

Abstract
In the present work, we described the activity of the thiosemicarbazone derived from 5,6-dimethoxy-1-indanone (TSC), which we previously characterized as a new compound that inhibits bovine viral diarrhea virus (BVDV) infection. We showed that TSC acts at a point of time that coincides with the onset of viral RNA synthesis and that it inhibits the activity of BVDV replication complexes (RCs). Moreover, we have selected five BVDV mutants that turned out to be highly resistant to TSC but still susceptible to ribavirin (RBV). Four of these resistant mutants carried an N264D mutation in the viral RNA-dependent RNA polymerase (RdRp). The remaining mutant showed an A392E mutation within the same protein. Some of these mutants replicated slower than the wild-type (wt) virus in the absence of TSC, whereas others showed a partial reversion to the wt phenotype over several passages in the absence of the compound. The docking of TSC in the crystal structure of the BVDV RdRp revealed a close contact between the indane ring of the compound and several residues within the fingers domain of the enzyme, some hydrophobic contacts, and hydrogen bonds with the thiosemicarbazone group. Finally, in the mutated RdRp from resistant BVDV, these interactions with TSC could not be achieved. Interestingly, TSC inhibited BVDV replication in cell culture synergistically with RBV. In conclusion, TSC emerges as a new nonnucleoside inhibitor of BVDV RdRp that is synergistic with RBV, a feature that turns it into a potential compound to be evaluated against hepatitis C virus (HCV).
INTRODUCTION
Bovine viral diarrhea virus (BVDV), an enveloped single-stranded positive-sense RNA virus, is one of the best characterized members of the Pestivirus genus. This genus and the genera Flavivirus and Hepacivirus constitute the family Flaviviridae. BVDV infection is distributed worldwide, and in many countries it tends to be endemic (3, 11). In Argentina, the prevalence of BVDV antibodies in adult cattle is around 70% (27, 31).
Because the genome organization, translation, replication strategy, and protein functions of pestiviruses closely resemble those of hepatitis C virus (HCV), the sole member of the Hepacivirus genus, BVDV has been adopted as a surrogate model for HCV studies (10).
The viral genomic RNA is neither capped nor polyadenylated and contains one large open reading frame (ORF) that is flanked by 5′ and 3′ nontranslated regions (NTRs). The ORF encodes a polyprotein of approximately 3,900 amino acids that is co- and posttranslationally processed to the mature viral proteins by cellular and viral proteases. The N-terminal third of the ORF encodes the viral autoprotease Npro, a nonstructural protein (NS), as well as the structural proteins, namely, the capsid protein C, the glycoprotein Erns (RNase soluble), and the envelope glycoproteins E1 and E2. The remaining part of the polyprotein is processed to the nonstructural proteins p7, NS2-3 (NS2 and NS3), NS4A, NS4B, NS5A, and NS5B (viral RNA-dependent RNA polymerase-RdRp) (24). Together with putative cellular cofactors, NS3 to NS5B are part of the viral replication complex (RC).
Thiosemicarbazones were the first antiviral compounds recognized to have broad-spectrum antiviral activity against a range of DNA and RNA viruses (6, 21). The use of N-methyl isatin-β-thiosemicarbazone (methisazone) as an effective antiviral drug in the chemoprophylaxis of smallpox virus was demonstrated in human volunteers in South India as early as 1965 (5). Moreover, other thiosemicarbazones have antiviral activity against Japanese encephalitis virus (JEV) and West Nile virus in vitro and have also been found to completely inhibit JEV replication in vivo in a mouse model (33). Similarly, other studies have shown that methylisatin-β-diethylthiosemicarbazone inhibits the replication of Moloney murine leukemia virus by interfering with the early phase of the viral life cycle (30) and that isatin-β-thiosemicarbazone inhibits human immunodeficiency virus type 1 (HIV-1) replication in vitro (4).
Previously, we reported on the anti-BVDV activity exerted by the thiosemicarbazone derived from 5,6-dimethoxy-1-indanone (TSC) (18, 19). Here, we describe a further characterization of the antiviral activity of TSC.
MATERIALS AND METHODS
Cells and virus.
Madin Darby bovine kidney (MDBK NBL-1; ATCC CCL-22), Vero cells (ATCC CCL-81), baby hamster kidney (BHK-21) clon 15 cells, A549 lung cancer cells (ATCC CCL-185), and MA104 embryonic African green monkey kidney epithelial cells were grown in a minimal essential medium (EMEM), supplemented with 10% fetal bovine serum (FBS; PAA) or γ-irradiated FBS for MDBK culture (GM). Bovine viral diarrhea virus (BVDV) type 1 strain NADL, cytopathic biotype (BVDV-1, ATCC VR 534), was provided by Laura Weber, INTA Castelar, Argentina. Adenovirus type 3 (AdV-3) was provided by Adriana Kajon. Dengue virus type 2 (DENV-2), the 16681 strain, was kindly provided by Andrea Gamarnik from Instituto Leloir, Argentina. Human rotavirus (HRV) strain WA was provided by Jorge Gomez from ANLIS- Malbrán, Argentina.
Compounds.
TSC (Fig. 1) was synthesized as previously described (19). Its solubility limit in infection medium (IM; EMEM supplemented with 2.5% donor horse serum [DHS]; Gibco) is 80 μM. Ribavirin (RBV) (1-β-d-ribavirinofuranosyl-1H-1,2,4-triazole-3-carboxamide) was purchased from MP Biomedicals, LLC.
Fig. 1.

Chemical structure of TSC.
Plaque assay for BVDV and DENV-2.
MDBK or BHK-21 clone 15 cells were seeded in GM at a density of 1.3 ×105 or 6.2 ×104 cells per well, respectively, in a 24-well plate. After 24 h in a 5% CO2 incubator at 37°C, they were infected with 100 PFU of BVDV or DENV-2. Following 1 h of adsorption at 37°C, the inoculum was removed, cell monolayers were washed twice with phosphate-buffered saline (PBS) and 1 ml of overlay medium supplemented with 2.5% DHS (or 2% FBS for the DENV-2 assay), and 0.8% methylcellulose with different concentrations of compound was added in each well. Mock-infected cells with and without compound and infected cells without compound were included as controls. After 72 h of incubation at 37°C with 5% CO2, cells were fixed with formalin 10% and stained with 0.75% crystal violet, and the viral plaques were counted. The 50% effective concentration (EC50) was calculated from the respective dose-response curves.
Reduction of CPE assay for AdV-3 and HRV.
A549 and MA104 cells were seeded in GM in microwell plates (96 wells) at a density of 2.2 × 104 and 1.6 × 104 cells per well, respectively. After 24 h in a 5% CO2 incubator at 37°C, they were infected with a dilution in IM (2% FBS) of Adv-3 (A549) or HRV (MA104) that induced an 80% cytopathic effect (CPE) on the sixth or fourth day postinoculation in the absence of the drug, respectively. For HRV infection, before inoculation, the viral stock was activated for 30 min at 37°C in the presence of 5 μg/ml of l-(tosylamido-2-phenyl)ethyl chloromethyl ketone (TPCK) trypsin (Sigma). Serial dilutions of TSC were tested in quadruplicate. Mock-infected cells with and without compound and infected cells without compound were included as controls. The cytopathic effect was determined by measuring cell viability by means of the MTS/PMS [5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt and phenazine methosulfate] method (CellTiter 96 AQueous) (Promega, Madison, WI) as previously described (19).
Virucidal assay for BVDV.
BVDV (7.0 × 105 PFU) was incubated at room temperature (rt) for 10 min in contact with 10× and 20× EC50 (17 μM and 34 μM, respectively) of TSC in IM. Viral stock incubated with IM without compound was included as a control. The residual infectious viruses were titrated by plaque assay in MDBK cells.
MDBK pretreatment assay.
MDBK cells were seeded in GM at a density of 1.3 ×105 cells per well of a 24-well plate. After 24 h in a 5% CO2 incubator at 37°C, they were washed three times with PBS, 1 ml of 10× and 20× EC50 of TSC was added, and cells treated with IM without compound were included as controls. After a 7-hour additional incubation at 37°C, the medium containing TSC was removed, and cells were washed thrice with PBS and infected with 100 PFU of BVDV. Following 1 h of adsorption at 37°C, the inoculum was removed, cell monolayers were washed twice with PBS, and 1 ml of overlay medium without compound was added. Mock-infected cells were included as controls. After 72 h of incubation at 37°C, cells were fixed and stained, and the viral plaques were counted.
Effect of TSC during viral adsorption-penetration.
Subconfluent MDBK cell monolayers grown in 96-well plates were infected with BVDV (multiplicity of infection [MOI] of 3) and incubated at 37°C. After 2 h, the inoculum was removed, and monolayers were washed twice with PBS and further incubated for 48 h. In order to evaluate the effect of TSC on the first steps of the viral cycle, three different treatments with the compound (34.0 and 17.0 μM) were carried out: (i) only during, (ii) only after, and (iii) during and after the 2-hour incubation period. Finally, at 48 h postinfection (p.i.), cell viability was determined by the MTS/PMS method (CellTiter 96 AQueous) (Promega, Madison, WI). Mock-infected and infected cells without compound were added as controls.
Time-of-drug-addition studies: effect on viral replication.
MDBK cells were seeded in GM at a density of 1.3 ×105 cells per well of a 24-well plate. After 24 h in a 5% CO2 incubator at 37°C, cells were precooled for 30 min on ice and then infected with BVDV (MOI of 3). Following 90 min of adsorption on ice, the inoculum was removed, and monolayers were washed twice with PBS and incubated in IM. At different time points postadsorption (p.a.), 17 μM TSC (10× EC50) was added. After 48 h of incubation, culture supernatants were collected, and the viral production was determined by an endpoint titration in 96-well plates. Fifty-percent tissue culture infective doses (TCID50) were calculated by the method of Reed and Muench. Mock-infected and infected cells without compound were added as controls.
Luciferase reporter constructs.
The DNA construction corresponding to the BVDV internal ribosomal entry site (IRES) followed by the firefly luciferase (Fluc) (pGL5′IRES plasmid) and linear DNA corresponding to Renilla luciferase (Rluc) (PRL AMV) were kindly provided by Andrea Gamarnik from Instituto Leloir, Argentina. These constructs were then in vitro transcribed with T7 RNA polymerase (Ambion) for 90 min at 37°C.
Transfection of MDBK cells and luciferase activity.
MDBK cells were grown in 24-well plates until 50% confluence and transfected with 1 μl of BVDV IRES-Fluc RNA, 1 μl of Rluc RNA, and 1 μl Lipofectamine 2000 (Invitrogen) diluted in Opti-MEM I (Gibco) according to the manufacturer's instructions. After a 2-h incubation at 37°C, TSC (10 μM and 50 μM) was added. Four hours later, when a maximum luciferase activity had been recorded in a previous optimization assay, the medium was discarded and cells were lysed using 100 μl of passive lysis buffer (Promega). The luciferase activity was determined using a dual-luciferase reporter assay system (Promega) and measured in a Veritas microplate luminometer (Turner BioSystems). Mock-transfected and transfected cells without treatment with TSC were included as controls. The IRES activity was monitored as a Fluc/Rluc ratio (relative light units [RLU]) to normalize for transfection efficiency, where the Fluc activity is the readout for IRES-dependent translation, while Rluc expression depends upon cap-dependent translation.
Time-of-drug-addition studies: effect on viral RNA synthesis.
MDBK cells were infected with BVDV (MOI of 3), TSC was added at different time points p.i., and the cells were further incubated to complete 24 h of infection. At that time, intracellular RNA (iRNA) was extracted, retrotranscribed, and quantified by real-time PCR. In addition, to determine the kinetics of viral RNA synthesis, the intracellular viral RNA (ivRNA) production from untreated infected cells was determined every 2 h for 24 h. A similar experiment was carried out, but with extraction of iRNA at 12 h p.i.
RNA extraction and RT.
Briefly, iRNA was extracted with TRIzol reagent (Gibco-BRL, CA) according to the manufacturer's instructions. RNA was denatured at 80°C for 5 min and primed with random hexamers (Biodynamics). The reverse transcription (RT) reaction was performed at 37°C for 90 min, using 0.5 U/μl Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI).
Real-time PCR.
In order to measure the ivRNA, a 108-bp fragment from the 5′ NTR was amplified with the following oligonucleotides: 5′NTR forward, 5′-GAGGCTAGCCATGCCCTTAGT-3′, and 5′NTR reverse, 5′-TCGAACCACTGACGACTACCCT-3′. The reaction was carried out in 2× SYBR green PCR master mix (Applied Biosystems, United Kingdom) in an ABI 7500 apparatus (Applied Biosystems), using an experimental run protocol of 10 min of activation at 95°C, followed by 45 cycles of amplification and quantification (15 s at 95.0°C, 1 min at 60.0°C, and 35 s at 78.5°C), when SYBR green I signal was measured. To normalize target amplification data, a 91-bp fragment of bovine β-actin mRNA was amplified simultaneously in each sample and used as endogenous control (BBA forward, 5′-CCCACACGGTGCCCATCTAT-3′, and BBA reverse, 5′-CCACGCTCCGTGAGGATCTTC-3′). Previously, validation assays had been performed in order to verify that efficiencies of BVDV 5′ NTR, and bovine β-actin amplifications were equivalent (data not shown). Data were expressed as 1/ΔCT, where CT is the threshold cycle and ΔCT = CT BVDV 5′ NTR − CT β-actin in each sample. The higher the value of 1/ΔCT, the higher the ivRNA level in the sample. Moreover, the 2−ΔΔCT method (1, 25) was used to carry out the relative quantitation of the ivRNA at 24 h p.i. of treated cells in relation to that of the untreated infected cells. Each sample was tested in triplicate, and both positive and negative controls were included.
Evaluation of the frequency of resistant mutants.
In order to detect and determine the likelihood of TSC-resistant variants within the wt BVDV P0, an estimated 1.0 ×106 PFU were screened for TSC-resistant plaques as described previously for HSV-1, with some modifications (7). MDBK cells were seeded in GM at a density of 3.0 ×106 cells per 10-cm-diameter dish and incubated for 24 h. Stringent conditions were used to prevent replication of sensitive virus. All the drug-treated cultures were preincubated with 10×, 20×, and 30× EC50 of TSC (17, 34, and 51 μM, respectively) in IM for 2 h before virus inoculation (1.0 ×106 PFU) and further incubated with overlay medium (supplemented with 2.5% DHS [Gibco] and 0.8% methylcellulose) containing these concentrations of compound. Untreated cultures were infected with 10-fold serial dilutions of BVDV P0 and then incubated after virus adsorption with overlay medium without compound. Mock-infected cells (with and without compound) were included as controls. After 72 h, infected monolayers were examined, clearly discernible viral plaques were marked, and the infected cells from treated cultures were aspirated from individual plaques. Then, cells monolayers were fixed with formalin 10% and stained with 0.75% crystal violet, and the plaques were counted. The aspirated viral plaques were amplified individually in cell culture, and then the inhibition level of TSC (34 μM) was determined by a plaque assay.
Selection of BVDV-TSCr.
First, three successive steps of biological cloning of wild-type (wt) BVDV in MDBK cells were carried out, and the virus grown from the third cloning was named P0. Eight MDBK cell monolayers were infected with BVDV P0 (MOI of 0.01). Five of them were incubated at 37°C in the presence of TSC (T1 to T5), whereas the other three (C1 to C3) were left in the absence of the compound, until cell monolayers presented 80 to 90% of the viral CPE. The virus produced was titrated in MDBK cells and used to infect new cell monolayers to generate the next passage. This procedure was repeated 10 times using increasing concentrations of TSC (1.7 to 136.0 μM) in T lines in order to select BVDV resistant to TSC (BVDV-TSCr).
Membrane preparation and RC assays.
The RC assays were essentially similar to that published by J. C. Tomassini et al. (38). Subconfluent monolayers of MDBK cells in 125-cm2 flasks were infected with either wt BVDV or BVDV-TSCr (T1 and T2; MOI of 0.1). After 24 h, cells were collected, pelleted (800 × g at 4°C for 10 min), and suspended in ice-cold lysis buffer A (10 mM Tris-HCl [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM phenylmethylsulfonyl fluoride [PMSF]) containing a protease inhibitor cocktail (Sigma). After 30 min on ice, cells were disrupted by passing them 20 times through both 20- and 27-gauge needles. The disrupted cells were pelleted (1,000 × g at 4°C for 5 min), and the supernatant fraction was concentrated (26,900 × g at 4°C for 30 min). The pellet was resuspended in buffer B (10 mM Tris-HCl [pH 8.0], 10 mM NaCl, and 15% glycerol) in a concentration of 2.5 × 105 cell equivalents/μl and used for an RC assay. Replicase reactions were carried out in a total volume of 50 μl in 50 mM HEPES (pH 7.5), 3.5 mM magnesium acetate, 50 mM KCl, 10 mM dithiothreitol, 14.3 mM creatine phosphate, 0.1 mg/ml creatine phosphokinase, 1 mM ATP, CTP, and UTP, 10 μM GTP, 20 μCi of [α-32P]GTP (3,000 Ci/mmol) (Perkin Elmer), 40 U of RNasin (Ambion), 40 μg/ml actinomycin D, 10 μl of the membrane preparation, and 1 μl of dimethyl sulfoxide (DMSO) or 1 μl of a 50× TSC solution in DMSO. Following incubation at 37°C for 1.5 h, the reactions were stopped by adding TRizol reagent (Gibco-BRL, CA), and RNA was extracted. RNA products were denatured with 93% formamide at 80°C for 3 min and analyzed on 3% PAGE-7 M urea. The radioactivity incorporated into the virus-specific RNA was quantified using ImageQuant software of the Storm 820 PhosphorImager (Amersham).
Sequencing.
The PCR fragments that cover the coding region of all the nonstructural proteins were generated using specific oligonucleotides (Table 1) by retrotranscription (ArrayScript reverse transcriptase; Ambion) and PCR amplification (Platinum DNA polymerase High Fidelity; Invitrogen). Both DNA strands were sequenced using an ABI ABI3130XL sequencer 131 (Applied Biosystems; Unidad de Genómica, Instituto de Biotecnología, INTA, Castelar, Argentina). The sequences were aligned using ClustalX 1.83 (37), edited with BioEdit, version 7.0.9.0, software (22), and compared with wt BVDV strain NADL (P0 and GenBank accession no. M31182.1, AJI33738.1, NP_776270.1, and NP_776271.1).
Table 1.
Sequences and positions in the genome of BVDV NADL of oligonucleotides used for real-time RT PCR and PCR amplification of the nonstructural protein coding region
| Oligonucleotide name | Sequence 5′–3′ | Position in the genomea |
|---|---|---|
| 5′NTR Fw | GAGGCTAGCCATGCCCTTAGT | 98–118 |
| 5′NTR Rv | TCGAACCACTGACGACTACCCT | 185–206 |
| NS2-3 Fw 0 | AATACTGGTTTGACCTGGAG | 3435–3454 |
| NS2-3 Rv 0 | CGCCAGTTCTTTCAGTTCAGT | 7646–7666 |
| NS2-3 Fw 1 | CGCTGAGTCCATATTAGTGGT | 3481–3501 |
| NS2-3 Rv 1 | ACTGAACTTATCCCGCCT | 5605–5622 |
| NS2-3 Fw 2 | CTGCCGTGTGTAAGAAGAT | 5427–5445 |
| NS2-3 Rv2 | GTCTTCTAGTCTCTGGTCCT | 7575–7594 |
| NS4A-B Fw9 | TAGACTGGCCTGATCCTG | 7401–7418 |
| NS4A-B Rv9 | GTTGGCAATCTGATCCTCT | 8826–8844 |
| NS5A Fw1 | CTGTATGGGGTTTACTACAAAGG | 8567–8589 |
| NS5A Rv3 | TTGGTAAGCTGATGCCATGTG | 10295–10315 |
| NS5B Fw11 | AGGAGATGTTGGAGAGGTAA | 10090–10109 |
| NS5B Rv16 | TCTTCTTGAGGTGGACGG | 12417–12434 |
According to BVDV NADL complete genome. GenBank accession no. AJI33738.1.
Molecular modeling.
The published X-ray structure of the BVDV RdRp (Protein Data Bank [PDB] entry 1S48) (13) was used in all docking experiments. Sulfur atoms in the methionine residues were modified back to selenium atoms to obtain selenomethionine residues. The inhibitor TSC was drawn using JChemPaint (23). The molecular geometry was fed into Gaussian 03 (20) for geometry optimization using a PM3 force field. Polar hydrogen atoms were added to the enzyme and the inhibitor structures using AutoDockTools (32). TSC was docked in the region of RdRp where mutations were observed for this compound in the BVDV-TSCr mutants by means of the AutoDock 3.0.5 software (26). The structures in the top 10 docking scores were retained for visual inspection and analysis. One docked TSC conformation was retained for further analysis. The criterion to withhold this docked complex was the interaction with Ala392 or the interaction with Asn264, which are mutated by Glu392 or by Asp264, respectively, in BVDV-TSCr strains. Pictures were generated using AutoDockTools (MGL Tools 1.3alpha2).
Anti-BVDV assay and drug combination experiment.
The anti-BVDV assay by viral CPE reduction and the cytotoxicity assay were performed by the MTS/PMS method (CellTiter 96 AQueous) (Promega, Madison, WI) as previously described (19). TSC and RBV were tested singly and in combination at 2-fold serial dilutions in quadruplicate. An initial evaluation of the cytotoxicity of individual compounds and combinations was performed. The tested combinations were chosen to cover a range of concentrations lower and higher than the TSC EC50 with fixed concentrations of RBV lower than its EC50 in order to identify a possible synergistic effect at a 50% CPE inhibition level. The concentrations tested had not showed cytotoxicity in the previous cytotoxicity assay. The relative dose-inhibition curves of TSC were plotted under each fixed concentration of RBV, which resulted in a series of relative TSC EC50s that represented the experimental combinations of TSC and RBV that led to a 50% viral CPE reduction.
Synergy analysis.
The analysis of the interaction of TSC with RBV was undertaken using two methods: the classic isobologram analysis and the combination index (CI) calculation (14, 35, 36). To generate the isobologram, individual EC50s of TSC and RBV were plotted on the x and y axes, respectively. The diagonal line that intercepts both points is defined as the additive isobole. If the combination data points fall on this isobole, an additive effect is indicated, whereas if the combination data points fall on the lower left or on the upper right of the isobologram, synergism or antagonism is suggested, respectively. The data from the isobologram analysis was replotted as a fraction plot as previously reported (35) and a statistical analysis (unpaired t test) was carried out (data not shown). In addition, the CI for each combination was calculated by the following formula: CI = (EC50 of TSC combined/EC50 of TSC alone) + (EC50 of RBV combined/EC50 RBV alone). In this case, the combined effects of the two drugs can be assessed as either an additive (CI = 1), synergistic (CI < 1), or antagonistic (CI > 1) effect.
RESULTS
Previously, we had reported on the selective activity of TSC against BVDV. Several DNA and RNA viruses, such as poliovirus type 1 (PV-1), human immunodeficiency virus type 1 (HIV-1), vesicular stomatitis virus type 1 (VSV-1), and herpes simplex virus type 1 (HSV-1) were not susceptible to this compound (19). In this work, TSC showed no relevant antiviral activity against viruses from nonrelated families, such as HRV and AdV-3 (EC50 > 50.0 μM and EC50 = 25.5 μM, respectively) or against a member of the same family, such as DENV-2 (EC50 > 50.0 μM). This compound did not show cytotoxicity in any of the cell lines until the highest concentration tested (50 μM).
A virucidal assay of TSC with BVDV was performed. There was no reduction of viral titers when the virus was treated with concentrations of TSC as high as 17 μM and 34 μM, suggesting that TSC did not exert virucidal activity (Fig. 2A). Moreover, the addition of these concentrations of TSC to MDBK monolayers 7 h before infection with BVDV did not result in any marked reduction in viral replication, suggesting that no antiviral activity had been induced during the pretreatment (Fig. 2B). When 34.0 or 17.0 μM TSC was present only during the first 2-h incubation period (when viral adsorption and penetration steps are supposed to be in course) and then removed, the compound failed to inhibit viral replication. However, the addition of TSC after this period resulted in almost complete protection from the virus-induced CPE, similar to that observed when the compound was present during the whole experiment (Fig. 3). These results suggest that TSC inhibits BVDV replication at a time point after the adsorption and penetration of the virus into the cell.
Fig. 2.

(A) Virucidal activity of TSC. BVDV was incubated 10 min at rt in contact with 10× and 20× EC50 (17 μM and 34 μM, respectively) of TSC. The residual infectious viruses were titrated by plaque assay in MDBK cells. (B) Effect on viral replication of the treatment of MDBK cells before infection. Subconfluent MDBK cells monolayers were treated with 10× and 20× EC50 of compound and further incubated for 7 h at 37°C before infection with BVDV. After removal of the medium, treated and untreated cells were infected with 100 PFU of BVDV. Following 72 h of incubation at 37°C, cells were fixed and stained, and the plaques counted.
Fig. 3.

Effect of TSC during viral adsorption-penetration. MDBK cells were infected with BVDV (MOI of 3). After 2 h at 37°C, the inoculum was removed, and cultures were further incubated for 48 h, when cell viability was determined by the MTS/PMS method. Three different treatments with the compound (34 and 17 μM) were carried out: (i) only during (white bars), (ii) only after (gray bars), and (iii) during and after the 2-h incubation period (black bars). Mock-infected and infected cells without compound were added as controls (open bars).
Time-of-drug-addition studies.
To determine the time of the viral cycle at which TSC interferes with viral replication, time-of-drug-addition experiments were carried out. A concentration of compound of 10× EC50 was added at different points of time after infection, and then viral replication was evaluated as the yield of viral infectious titers. Viral titers were markedly reduced when the compound was added during the first 8 h, after which a gradual reduction in drug inhibition was observed, suggesting that TSC may be acting at a point of time between 8 and 10 h p.i. (Fig. 4).
Fig. 4.

Time-of-drug-addition studies. MDBK cells were infected with BVDV (MOI of 3), and 17 μM TSC was added at different times p.i. After 48 h, supernatants were collected and endpoint titrated in 96-well plates. TCID50 were calculated by the method of Reed and Muench. VC, virus control (untreated infected cells).
Effect of TSC on the BVDV IRES.
The effect of TSC on the IRES of BVDV was evaluated. We analyzed the translation efficiency in MDBK cells of a reporter RNA encoding Fluc under the BVDV IRES control in the presence or absence of the compound. The luciferase activity at 4 h after transfection (when maximum activity had been detected in untreated cultures) in the presence of 10 and 50 μM TSC was similar to that observed in the absence of the drug (Fig. 5). These results suggest that TSC does not inhibit the translation process mediated by the BVDV IRES.
Fig. 5.

Effect of TSC on the BVDV IRES. MDBK cells were cotransfected with an in vitro synthesized BVDV IRES-Fluc and Rluc RNAs and treated with 10 μM and 50 μM TSC. Untreated transfected cells were included as controls. The luciferase activity was determined at 4 h posttransfection, and the IRES activity was monitored as a Fluc/Rluc ratio (RLU) to normalize for transfection efficiency.
TSC interferes with BVDV RNA synthesis.
To investigate the effect of the compound on vRNA synthesis, first, the kinetics of this process in MDBK cells was analyzed. In agreement with previous reports (29), an increase in intracellular BVDV RNA (ivRNA) was detected between 8 and 10 h p.i. by real-time RT-PCR (Fig. 6A). When TSC was added during the first 8 h p.i., an almost complete reduction of ivRNA (97 to 99%) was observed (Fig. 6B). Further times of addition resulted in a gradual loss of inhibition. Similar results were obtained when ivRNA was measured at 12 h p.i. (data not shown). The main observation of these experiments was that when the drug was added at each point of time, the yield of ivRNA detected at 24 h was similar to that measured at these time points from untreated infected cells, suggesting that when the TSC was added to the culture a blockade of viral RNA synthesis occurred. Similar to the results obtained in viral replication experiments, the inhibition of vRNA synthesis was dose dependent (data not shown).
Fig. 6.

Effect of TSC addition on viral RNA synthesis. (A) Kinetics of BVDV RNA synthesis. MDBK cells were infected with BVDV (MOI of 3), and the ivRNA was measured by real-time PCR at different times p.i. (the inset presents the ivRNA levels during the period from 4 to 12 h, on a more detailed scale). (B) Simultaneously, MDBK cells were infected with BVDV (MOI of 3), and 34 μM TSC was added every 2 h during the first 12 h p.i. The incubation period continued until 24 h p.i., when intracellular RNA was extracted and ivRNA measured by real-time RT-PCR (the inset presents the 24-h ivRNA levels during which the compound was added from 0 to 12 h, on a more detailed scale). An infected untreated control (IUT) was included, and its 24-h-p.i. ivRNA was measured. Values are expressed as 1/ΔCT, where ΔCT = CT BVDV 5′ NTR − CT β-actin. NIUT, nontreated uninfected control.
Viral RC assays.
In order to further study the inhibitory activity of TSC on viral RNA synthesis, the effect of the compound on membrane-associated replication complexes isolated from BVDV-infected cells was evaluated. These membrane extracts were active for RNA synthesis in the presence of actinomycin D and incorporated radiolabeled GTP into the synthesized vRNA inside the RCs without the need of the addition of external templates. Incubation of these complexes at 37°C resulted in the synthesis of vRNA, and when denaturing conditions were used, a single band of full-length single-stranded RNA of about 12 kb, which was absent with mock-infected extracts, was observed (Fig. 7A). Different concentrations of TSC were added to the extracts and incubated as described above. The results obtained demonstrated that TSC inhibited the activity of the RCs from wt BVDV in a dose-dependent manner with an EC50 of 8.3 ± 2.7 μM, showing an almost complete inhibition when the complexes were incubated with 80 μM compound (Fig. 7B).
Fig. 7.

Effect of TSC on BVDV RC activity. MDBK cells were mock infected or infected with BVDV. After 24 h, RCs were collected, and RNA synthesis was evaluated by the in vitro incorporation of [α-32P]GTP into vRNA. Reaction product of the RC assay was denatured and analyzed on 3% PAGE-7 M urea. (A) RNA product from infected and mock-infected MDBK cells. (B) RNA product from RCs of wt BVDV-infected cells in the presence of a series of TSC concentrations. (C) RNA product from RCs of wt BVDV- or BVDV-TSCr T2-infected cells in the presence or absence of TSC (80 μM). Similar results were obtained for the RCs BVDV-TSCr T1-infected cells.
Frequency of TSC-resistant variants.
In order to evaluate the likelihood of the emergence of drug-resistant mutants, a large number of PFU of wt BVDV were screened for TSC-resistant plaques. At the first virus dilution without TSC, the viral plaques could not be distinguished because of a confluent viral CPE, but an estimated 1.5 × 105 PFU/dish were infecting the cells monolayer (according to the number of PFU enumerated in the cultures infected with serial 10-fold-lower dilutions). When cultures were treated with 20× and 30× EC50, 13 and 3 viral plaques were counted, respectively, indicating a frequency of resistant variants of 0.009% and 0.002% within the wt BVDV. When the viral plaques aspirated from the 20× and 30× EC50 treatments were amplified in cell culture and then tested against TSC, with a concentration of compound that reduced at least by 1,000 times the number of wt BVDV PFU, a reduction only around 10 times was observed.
Selection and phenotypic characterization of resistant mutants.
In order to ascertain the molecular target of our compound, we proceeded to isolate TSC-resistant viruses. To this end, five independent BVDV-TSCr lines were selected after 10 successive passages in MDBK cells in the presence of increasing concentrations of compound. To characterize these viruses, the antiviral activity of TSC and RBV was evaluated against both treated lines (T) and control lines (C) from the tenth passage (Table 2). While control lines (C1 to C3) presented EC50s similar to those of wt BVDV, the treated lines (T1 to T5) were not inhibited by the use of TSC concentrations as high as 80 μM (BVDV-TSCr). In addition, both resistant and control viruses remained as susceptible to RBV as wt BVDV (EC50 of 5 to 10 μM). Finally, replicase activity assays were carried out from RCs isolated from BVDV-TSCr T1 and T2 infected cells (Fig. 7C). As expected, the replicase activity of resistant BVDV was not affected by the compound at a concentration that blocked the wt virus almost completely. When all resistant mutants were further passed in MDBK cells in the absence of the compound, it was observed that T2 and T5 replicated more slowly than T1, T3, and T4, requiring 96 to 120 h to reach 80 to 90% of the viral CPE (instead of 48 h), and over seven passages did not show any reversion to the wt phenotype. Although they did not show any changes regarding replication in cell culture, T1, T3, and T4 showed a partial change in the resistant phenotype from the fourth to the tenth passage in the absence of compound, being inhibited by 77% ± 11% with 34 μM compound (BVDV wt was inhibited by 99.9%). Further studies of these viral populations will be necessary to define the characteristics of the phenotype reversion.
Table 2.
Antiviral activity of TSC and RBV against different BVDV linesa
| BVDV line | TSC EC50 (μM) | RBV EC50 (μM) |
|---|---|---|
| P0 | 7.9 ± 3.4 | 9.8 ± 1.2 |
| C1 | 1.7 ± 0.7 | 6.0 ± 1.4 |
| C2 | 5.1 ± 1.8 | 8.3 ± 2.0 |
| C3 | 4.9 ± 2.5 | 8.8 ± 2.0 |
| T1 | >80 | 10.0 ± 2.2 |
| T2 | >80 | 8.8 ± 2.0 |
| T3 | >80 | 8.5 ± 1.2 |
| T4 | >80 | 6.9 ± 1.6 |
| T5 | >80 | 10.7 ± 2.4 |
Antiviral activity was tested by plaque reduction assays in MDBK cells infected with C (control) and T (treated) lines from the tenth passage (C1 to C3 and T1 to T5). EC50s were calculated from dose-response curves.
BVDV-TSCr molecular characterization.
In order to find any mutation that could be associated with the resistant phenotype and use that information in our studies of molecular docking, the sequences of the nonstructural proteins of the BVDV genome were obtained from the control and resistant mutants from the tenth passage. No nucleotide changes were detected in the viral NS2, NS3, and NS4A proteins. In the NS4B protein, a C-to-G mutation was found at position 8224 of the viral RNA in T3 and a U-to-G mutation at position 8410 in T4. Only the last mutation resulted in an amino acid change of Ser to Arg at amino acid residue 294. On the other hand, no nucleotide changes were observed in the NS5A protein, whereas an A-to-G mutation at position 10701 of the NS5B coding region in the viral genome was identified in four out of five BVDV-TSCr (T2 to T5), and a C-to-A mutation at position 11086 was identified in the remaining line (T1). These point mutations resulted in an amino acid change of Asn to Asp at amino acid residue 264 and in an Ala to Glu change at amino acid residue 392 in the RdRp, respectively. No other nonsynonymous mutations were detected throughout the NS5B region of the genome of the drug-resistant viruses, except for an A-to-G mutation at position 10150 in the T3-resistant mutant that resulted in an amino acid change of Glu to Gly at residue 80 in the RdRp. In addition, synonymous mutations were selected in two resistant lines, T1 (A9,980G) and T3 (G11,565A). No mixed populations were noted on the sequence chromatogram. Because of these results, nonsynonymous mutations at amino acids 264 and 392 in the NS5B protein were screened throughout the previous passages. Position 10701 of the viral genome (which defined the amino acid change at position 264) showed mixed populations of nucleotides A/G at the fifth passage, when 6.8 μM TSC was used. However, position 11086 was unchanged until the ninth passage (68 μM TSC), when mixed populations of C/A were detected on the sequence chromatograms.
Docking of TSC in the BVDV RdRp crystal structure.
The two amino acids that are mutated in the TSC-BVDVr strains are located near each other in the BVDV RdRp (PDB entry 1S48) (13), with a gap of 13 Å between them. This distance is similar to the long diagonal distance of the TSC studied as a ligand (Fig. 8A). The docking of TSC close to Asn264 showed some possible interactions between the RdRp and TSC (Fig. 8B): (i) a close contact between the indane ring and Leu530 and Lys 266; (ii) a close contact between the 6-methoxy group of indane moiety and Asn264; (iii) two hydrogen bonds between the H atoms on the N-4 atom of the thiosemicarbazone group and the carbonyl groups of Glu265 and Lys266, respectively; (iv) a hydrogen bond between the N-1 atom of the thiosemicarbazone group and the α-NH2 group of Lys266; and (v) a hydrogen bond between the H atom on the N-2 atom of the thiosemicarbazone group and the carbonyl group of Lys266. Docking studies close to Asp264 in the mutated BVDV RdRp showed that TSC interacts with other amino acids far away from the mutated site. On the other hand, the docking of TSC close to Ala392 showed the following possible interactions between the RdRp and TSC (Fig. 8C): (i) hydrophobic contacts of the thiocarbonyl group of TSC with Phe165 and Ala392, (ii) two hydrogen bonds between the H atoms of the N-4 atom of the thiosemicarbazone group and the amino acids Thr152 and Thr160, and (iii) a close contact between the indane ring and Pro262, Ile261, and Tyr289. The Glu residue in position 392 in BVDV-TSCr did not allow the compound to access the studied site (Fig. 8D).
Fig. 8.

Docking of TSC in the BVDV RdRp crystal structure. (A) PDB image of BVDV RdRp (PDB entry 1S48) showing Ala392 and Asn264. (B) Stereo picture of the docking of TSC near Asn264. All surrounding amino acids having closed contact with the inhibitor are shown, as are the H bonds between the H atoms of N-4 atom of the thiosemicarbazone group and carbonyl group of Glu265 and Lys266, between the N-1 atoms of the thiosemicarbazone group and α-NH group of Lys266, and between the H atoms on the N-2 atom of the thiosemicarbazone group and carbonyl group of Lys266. (C) Stereo picture of the docking of TSC near Ala392. All surrounding amino acids having a hydrophobic contact interaction with the inhibitor are shown, as are the H bonds between the H atoms of the N-4 atom of the thiosemicarbazone group and the OH group of Thr152 and Thr160. (D) In red is shown mutated Ala392 by a Glu residue, illustrating the low possibility of TSC accessing the studied site.
TSC acts synergistically with RBV.
Because RBV is the anti-BVDV compound of reference, the interaction between TSC and RBV as anti-BVDV agents was evaluated. To this end, cytotoxicity assays for individual and combined compounds were performed in order to determine the range of concentrations to test (Fig. 9), and none of these combinations were shown to be cytotoxic in MDBK cells. BVDV-infected MDBK cells were treated with different concentrations of TSC combined with different fixed concentrations of RBV, and the viral CPE was measured (Fig. 10). The results obtained were then analyzed by two methods: the isobologram and the CI (Fig. 11A and B), both for a 50% CPE viral reduction. TSC and RBV individual EC50s (EC50 TSC [3.4 ± 0.2 μM] and EC50 RBV [4.2 ± 0.1 μM]) were plotted on the x and y axes, respectively, to generate an isobologram. The experimental combinations that led to a 50% CPE reduction fell clearly below the additive isobole. When the CI for each combination was calculated, CI values lower than 1 were obtained. On the whole, these results suggest that TSC caused a synergistic antiviral effect when combined with the range of concentrations of RBV tested. The statistical analysis of the results showed a significant difference between the theoretical additive combinations and those experimentally obtained (P < 0.0001).
Fig. 9.
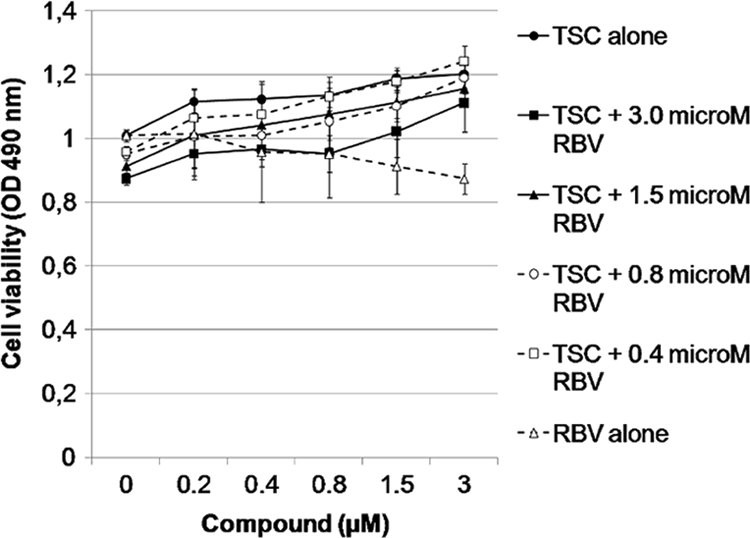
Cytotoxic effect of the combination of TSC and RBV. MDBK cells treated with different concentrations of TSC and RBV alone or in combination, and cell viability was measured by MTS/PMS method.
Fig. 10.

Effect of the combination of TSC and RBV on BVDV replication. Culture was infected with BVDV and treated with different nontoxic concentrations of TSC alone and RBV alone or in combination. Antiviral activity was measured by CPE reduction method (MTS/PMS) after 72 h at 37°C.
Fig. 11.

Isobologram analysis and CI. (A) Isobologram analysis. Individual TSC and RBV EC50s were plotted on the x and y axes, respectively (white circles). The diagonal line that intercepts these points is defined as the additive isobole. Each full circle on the plot represents each experimental drug combination that produced the same antiviral effect (50% inhibition of viral CPE). Synergism or antagonism is indicated when experimental combinations fall on the lower left or on the upper right of this isobole, respectively. (B) The CI was calculated for each experimental combination that led to a 50% inhibition of viral replication (see Materials and Methods). A CI value equal, below, or above 1 represents additive interaction, synergism, or antagonism, respectively.
DISCUSSION
In a previous study we reported on the synthesis and antiviral activity of a series of 1-indanone thiosemicarbazone derivatives as well as the effect of the substituents in the aromatic ring and of the thiosemicarbazone group on their activity (18, 19). In the present study we characterized the antiviral activity of the most active derivative, TSC, which showed highly selective activity against BVDV by blocking the viral RNA synthesis in cell culture and in isolated BVDV RCs.
BVDV-TSCr strains were selected after several passages under the pressure of the compound in order to evaluate the molecular target. The genetic characterization of these resistant viruses showed that four out of five lines carried an N264D mutation in the NS5B, whereas the remaining one showed an A392E mutation in the same protein, suggesting that these amino acids are necessary for the sensitivity to the drug and that this protein is necessary for at least part of the selective action of the drug. BVDV-TSCr strains carrying mutations affecting the activity of TSC remained susceptible to RBV.
In order to evaluate the development of drug resistance, the frequency of BVDV-TSCr variants within the wt virus population (P0) and the rapidity of the resistance selection were studied. We detected a low frequency of virus capable of replicating in the presence of high concentrations of compound within P0. These results suggested that the likelihood of the emergence of TSC-resistant mutants should be low.
The N264D mutation in the NS5B, which was present in the majority of the BVDV-TSCr mutants, was detected at the fifth passage in the presence of 6.8 μM TSC, whereas the A392E mutation was lately selected at the ninth passage in the presence of 68 μM compound.
The frequency of drug-resistant mutants in a population and how rapidly they arise depend on several factors, including: (i) the mutation rate (BVDV is an RNA virus, and therefore, its mutation rate is high; the higher this rate, the higher the genetic diversity in the viral population and the more rapidly resistance can develop); and (ii) the genetic barrier to resistance (the lower the number of mutations required for the loss of antiviral activity, the more rapidly resistant variants can be selected). The rapid selection of TSC resistance and the low genetic barrier (only one mutation was required for resistance to the drug) are consistent with the existence of a specific viral target for TSC and reflect the compound selectivity (15).
The crystal structure of the BVDV RdRp presents the shape of a right hand with fingers, palm, and thumb domains, and the inner surfaces of the three domains form a central template-binding channel. Unique to RdRp, the N terminus of the fingers domain and a long insert in this domain (residues 260 to 288) form the fingertip region (12, 13). The N264 residue, which is mutated in most TSCr viruses, is located in this region, whereas A392 was found located in the fingers domain.
Recently, in another study, three 2-phenylbenzimidazole derivatives: 50, 51, and 53 were reported to have selected BVDV resistant mutants that carried exactly the same amino acid mutations (39). In that study, the authors showed by a hierarchical molecular modeling procedure that the A392E mutation decreases the net electrostatic component greatly, whereas N264D caused a substantial decrease in both electrostatic and van der Waals energies in the BVDV RdRp. These results are in accordance with our docking studies with the D264 mutant RdRp in which TSC-BVDV RdRp interactions were modified and with the E392 RdRp mutant in which the access of the compound to the studied site was not allowed.
In the BVDV RdRp crystal structure, both N264 and A392 are located in the vicinity of F224, a residue that was mutated in BVDV resistant mutants selected with other nonnucleoside inhibitors (NNIs), such as BPIP (29), VP32947 (2), and LZ37 (28). Molecular modeling revealed that F224 is located near the fingertip domain of the RdRp and that F224, I390, A392, and L225 are amino acids that limit the cavity where these compounds bind. This cavity represents one allosteric site of inhibition for BVDV RdRp, and its hydrophobicity matches physicochemically the lipophilicity of TSC and of the other NNIs.
The fingertip region associates with the thumb domain, with which it forms an entrance to the template-binding channel, and it is believed to be involved in template/product translocation, dimerization of the RdRp within the RC, or protein-protein interactions, enabling the assembly of an active RC (12, 13). Moreover, the fingertip region contains the polymerase motifs I and II, which are involved in RNA template and nucleoside triphosphate (NTP) binding. Motif I has been reported to be close to the initiation NTP (NTPi) +1 binding site and to bind the incoming NTP (13). Interestingly, N264, which is mutated in most of the TSC-resistant viruses, is located in the fingertip region within this motif.
Therefore, TSC might affect the movement of the fingertip region and/or the binding of each subsequent NTP that is to be incorporated into the growing RNA product (where motif I plays a role). Both processes may help to translocate the template and product for each elongation step. However, a direct interaction between TSC and BVDV RdRp would be the key experimental approach to confirm this hypothesis, and the direction of future research lies in this characterization.
In addition, we demonstrated that TSC acts synergistically with RBV as anti-BVDV agents. To our knowledge, besides alpha interferon (IFN-α) (8) and the α-glucosidase I inhibitor 6 o-butanoyl castanospermine (Celgosisvir) (40), which do not target any viral protein, TSC is the first NNI of viral RdRp that is reported to show synergism against BVDV replication in cell culture.
RBV exhibits antiviral activity against a variety of RNA and DNA viruses in vitro and is currently used in a combination with IFN-α—in which the synergism action with IFN-α is responsible for the antiviral efficacy in vivo—to treat chronic HCV infections. The RBV mechanism of action is uncertain, and several mechanisms, including an immunomodulatory effect, the inhibition of the IMP dehydrogenase (which leads to a reduction in the intracellular GTP pool), the direct inhibition of viral RNA polymerase, and viral genome mutagenesis, have been proposed since its discovery (17). The direct effect of RBV, alone or in combination with IFN-α, on BVDV replication in cell culture has also been reported elsewhere (9, 16).
The synergistic effect and the fact that BVDV-TSCr mutants are still susceptible to RBV strongly suggest different viral targets. The synergism between both compounds could be due to a collaborative action in the inhibition of the BVDV RdRp. Further studies are necessary to determine the true mechanism of the synergy between both compounds against BVDV replication.
In conclusion, TSC emerges as a new potent NNI of BVDV and, to our knowledge, as the first thiosemicarbazone derivative whose mechanism of action as a viral polymerase inhibitor is reported. As a specific antiviral agent, TSC could provide an interesting tool for the study of the BVDV polymerase function and viral replication as well as for the development of new compounds against pestiviruses. The synergistic TSC-RBV interaction observed in vitro against BVDV suggests that TSC could be used in future combination therapies, and it should be evaluated against HCV replication.
Acknowledgments
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica grants PICT 34386 and 25355, Universidad de Buenos Aires grants B045 and B037, and Consejo Nacional de Investigaciones Científicas y Tecnológicas grants PIP 112-200801-01169 from Argentina.
We gratefully acknowledge Andrea Gamarnik, Diego Alvarez, and coworkers for providing the IRES BVDV construct, facilitating the laboratory and reagents for in vitro RNA synthesis, and encouraging discussions and advice.
Footnotes
Published ahead of print on 23 March 2011.
REFERENCES
- 1. Applied Biosystems 2004. 7900HT Fast Real-Time PCR System and 7300/7500/7500 Fast Real-Time PCR Systems. Chemistry Guide, p. 3.1–3.46 Applied Biosystems, Foster City, CA [Google Scholar]
- 2. Baginski S. G., et al. 2000. Mechanism of action of a pestivirus antiviral compound. Proc. Natl. Acad. Sci. U. S. A. 97(14):7981–7986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker J. C. 1995. The clinical manifestations of bovine viral diarrhea infection. Vet. Clin. North Am. 11:425–445 [DOI] [PubMed] [Google Scholar]
- 4. Bal T. R., Anand B., Yogeeswari P., Sriram D. 2005. Synthesis and evaluation of anti-HIV activity of isatin beta-thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 15:4451–4455 [DOI] [PubMed] [Google Scholar]
- 5. Bauer D. J. 1965. Clinical experience with the antiviral drug marboran (1-methylisatin 3-thiosemicarbazone). Ann. N. Y. Acad. Sci. 130:110–117 [DOI] [PubMed] [Google Scholar]
- 6. Bauer D. J., Sadler P. W. 1960. The structure-activity relationships of the antiviral chemotherapeutic activity of isatin β-thiosemicarbazone. Br. J. Pharmacol. Chemother. 15:101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biswas S., Smith C., Field H. J. 2007. Detection of HSV-1 variants highly resistant to helicase-primase inhibitor BAY 57-1293 at high frequency in 2 of 10 recent clinical isolates of HSV-1. J. Antimicrob. Chemother. 60:274–279 [DOI] [PubMed] [Google Scholar]
- 8. Buckwold V. E., Wei J., Wenzel-Mathers M., Russell J. 2003. Synergistic in vitro interactions between alpha interferon and ribavirin against bovine viral diarrhea virus and yellow fever virus as surrogate models of hepatitis C virus replication. Antimicrob. Agents Chemother. 47:2293–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buckwold V. E. 2004. Implications of finding synergic in vitro drug-drug interactions between interferon-alpha and ribavirin for the treatment of hepatitis C virus. J. Antimicrob. Chemother. 53(3):413–414 [DOI] [PubMed] [Google Scholar]
- 10. Buckwold V. E., Beer B. E., Donis R. O. 2003. Bovine viral diarrhea virus as a surrogate model of hepatitis C virus for the evaluation of antiviral agents. Antiviral Res. 60:1–15 [DOI] [PubMed] [Google Scholar]
- 11. Chimeno Zoth S., Taboga O. 2006. Multiple recombinant ELISA for the detection of bovine viral diarrhea virus antibodies in cattle sera. J. Virol. Methods 138:99–108 [DOI] [PubMed] [Google Scholar]
- 12. Choi K. H., Gallei A., Becher P., Rossmann M. G. 2006. The structure of bovine viral diarrhea virus RNA-dependent RNA polymerase and its amino-terminal domain. Structure 14(7):1107–1113 [DOI] [PubMed] [Google Scholar]
- 13. Choi K. H., et al. 2004. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc. Natl. Acad. Sci. U. S. A. 101:4425–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou T. C., Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effect of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27–55 [DOI] [PubMed] [Google Scholar]
- 15. Coen D. M., Richman D. D. 2007. Antiviral agents, p. 447–485 In Knipe D. M., et al. (ed.), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 16. Cosnes J., et al. 2002. Maladie coeliaque méconnue dans l'enfance. Gastroenterol. Clin. Biol. 26:616–623 [PubMed] [Google Scholar]
- 17. Crotty S., Cameron C., Andino R. 2002. Ribavirin's antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. 80:86–95 [DOI] [PubMed] [Google Scholar]
- 18. Finkielsztein L. M., et al. 2010. What is known about the antiviral agents active against bovine viral diarrhea virus (BVDV)? Curr. Med. Chem. 17(26):2933–2955 [DOI] [PubMed] [Google Scholar]
- 19. Finkielsztein L. M., et al. 2008. New 1-indanone thiosemicarbazone derivatives active against BVDV. Eur. J. Med. Chem. 43:1767–1773 [DOI] [PubMed] [Google Scholar]
- 20. Frisch M. J., et al. 2003. Gaussian 03, Revision B.05. Gaussian, Inc., Pittsburgh, PA [Google Scholar]
- 21. Glover V., Bhattacharya S. K., Sandler M. 1991. Isatin: a new biological factor. Indian J. Exp. Biol. 29:1–5 [PubMed] [Google Scholar]
- 22. Hall T. A. 1999. Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/nt. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 23. Krause S., Willighagen E., Steinbeck C. 2000. JChemPaint: using the collaborative forces of the internet to develop a free editor for 2D chemical structures. Molecules 5:93–98 [Google Scholar]
- 24. Lindenbach B. D., Heinz-Jürgen T., Rice C. M. 2007. Flaviviridae: the viruses and their replication, p. 1126–1131 In Knipe D. M., et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 25. Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 26. Morris G. M., et al. 1998. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 19:1639–1662 [Google Scholar]
- 27. Pacheco J., Lager I. 2003. Indirect method ELISA for the detection of antibodies against bovine diarrhea virus in bovine serum. Rev. Argent. Microbiol. 35:19–23 (In Spanish.) [PubMed] [Google Scholar]
- 28. Paeshuyse J., et al. 2009. A pyrazolotriazolopyrimidinamine inhibitor of bovine viral diarrhea virus replication that targets the viral RNA-dependent RNA polymerase. Antiviral Res. 82(3):141–147 [DOI] [PubMed] [Google Scholar]
- 29. Paeshuyse J., et al. 2006. A novel, highly selective inhibitor of pestivirus replication that targets the viral RNA-dependent RNA polymerase. J. Virol. 80:149–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ronen D., Nir E., Teitz Y. 1985. Effect of N-methylisatin-β-4′:4′-diethylthiosemicarbazones on intracellular Moloney leukemia virus constituents. Antiviral Res. 5:249–254 [DOI] [PubMed] [Google Scholar]
- 31. Rweyemamu M. M., et al. 1990. Incidence, epidemiology and control of bovine viral diarrhea virus in South America. Rev. Sci. Tech. 9:207–214 [DOI] [PubMed] [Google Scholar]
- 32. Sanner M. F. 1999. Python: a programming language for software integration and development. J. Mol. Graph. Model. 17:57–61 [PubMed] [Google Scholar]
- 33. Sebastian L., et al. 2008. N-methylisatin-beta-thiosemicarbazone derivative (SCH 16) is an inhibitor of Japanese encephalitis virus infection in vitro and in vivo. Virol. J. 5:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reference deleted.
- 35. Tallarida R. J. 2001. Drug synergism: its detection and applications. J. Pharmacol. Exp. Ther. 298:865–872 [PubMed] [Google Scholar]
- 36. Tallarida R. J. 2006. An overview of drug combination analysis with isobolograms. J. Pharmacol. Exp. Ther. 319:1–7 [DOI] [PubMed] [Google Scholar]
- 37. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tomassini J. E., et al. 2003. An in vitro Flaviviridae replicase system capable of authentic RNA replication. Virology 313:274–285 [DOI] [PubMed] [Google Scholar]
- 39. Tonelli M., et al. 2010. Pharmacophore modeling, resistant mutant isolation, docking, and MM-PBSA analysis: combined experimental/computer-assisted approaches to identify new inhibitors of the bovine viral diarrhea virus (BVDV). Bioorg. Med. Chem. 18(6):2304–2316 [DOI] [PubMed] [Google Scholar]
- 40. Whitby K., Taylor D., Patel D., Ahmed P., Tyms A. S. 2004. Action of celgosivir (6 O-butanoyl castanospermine) against the pestivirus BVDV: implications for the treatment of hepatitis C. Antivir. Chem. Chemother. 15(3):141–151 [DOI] [PubMed] [Google Scholar]