/-, and NOD.CD38-/-.P2X7-/- mice is significantly different (P=0.0304, Kaplan–Meier log-rank analysis).
P2X7 deficiency rescues the depleted iNKT and Treg compartments in NOD.CD38-/- mice
T1D acceleration in NOD.CD38-/- mice is associated with a loss of peripheral CD4+ iNKT-cells (18). Therefore, we asked if the frequencies and numbers of CD4+ iNKT-cells in the NOD.CD38-/-.P2X7-/- stock are restored to the levels in standard NOD mice. We determined the numbers of CD4+ and DN iNKT-cells in the spleens and pancreatic lymph nodes (PLNs) of standard NOD, NOD.CD38-/-, NOD.P2X7-/-, and NOD.CD38-/-.P2X7-/- mice. As expected, the NOD.CD38-/- stock had significantly reduced numbers of splenic CD4+ iNKT-cells compared to standard NOD mice (Fig. 4A). Interestingly, numbers of splenic CD4+ iNKT-cells were not only restored in the NOD.CD38-/-.P2X7-/- stock but actually greater than in standard NOD mice (Fig. 4A). This was also true for NOD.P2X7-/- mice (Fig. 4A). Hence, the loss of immunoregulatory CD4+ iNKT-cells in NOD.CD38-/- mice is a P2X7 dependent process. However, P2X7 ablation also allows for an expansion of CD4+ iNKT-cells in CD38 intact NOD mice. Consistent with our previous results (18), standard NOD and NOD.CD38-/- mice had similar levels of splenic DN iNKT-cells (Fig. 4B). Compared to standard NOD mice, the NOD.CD38-/-.P2X7-/- stock, but not NOD.P2X7-/- mice, also had increased numbers of splenic DN iNKT-cells (Fig. 4B). Similarly, the loss of CD4+ iNKT-cells in the PLNs of NOD.CD38-/- mice was also rescued by the concomitant deficiency of P2X7 molecules (Fig. 4C). No differences in the numbers of PLN DN iNKT-cells was observed in standard NOD mice and those deficient in CD38 and/or P2X7 molecules (Fig. 4D). While CD38-deficient mice have fewer iNKT cells, we previously showed that the remaining ones responded normally to α-GalCer stimulation (18). In the current study, we also tested the function of CD38 and P2X7 double deficient iNKT cells. Upon α-GalCer stimulation, iNKT cells in standard NOD mice and the stock deficient in both CD38 and P2X7 responded similarly as determined by intracellular IFN-γ and IL-4 staining (data not shown).
Figure 4.

Co-ablation of P2X7 improves the survival of CD4+ iNKT-cells in CD38 deficient NOD mice. The numbers of iNKT-cell subsets were determined in 6-8 week-old female mice. Splenic (A and B) and pancreatic lymph node (C and D) iNKT-cells were identified by CD1d tetramer staining and further separated into CD4+ (A and C) and DN (B and D) subsets. * P<0.05, ** P<0.01, *** P<0.005 compared to NOD mice (Mann-Whitney test, n=6-9 per genotype).
NOD.CD38-/- mice also have reduced frequencies of Tregs (CD4+CD25+Foxp3+) in PLNs (17). Therefore, we determined if Tregs in the NOD.CD38-/-.P2X7-/- stock also return to the level characterizing standard NOD mice. While the frequencies of Tregs were not significantly different in the spleens of NOD and NOD.CD38-/- mice, we observed a trend of reduced Foxp3 protein expression (based on mean fluorescence intensity (MFI) of specific antibody staining) in the latter stock, consistent with our previous analysis at the RNA transcript level (17) (Fig. 5A). In contrast, as shown in Figure 5A, the ablation of P2X7 resulted in marginal, but a statistically significant increase in the percentages of splenic Tregs in both CD38-deficient and intact NOD mice. Also in agreement with previous studies (17), the NOD.CD38-/- stock had reduced percentages of Tregs in PLNs compared to standard NOD mice (Fig. 5B). Similarly, Foxp3 expression in CD38-deficient Tregs was also reduced compared to those in standard NOD mice (Fig. 5B). These defects were completely reversed in the concurrent absence of P2X7 molecules (Fig. 5B). To compare the function of Tregs in standard NOD mice and those deficient in CD38 and/or P2X7 molecules, we determined their suppressive activity in vitro. As previously described (21), Tregs from the NOD.CD38-/- stock had reduced suppressive activity compared to those in standard NOD mice (Fig. 5C). While P2X7 deficiency alone did not alter the suppressive activity of Tregs (Fig. 5D), it completely reversed the defect of those from CD38 deficient NOD mice (Fig. 5E vs. 5C). These results indicate that the impaired function of Tregs in CD38 deficient NOD mice depends on the presence of intact P2X7 molecules.
Figure 5.
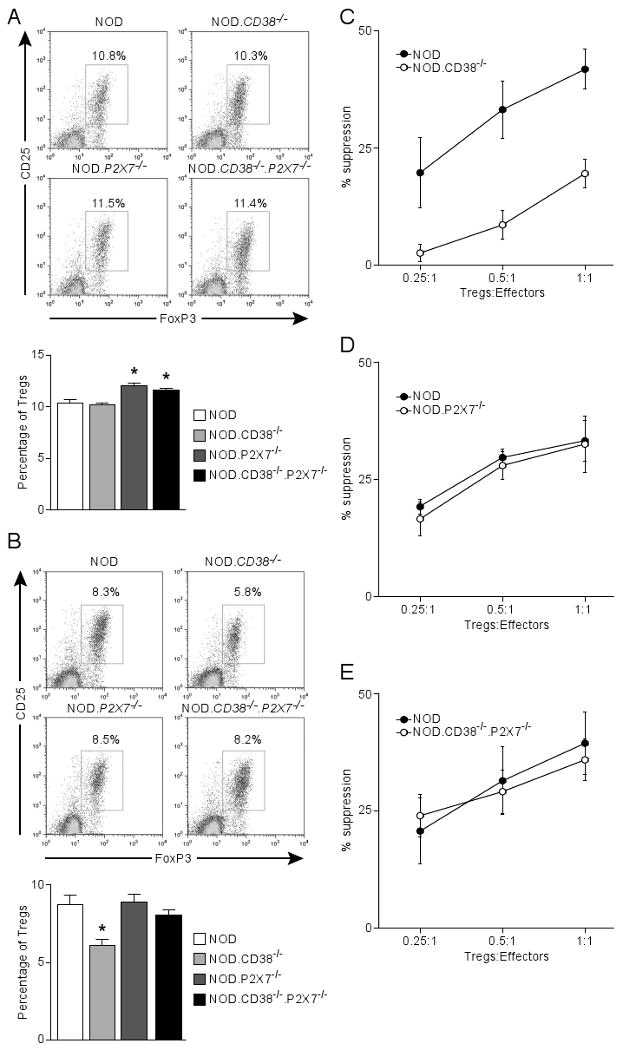
Co-ablation of P2X7 improves the survival and function of Tregs in CD38-deficient NOD mice. The frequency of CD4 Tregs (CD25+Foxp3+) was determined in the spleens (A) and PLN (B) of 10-11 week-old male NOD mice and those deficient in CD38 and/or P2X7. (A) The upper panel shows the representative CD25 and Foxp3 staining (gated on CD4 T cells). Results are summarized in the lower panel. *P<0.05 compared to NOD mice (Mann-Whitney test, n=5-6 per genotype). (B) The upper panel shows the representative CD25 and Foxp3 staining (gated on CD4 T cells). Results are summarized in the lower panel. *P<0.05 compared to NOD mice (Mann-Whitney test, n=5-6 per genotype). (C, D, E) In vitro Treg assay comparing the suppressive activity between indicated strains (age-matched females at 6-8 weeks). NOD effector T-cells (CD4+CD25-) were labeled with CFSE and co-cultured at indicated ratios with Tregs (CD4+CD25+) in triplicate in a 96-well plate in the presence of NOD.scid splenocytes (2×105) and 5 μg/ml anti-CD3 for 3 days. Proliferation of effector T-cells was determined by CFSE dilution. The percentage of suppression is defined by the reduction in the proportion of divided effector T-cells relative to that of the control without Tregs. Results indicate the mean±sem of 3 independent experiments.
A locus closely linked to CD38 regulates T1D development
We previously showed that further depletion of the iNKT and Treg compartments apparently contributing to accelerated T1D development in NOD.CD38-/- mice depended on the expression of ART2 molecules (17, 18). Together with the results reported herein, we concluded that the further loss of iNKT-cells, particularly the CD4+ subset, and Tregs in NOD.CD38-/- mice are due to P2X7 dependent NICD. However, in contrast to the observation in the current study that co-depletion of CD38 and P2X7did not alter the rate of T1D onset in NOD background mice, disease development was strongly suppressed in a previously reported stock lacking both CD38 and ART2 (denoted as NOD.CD38-/-<long>.ART2-/- in the current study) (17). This suggested that in the absence of both CD38 and ART2-mediated NAD-degrading activities, elevated levels of extra- and/or intracellular NAD may exert T1D suppressive effects. It was also possible that a 129/Sv or C57BL/6 (B6)-derived T1D resistance gene(s) closely linked to the targeted Cd38 and/or Art2 alleles was inadvertently introduced into the previously reported NOD DKO stock during the backcrossing process. The latter possibility is supported by a recent study identifying a novel 129/Sv-derived T1D resistance locus on Chr. 5 in close linkage to the Cd38 gene (28). To test these possibilities, we used the currently reported NOD.CD38-/- mice with a shortened congenic interval (Fig. 6A) to generate a new stock lacking both CD38 and ART2 (denoted as NOD.CD38-/-<short>.ART2-/-) and monitored them for T1D development. It should be noted that the Chr. 5 congenic interval carrying the P2rx7 targeted allele does not overlap the 129/Sv and/or B6 derived region linked to the ablated Cd38 gene present in the original NOD.CD38-/-<long>.ART2-/- stock (Fig. 6A).
Consistent with previous results, NOD.CD38-/- and NOD.CD38-/-<long>.ART2-/- stocks were respectively more susceptible or resistant to T1D compared to standard NOD mice (17) (Fig. 6B). Also as shown previously, ART2 deficiency alone did not alter the course of T1D development in NOD mice (17) (Fig. 6B). Interestingly, the newly generated NOD.CD38-/-<short>.ART2-/- stock displayed a similar T1D incidence as standard NOD mice (Fig. 6B). Collectively, these results indicate that T1D resistance observed in NOD.CD38-/-<long>.ART2-/-mice is largely due to the presence of a 129/Sv and/or B6 derived disease protective gene(s) closely linked to the inactivated Cd38 allele.
Discussion
A P2X7 receptor variant has been previously suggested as a candidate T1D susceptibility gene in NOD mice (16). In the current study, we found P2X7 deficiency alone does not alter T1D development in NOD background mice. However, P2X7 is required to promote the accelerated rate of T1D onset characterizing NOD.CD38-/- mice. This was demonstrated by the finding that a newly generated NOD.CD38-/-.P2X7-/- stock developed T1D at the same rate as standard NOD mice. In the T-cell compartment, CD4+ iNKT-cells and Tregs are highly sensitive to P2X7-dependent ATP or NAD-induced cell death (18, 22). While tight control mechanisms normally do not allow plasma ATP and NAD to reach levels sufficient to activate P2X7 mediated cell death, local concentrations of these small molecules could be significantly increased as a result of inflammation or tissue destruction (29). Indeed, it has recently been shown that high concentrations of extracellular NAD depleted Tregs in vivo and promoted immunogenic responses (30). Therefore, it is tempting to speculate that the suboptimal survival of Tregs in inflamed pancreatic islets in NOD mice is in part due to P2X7 dependent NICD in addition to the previously reported reduction in IL2-IL2R signaling (31). As a result of intra-islet Treg loss, the pathogenic functions of β-cell specific effector T-cells are further unleashed, possibly through enhanced accumulation of islet infiltrated and fully activated dendritic cells (32). Activated T-cells shed ART2 molecules and become resistant to NICD (33). Therefore, P2X7 dependent NICD can also promote effector T-cell activity by eliminating bystander naïve T-cells that express high levels of ART2 (29). P2X7 receptors can also regulate T-cell activation directly. It has recently been demonstrated that stimulated T-cells release ATP, which in turn stimulates P2X7 molecules in an autocrine fashion to further promote their activation (5, 6).
The previously reported findings described above would seemingly have predicted suppressed T1D development in NOD. P2X7-/- mice, a result that was not observed. On the other hand, the P2X7 dependent cell death induction mechanism may also limit the initial activation of effector T-cells. It has been reported that exacerbation of experimental autoimmune encephalomyelitis (EAE) in P2X7 deficient mice is associated with reduced lymphocyte apoptosis in the inflamed tissues (34). Therefore, it is possible that pathogenic T-cells remain susceptible to P2X7 dependent T-cell death induction during the initial phase of their activation after encountering self-antigens and prior to shedding of ART2 molecules. Lack of P2X7 may allow β-cell autoreactive T-cells to escape from this tolerance induction mechanism that normally limits the extent of self-reactivity. In this case, one would predict accelerated T1D onset in NOD.P2X7-/- mice. However, our results demonstrate that T1D development in NOD.P2X7-/- mice is not altered. It is possible that a combination of multiple T1D promoting and suppressing functions of the P2X7 receptor on various cell populations in NOD mice mask its regulatory effects in this disease model.
In the absence of CD38 molecules, the balance is tipped to favor the pro-inflammatory functions of P2X7 receptors as a result of elevated extracellular levels of NAD. At the cellular level, the most apparent outcome of CD38 deficiency on the NOD background is the loss of CD4+ iNKT-cells and Tregs, which in turn contributes to accelerated T1D development in these mice. Both NOD.CD38-/-.P2X7-/- and NOD.CD38-/-<short>.ART2-/- exhibited similar T1D levels as standard NOD mice, indicating an important role of NICD in the regulation of disease development. In addition, elevated levels of extracellular NAD may also increase ADP-ribosylation of P2X7 receptors on macrophages, which in turn promotes their production of the pro-inflammatory cytokine IL-1β (4).
In the current study, we also revealed a NOD-derived T1D susceptibility locus closely linked to the Cd38 gene. The most apparent candidate region to harbor this T1D susceptibility locus is defined by the Cd38 gene (44.3 Mb) and the microsatellite marker D5Mit259 (89.7 Mb) (Fig. 6A). However, we could not rule out the possibility that the region proximal to the Cd38 gene was also truncated but not revealed by the markers used to define the congenic region. This possibility is supported by the discovery of a 129/Sv-derived T1D resistance locus with the LOD score peaking at 31 Mb, proximal to the Cd38 gene, in a previous QTL analysis (28). Therefore, the location of this T1D susceptibility gene(s) reported in the current study can only be defined by the markers D5Mit75 (36.6 Mb) and D5Mit259 (89.7Mb). It is also possible that the congenic region carrying the Art2 mutant allele on Chr. 7 was also truncated without being revealed by the microsatellite markers used to type the interval in the newly generated NOD.CD38-/-<short>.ART2-/- mice. However, this is not likely to be the reason why the newly generated NOD.CD38-/-<short>.ART2-/- stock became T1D susceptible in contrast to the marked T1D resistance in the previously reported DKO line. This is because NOD.ART2-/- and standard NOD mice developed T1D at the same rate, indicating the 129/Sv-derived congenic region carrying the Art2 mutant allele alone does not suppress disease development. In addition, no 129/Sv T1D resistance loci were found on Chr. 7 in the previously reported QTL analysis [28].
In conclusion, we demonstrated that P2X7 deficiency alone does not alter T1D incidence in NOD mice. However, T1D acceleration in NOD.CD38-/- mice requires intact P2X7 molecules. Similarly, expression of ART2 is also essential for accelerated T1D development in CD38-deficient NOD mice. Collectively, these results indicate an important role of NICD in the regulation of T1D in NOD mice. Our study also provides an example of how molecules in the same pathway can interactively modulate T1D development.
Footnotes
We thank Brenice Timms and Pam Stanley for their technical assistant. This work was supported by National Institutes of Health grants DK46266 and DK51090 (to D.V. Serreze), DK36175 and DK27722 (to E.H. Leiter), and DK077443 (to Y.-G. Chen); as well as by grants from the Juvenile Diabetes Research Foundation International (to D.V. Serreze).
References
- 1.Chen L, Brosnan CF. Regulation of immune response by P2X7 receptor. Crit Rev Immunol. 2006;26:499–513. doi: 10.1615/critrevimmunol.v26.i6.30. [DOI] [PubMed] [Google Scholar]
- 2.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 3.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 5.Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, Ferrari V, Insel PA, Junger WG. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. Faseb J. 2009;23:1685–1693. doi: 10.1096/fj.08-126458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, Verderio C, Buer J, Scanziani E, Grassi F. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal. 2008;1:ra6. doi: 10.1126/scisignal.1160583. [DOI] [PubMed] [Google Scholar]
- 7.Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, Deterre P, Haag F, Koch-Nolte F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity. 2003;19:571–582. doi: 10.1016/s1074-7613(03)00266-8. [DOI] [PubMed] [Google Scholar]
- 8.Seman M, Adriouch S, Hagg F, Koch-Nolte F. Ecto-ADP-ribosyltransferases (ARTs): emerging actors in cell communication and signaling. Curr Med Chem. 2004;11:857–872. doi: 10.2174/0929867043455611. [DOI] [PubMed] [Google Scholar]
- 9.Koch-Nolte F, Duffy T, Nissen M, Kahl S, Killeen N, Ablamunits V, Hagg F, Leiter EH. A new monoclonal antibody detects a developmentally regulated mouse ecto-ADP-ribosyltransferase on T cells: subset distribution, inbred strain variation, and modulation upon T cell activation. J Immunol. 1999;163:6014–6022. [PubMed] [Google Scholar]
- 10.Adriouch S, Ohlrogge W, Haag F, Koch-Nolte F, Seman M. Rapid induction of naive T cell apoptosis by ecto-nicotinamide adenine dinucleotide: requirement for mono(ADP-ribosyl)transferase 2 and a downstream effector. J Immunol. 2001;167:196–203. doi: 10.4049/jimmunol.167.1.196. [DOI] [PubMed] [Google Scholar]
- 11.Haag F, Freese D, Scheublein F, Ohlrogge W, Adriouch S, Seman M, Koch-Nolte F. T cells of different developmental stages differ in sensitivity to apoptosis induced by extracellular NAD. Dev Immunol. 2002;9:197–202. doi: 10.1080/10446670310001593514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohlrogge W, Haag F, Lohler J, Seman M, Littman DR, Killeen N, Koch-Nolte F. Generation and characterization of ecto-ADP-ribosyltransferase ART2.1/ART2.2-deficient mice. Mol Cell Biol. 2002;22:7535–7542. doi: 10.1128/MCB.22.21.7535-7542.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong S, Brass A, Seman M, Haag F, Koch-Nolte F, Dubyak GR. Lipopolysaccharide, IFN-gamma, and IFN-beta induce expression of the thiol-sensitive ART2.1 Ecto-ADP-ribosyltransferase in murine macrophages. J Immunol. 2007;179:6215–6227. doi: 10.4049/jimmunol.179.9.6215. [DOI] [PubMed] [Google Scholar]
- 14.Hong S, Schwarz N, Brass A, Seman M, Hagg F, Koch-Nolte F, Schilling WP, Dubyak GR. Differential regulation of P2X7 receptor activation by extracellular nicotinamide adenine dinucleotide and ecto-ADP-ribosyltransferases in murine macrophages and T cells. J Immunol. 2009;183:578–592. doi: 10.4049/jimmunol.0900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adriouch S, Dox C, Welge V, Seman M, Koch-Nolte F, Haag F. Cutting edge: a natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2X7 receptor. J Immunol. 2002;169:4108–4112. doi: 10.4049/jimmunol.169.8.4108. [DOI] [PubMed] [Google Scholar]
- 16.Elliott JI, Higgins CF. Major histocompatibility complex class I shedding and programmed cell death stimulated through the proinflammatory P2X7 receptor: a candidate susceptibility gene for NOD diabetes. Diabetes. 2004;53:2012–2017. doi: 10.2337/diabetes.53.8.2012. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Chen YG, Reifsnyder PC, Schott WH, Lee CH, Osborne M, Scheuplein F, Haag F, Koch-Nolte F, Serreze DV, Leiter EH. Targeted disruption of CD38 accelerates autoimmune diabetes in NOD/Lt mice by enhancing autoimmunity in an ADP-ribosyltransferase 2-dependent fashion. J Immunol. 2006;176:4590–4599. doi: 10.4049/jimmunol.176.8.4590. [DOI] [PubMed] [Google Scholar]
- 18.Chen YG, Chen J, Osborne MA, Chapman HD, Besra GS, Porcelli SA, Leiter EH, Wilson SB, Serreze DV. CD38 is required for the peripheral survival of immunotolerogenic CD4+ invariant NK T cells in nonobese diabetic mice. J Immunol. 2006;177:2939–2947. doi: 10.4049/jimmunol.177.5.2939. [DOI] [PubMed] [Google Scholar]
- 19.Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262:1056–1059. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 20.Krebs C, Adriouch S, Braasch F, Koestner W, Leiter EH, Seman M, Lund FE, Oppenheimer N, Haag F, Koch-Nolte F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J Immunol. 2005;174:3298–3305. doi: 10.4049/jimmunol.174.6.3298. [DOI] [PubMed] [Google Scholar]
- 21.Scheuplein F, Rissiek B, Driver JP, Chen YG, Koch-Nolte F, Serreze DV. A recombinant heavy chain antibody approach blocks ART2 mediated deletion of an iNKT cell population that upon activation inhibits autoimmune diabetes. J Autoimmun. 2010;34:145–154. doi: 10.1016/j.jaut.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aswad F, Kawamura H, Dennert G. High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: a role for P2X7 receptors. J Immunol. 2005;175:3075–3083. doi: 10.4049/jimmunol.175.5.3075. [DOI] [PubMed] [Google Scholar]
- 23.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 25.Serreze DV, Leiter EH. Defective activation of T suppressor cell function in Nonobese Diabetic mice. Potential relation to cytokine deficiencies. J Immunol. 1988;140:3801–3807. [PubMed] [Google Scholar]
- 26.Chen YG, Scheuplein F, Osborne MA, Tsaih SW, Chapman HD, Serreze DV. Idd9/11 Genetic Locus Regulates Diabetogenic Activity of CD4 T-Cells in Nonobese Diabetic (NOD) Mice. Diabetes. 2008;57:3273–3280. doi: 10.2337/db08-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheuplein F, Schwarz F, Adriouch S, Krebs C, Bannas P, Rissiek B, Seman M, Haag F, Koch-Nolte F. NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol. 2009;182:2898–2908. doi: 10.4049/jimmunol.0801711. [DOI] [PubMed] [Google Scholar]
- 28.Leiter EH, Reifsnyder PC, Wallace R, Li R, King B, Churchill GC. NOD × 129.H2(g7) backcross delineates 129S1/SvImJ-derived genomic regions modulating type 1 diabetes development in mice. Diabetes. 2009;58:1700–1703. doi: 10.2337/db09-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adriouch S, Hubert S, Pechberty S, Koch-Nolte F, Haag F, Seman M. NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells in vivo. J Immunol. 2007;179:186–194. doi: 10.4049/jimmunol.179.1.186. [DOI] [PubMed] [Google Scholar]
- 30.Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, Koch-Nolte F, Boyer O, Seman M, Adriouch S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J Exp Med. 2010;207:2561–2568. doi: 10.1084/jem.20091154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MH, Lee WH, Todorov I, Liu CP. CD4+ CD25+ regulatory T cells prevent type 1 diabetes preceded by dendritic cell-dominant invasive insulitis by affecting chemotaxis and local invasiveness of dendritic cells. J Immunol. 2010;185:2493–2501. doi: 10.4049/jimmunol.1001036. [DOI] [PubMed] [Google Scholar]
- 33.Kahl S, Nissen M, Girisch R, Duffy T, Leiter EH, Haag F, Koch-Nolte F. Metalloprotease-mediated shedding of enzymatically active mouse ecto-ADP-ribosyltransferase ART2.2 upon T cell activation. J Immunol. 2000;165:4463–4469. doi: 10.4049/jimmunol.165.8.4463. [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Brosnan CF. Exacerbation of experimental autoimmune encephalomyelitis in P2X7R-/- mice: evidence for loss of apoptotic activity in lymphocytes. J Immunol. 2006;176:3115–3126. doi: 10.4049/jimmunol.176.5.3115. [DOI] [PubMed] [Google Scholar]