

Abstract
Microfluidic devices for cell culture based assays provide new types of engineered microenvironments and new methods for controlling and quantifying cellular responses to these microenvironments. However, without an understanding of the effects of the microenvironments present in microdevices from a cellular perspective, it will be challenging to integrate work done in microdevices with biological data obtained via traditional methods. With the adaptation and validation of In Cell Westerns (ICWs) and in situ analysis techniques to microfluidic devices, we can begin to look at a variety of cellular responses to microcultures. Here we observe several differences in proliferation, glucose metabolism, signaling pathway activation and protein expression levels between cells cultured in traditional macroscale cultures and in microfluidic cultures. The issues of glucose starvation, growth factor restriction, volume density and effects of interactions with poly(dimethylsiloxane) (PDMS) were examined to determine the relative importance of each to cell behavior. Changes in glucose metabolism, insensitivity to volume density or media supplementation, and finally reduced proliferation as the exposure to PDMS increased, suggests that perhaps interactions between media/cells and this commonly employed polymer may be significant for some cell based assays. The differences between cells in macroscale and microfluidic cultures suggest that the cellular baseline may be substantially altered in microcultures due to both inherent differences in scale as well as material differences. The observations highlight the need to biologically validate micofluidic devices for cell based assays in order to accurately interpret the data obtained with them in the context of traditional macroculture data. Additional areas of study that will further characterize and validate microscale culture are discussed.
Introduction
Microscale experimental techniques have been applied to biological assays for nearly two decades,1,2 but microfluidic devices for cell based assays have not been widely integrated as common tools in biological laboratories. The significant differences between several physical phenomena at the microscale versus the macroscale have been exploited to provide a wide variety of new types of assays not previously possible using macroscale techniques, by allowing existing assays to be performed on significantly smaller samples (down even to the single cell level) or by reducing reagent costs. Microscale techniques for cell biology range from single cell analyses and flow cytometry-like techniques,3 to treating fields of cells in gradient generating devices,4 patterned 3-dimensional cultures,5–7 to microscale versions of more traditional assay types such as cell culture (via perfusion,8,9 or static cultures5,10–12). Temporal and spatial control on the micrometre scale (0.1–100 μm) has been used in fundamental studies from the subcellular13 to the organismal14 level in studies of cell division axis orientation15 and geometric influence on cell survival.16 Thus it is clear that at its core, microfluidics has the potential to have a great impact in cell biology as many of the leading questions in cell biology are well suited to study using these functionalities. Although microfluidics holds enormous potential to provide a platform for new and more relevant cellular assays, more in depth investigation of the biological influence of the engineered microenvironments will be required for this potential to be fully realized. Aspects of these engineered microenvironments that are new and different from the more traditional issues associated with traditional cell culture and treatment should be considered and understood.
Influences of microfluidic microenvironments on cellular behavior
Gradient generating microfluidic devices have illustrated why an understanding of the effects of specific microfluidic devices from a cellular perspective is critical for further implementation of the devices in biological research. Stimulating a field of cells with a controlled gradient of a soluble factor is a unique type of microfluidic assay that can effectively produce different microenvironments in a single device.4 Few traditional techniques for gradient production, such as the Zigmond chamber,17 have been able to produce as defined, controlled and repeatable gradients as those produced using microfluidic techniques. However, many of these devices rely upon continuous flow of the exogenous compound for gradient formation. The effects of flow alone on neutrophils has been addressed via studies of mechanical activation by shear stresses from laminar flow in microchannels.18 Walker and colleagues have also shown that the flow rate used to create gradients can bias the migratory behavior of these cells.19 The validity of cell based assays done in microfluidic devices will rely upon addressing these types of issues that are inherent to each specific microfluidic device design used.
Another application of microfluidics that, while seemingly simple, holds immense promise is cell culture. Microfluidic devices for cell culture provide a platform for higher throughput analyses of cellular responses to soluble stimuli with a variety of cost and resource benefits.5,20,21 Because each assay can be performed on a smaller total number of cells when done in microfluidic devices, more assays can be performed with the same sample size. However, in several cell types, differences in various aspects of cell behavior and functioning have been observed in microcultures from the phenotypes seen in macroscale cultures.10,12,22–24 It is not clear to what extent or why these microfluidic environments seem to influence cell behavior, nor whether these effects are device- or cell type-specific (or both).
Cell proliferation is a common readout from a microfluidic culture, as often entire culture areas can be imaged and analyzed via imaging software or plate reader assay, and total adherent or non-adherent cell numbers per channel can be obtained and tracked over time.10–12,24 While many cell types have been shown to be compatible with a wide variety of microdevices, proliferation kinetics are not always the same in microcultures versus macrocultures.10,12,24,25 Differences in the responses of cells to the engineered microenvironments of microfluidic devices to those in macroscale techniques has not only been reflected in proliferation, but has also been assayed via microarrays. A notable study done to analyze the artifacts imparted by a microfluidic culture chamber via the analysis of cellular expression profiles by microarray showed differences between the expression profiles of macro- and microscale cultures, though most were less than 3 fold induction or reduction.22,23 This work is the most comprehensive analysis of the differences in cellular behavior (in this case mRNA expression) in microfluidic devices to date. The addition of more analyses of differences in expression and activation at the protein level will complement these studies and aid in interpreting whether these changes are significant with respect to cellular function.
As these devices begin to be integrated into biological research, we face the issues regarding biological validation of these devices. While many devices have been used for cell based assays in microfluidics, relatively little has been done to investigate the characteristics of microcultures that influence the behavior and phenotypes of the cells they aim to study. Reduced proliferation,10,12,24 reduced seeding efficiencies or plating delays (data not shown), changes in sensitivities to soluble factors,26 and small differences in mRNA expression via microarray22,23versus macroculture have all been shown in microcultures. If results from cell based assays performed in microfluidic devices are to be incorporated into current work using traditional techniques, it will be important to know that the culture conditions alone do not predispose cells to specific responses (i.e., responses different from those observed using other techniques).26 It is clear that microfluidic cell culture alone may impart a range of influences on the behavior of the cells and the effects of microculture may be more wide ranging than previously thought. If significant differences in cellular responses occur in microfluidic devices, knowing what the specific effects on the cellular responses are for the device and cell types of interest is an essential step in validating new microfluidic culture assays.
Characteristics of microfluidic devices that influence the cellular microenvironment
There are a range of physical phenomena that are known to be substantially different as the scale of the culture vessel is reduced that may influence the microenvironment and subsequently cell behavior in microcultures. Some phenomena are particularly interesting for devices that are used in cell based assays such as volume density and surface and material interactions. In this review, we will address some of these phenomena from a cellular perspective, specifically:
-
(1)
Increased volume densities—microcultures often use significantly less reagents, with the result being a higher volume density (more cells for each unit of medium). In turn, this results in reduction of nutrients/growth factors and buffers and presumably more rapid buildup of waste products, per cell. Effects of volume density and microchannel height have been shown to be important modulators of cell proliferation in microdevices.12,24 As existing media compositions have been optimized for macroscale cultures, microfluidic culture devices may require slightly different compositions because of the differences in volume density often found in these devices.
-
(2)
Surface area to volume ratios and polymer interactions—smaller total volumes makes surface area to volume ratios (SA/V) with polymers involved much larger, resulting in a higher sensitivity to any surface interactions that may influence cell behavior. Also, as the ratio between the surface area of the liquid-air interface to the total volume increases, evaporation induced shifts in media osmolarity (and thus media component concentrations) can become a significant limitation.27–29 When the higher SA/V are combined with the unknown effects on media composition from polymer components, and potential cytotoxicity of less well known, though commonly used polymers, such as poly(dimethylsiloxane) (PDMS), these effects may be multiplied. Initial work has suggested that PDMS in particular may not be as “inert” as it has been previously considered,30 though its effects on cell behavior are largely unknown.
Analysis of cellular responses and the cellular baseline in microfluidic cultures
An existing roadblock to further validation and understanding of the microenvironments in microfluidic devices is the relative lack of methods for quantitative analysis of more complex cellular functions applicable to devices of this scale (i.e. protein expression or signal transduction). While a strength of microfluidic devices is the small sample sizes, this is also a limiting factor for many different traditional assay types (e.g. Western blots, flow cytometry) that could potentially be used to better understand the cellular response to microculture. Without the ability to probe more complex aspects of cellular responses beyond viability or proliferation assays, the adaptation and implementation of more cell based assays to microfluidic devices will be challenging. Additionally, the relative lack of validation of the biological responses to microculture is a sizeable hurdle to the integration of these devices into current cell biology methods.
To begin to address this issue, we have demonstrated that the In Cell Western (ICW) technique can accurately quantify protein expression changes in microfluidic cultures, in situ, allowing us to compare microfluidic cultures with macroscale cultures.31 Here, we applied this technique in conjunction with several known readouts of stress and proliferation to better understand whether cells cultured in microfluidic channels may be under stress, or have significantly different baseline levels of signaling and expression as compared to macroscale cultures. An array of microfluidic channels currently in use in our laboratory (ref. 5 and described in detail in Fig. 1) for cell based assays was used to study the responses of cells to microculture as it is a relatively simple microfluidic system with (theoretically) minimal sources of potential influences to cell behavior. First, an analysis of a static microfluidic culture is presented in order to identify the major phenomena influencing cell behavior in microcultures as compared to traditional macroscale cultures, in this case, 96 well plates. Experiments examining how altering media composition, volume density and PDMS exposure in both macro- and microcultures influences proliferation, glucose consumption and activation of metabolic and growth factor signaling cascades elucidates how microcultures can affect cell behavior. To study the changes in signaling and protein expression that might result in the reductions in proliferation seen in microcultures, we identified a range of signaling pathways and housekeeping proteins that are both universal and that are critical to the core functions of mammalian cells. Using this range of universal readouts of stress from energy and metabolism to endoplasmic reticulum (ER) stress, to DNA damage, we compared micro and macroscale cultures for signs of stress. These experiments are presented to illustrate how even in a relatively simple microfluidic culture device, the extent of influence of microculture can be wide-ranging and significant, though the results we have shown are likely to be specific to this cell type and device design. In addition, the potential influence of PDMS on cellular behavior is demonstrated to be dependent on the PDMS surface area to volume ratio, a parameter that is likely quite variable in microfluidic device designs. These results indicate that careful validation of microfluidic devices for cell based assays needs to be performed and presented prior to mounting larger scale experiments employing them in order to understand the biases that may be imparted by microculture alone. By understanding the artifacts and biases of the system one can then begin to examine and interpret the data obtained in a more biologically meaningful way.
Fig. 1.
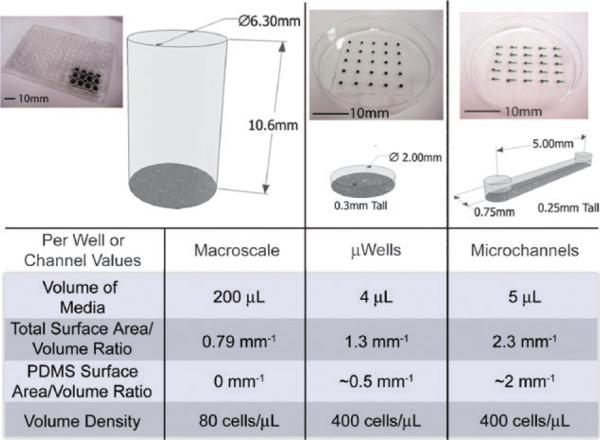
Important differences between micro- and macroscale cultures range from total media volume, SA/V ratios and volume densities. Macroscale cultures represent what is typically used in biological laboratories and have low volume densities, and SA/V ratios, with well-studied polymers (typically polystyrene). Microwells (μWells) are a PDMS stencil placed on tissue culture plastic similar to microchannels. They provide the same small volumes and high volume densities, but with less contact with PDMS and a smaller SA/Vtotal than microchannels. These three culture devices were utilized to study how cell behavior changes in each, to illustrate the impact of these various characteristics (volume density differences, effects of small volumes, and interactions with PDMS).
Results and discussion
Three different conditions with different sets of physical parameters were employed to demonstrate the potential influences of the physical phenomena mentioned above (summarized in Fig. 1). Each culture device (96 well plates, subsequently labeled Macro; microwells, labeled μWells; and microchannels labeled Micro) represents a potential culture platform that produces different microenvironments, to which cells may react differently depending on the relative importance of the different characteristics on cell behavior. In the case of 96 well plates, relatively large total volumes are used (as compared to microdevices), and they have low total SA/V (SA/Vtotal) consisting only of polystyrene (no PDMS) and low volume densities. Meanwhile, microchannels have low volumes, higher SA/Vtotal, mostly consisting of PDMS contact (SA/VPDMS), and higher volume densities. Microwells share the low SA/Vtotal with 96 well plates, have a small amount of PDMS contact area, but have the low volumes and high volume densities of microchannels. SA/V ratios similar to those found in our microchannel devices cannot easily be replicated in macroscale cultureware (even in 384+ well plates due to the low “ceiling” of these devices) due to how SA/V ratios scale with volume, and thus because the properties of microwells are intermediate to those of either macroscale or microchannel cultures, they can be employed to more fully investigate cellular interactions with the microenvironment (and more specifically PDMS). For the purposes of this review, microwells are simply an illustrative platform, and data using this construct will be included only as a reference when required to clarify which specific aspects of microchannel culture is responsible for a specific cellular response.
Baseline cell behavior assays
Mouse mammary fibroblasts grown in microchannels were compared to those in macroscale cultures over a typical culture period for this cell type (2 days) to determine if, for this cell type and microfluidic channel geometry, there were differences in cellular function as compared to macroscale cultures. No media changes or perfusion of the microcultures were used in order to compare more completely macro- and microscale cultures via identical protocols. Typical microfluidic devices require perfusion or frequent media changes (media change frequency has been shown to influence proliferation in microcultures12), but it is not clear why this is required in microcultures but not in macroscale techniques, and the reasons for which we hoped to elucidate. For example, simplified calculations suggest that nutrient depletion is not significant12 but little quantitative biological data exists to demonstrate if this is truly the case. To assay several aspects of the cellular response to microculture, we employed proliferation assays, measurements of glucose concentration in the medium, and ICWs to quantitate activation of two metabolism and growth relating signaling pathways, namely AMP activated protein kinase (AMPK) and S6 ribosomal protein (S6).
AMPK responds to changes in intracellular AMP/ATP availability and thus changes in glucose and energy availability in the microenvironment, and is an important mediator of the cell's adaptation mechanisms in response to the cellular energy status. The function of AMPK is to regulate the balance between anabolic and catabolic cellular functions in response to the microenvironment.32,33 When it is activated, catabolic pathways, which create ATP, are activated, while anabolic pathways, which consume ATP, are inhibited. Catabolic pathways include glucose uptake and glycolysis, which are required for basic cellular respiration and function. Anabolic pathways include protein and fatty acid syntheses and are required for cell growth and proliferation. Thus levels of phosphorylated AMPKα (the catalytic subunit of AMPK), serves as an excellent readout of whether microfluidic cultures are comparatively energy deprived versus macrocultures.
The AMPK pathway and many other growth factor sensitive pathways converge and feed into the mammalian target of rapamycin (mTOR) pathway via shared signaling targets. The mTOR pathway is responsible for regulation of cell size and translation rates, and integrates a wide range of signaling cascades, many of which are influenced by growth factors and nutrient availability.34 As part of the mTOR complex 1 (mTORC1), mTOR activates downstream targets such as S6 via phosphorylation. S6 activation correlates with increases in protein translation levels and also expression of cell cycle related proteins. The levels of phosphorylated S6 will provide a view of the collaborative activation status of several growth and metabolism regulating pathways in response to many different cellular stressors that may be present in microcultures.
The activation of these signaling cascades and the resulting glucose consumption and proliferation rates were assayed in microchannel and macroscale cultures over the two day culture period (Fig. 2). A significant inhibition of proliferation was seen in microchannel cultures, consistent with previous data comparing macro- and microscale cultures. Although proliferation rates were significantly reduced, glucose consumption in microchannel cultures was over 3 fold higher than in macroscale cultures. Additionally, significant differences in S6 signaling were seen, and while at 24 h after seeding microchannel cultures had over 3 fold higher levels of phosphorylated S6 than macroscale, at 48 h after seeding, the levels had reduced to below those seen in macrocultures. Meanwhile, AMPK phosphorylation was not significantly different between the culture scales. These data suggest that even though microchannel cultures do not proliferate as much as their macroscale counterparts, they require many fold more glucose for the same level of AMPK phosphorylation to be established. Also, while early on (24 h) growth factor signaling is 3 fold higher in microchannel cultures than macroscale, this does not translate into the decision to proliferate, and levels reduce substantially by 48 h. These results illustrate that the cellular baseline in microchannel cultures may be substantially altered early in the culture period (before 24 h as compared to their macroscale counterparts, given the same culture protocols (same seeding density, medium and lack of media adjustments). The following sections will use similar assays to begin to evaluate which of the differences between these simple microchannel culture and macroscale culture conditions are most influential in determining cell behavior.
Fig. 2.

Comparison of proliferation rates, glucose consumption, and S6 and AMPK phosphorylation levels between macroscale culture and microchannels. Proliferation over the two day culture period in each culture type (microchannel, micro; 96 well plate, macro) was normalized to the cell number after attachment (3 h) (A). Via glucose concentration monitoring and proliferation data, an average glucose consumption rate per cell was calculated for each culture type (B). ICWs for important energy and growth factor signaling molecules were performed at 24 and 48 h after seeding for S6 (C) and AMPK (D). Error bars represent standard deviations, with n > 3 for both macro- and microscale cultures.
Increases in volume densities
Volume density is not a commonly addressed variable in cell culture, although in traditional macroscale cultures volume densities are typically not largely different between different culture platforms (e.g., 96 well plate vs. Petri dish). As the scale of the culture is reduced and confined in a microfluidic device, the volume densities can increase substantially (5 fold or more depending on device size), unless the devices are perfused continually with medium, or have frequent and regular media changes. The effects of volume density changes alone are not well understood, although an increase in volume density conceivably would reduce the total amount of all media components available to each cell, including growth factors, nutrients, amino acids, sugars and buffers. Additionally, the rate of accumulation of waste products and signaling molecules produced by the cells would be higher when less fluid is available per cell. These influences are likely reasons why perfusion or media changes are generally required for long term monolayer cell culture in microfluidic devices, though this has not been proven explicitly. We have shown previously that media changes and channel height can significantly impact the proliferation rates seen in microfluidic channels as compared to macroscale cultures.12,24 However, as channel height is changed, the SA/V ratio and the relative amount of nutrients/waste per cell are influenced as well. Thus while these experiments demonstrate how channel design and protocols can influence cell behavior, they do not identify what specific characteristics cause these changes. A complete picture of the effects on cellular functions of these changes is yet not clear, but current media formulations have been developed using macroscale techniques, and could potentially be less appropriate for the specific needs of microscale cultures.
Because volume densities are 5 fold higher in our micro-channel cultures than typical macroscale cultures, it is possible that simply the reduction in the amount of media components such as glucose or growth factors per cell is significantly affecting cell behavior. This could potentially be remedied by appropriate media supplementation with either glucose or serum, or simply by adding extra medium to reduce the volume density. To analyze what the effects of media supplementation and changes in volume density are in both microchannel and macroscale cultures, cells were seeded in media either with 10% serum and a range of glucose concentrations (from 0 g L−1 to 9 g L−1, the typical medium for these cells contains 4.5 g L−1), or media with 4.5 g L−1glucose and a range of serum concentrations (from 3 to 20%). With these dilutions, the per cell amount of glucose or serum in macro- and microchannel cultures would be very similar in the appropriate pairs of conditions, i.e. macroscale cultures with 1.8 g L−1 glucose and microscale cultures with 9 g L−1glucose or macroscale cultures with 3% serum and microscale cultures with 20%. If media components alone were the predominating factor, then these cultures would be expected to show similar proliferation, glucose consumption and S6/AMPK signaling. Media supplementation does not, however, influence other factors such as depletion of amino acids (and other media components), buffering capabilities or the buildup of waste products. These factors can be altered by changing the volume density in the cultures by either reducing the total media volume used in 96 well plates, or by adding supplemental medium drops to microchannels.
The proliferation results from simply changing the volume densities of the macro- and microscale cultures are shown in Fig. 3A, where 1× density is that of the typical macroscale cultures (a volume density of 80 cells per μL, 200 cells per μL for 2.5× and 400 cells per μL for 5×, though surface density remained the same). In both 2.5× and 5× conditions, microchannel culture proliferation was significantly reduced as compared to the corresponding density in macroscale cultures. Also, while a significant increase in proliferation was seen in macroscale cultures when the volume density was decreased from 5× to 2.5× (p < 0.01), the increase in microchannel cultures was not significant. These data suggest that volume density alone is not a predominating factor in the reduction in proliferation seen in microchannels, as proliferation rates seem to be less sensitive to decreases in volume density than in macroscale cultures.
Fig. 3.

Volume density and media supplementation assays. Volume density changes alone were not sufficient to increase microchannel proliferation, and in both 2.5× and 5× densities (1× density being the typical macroscale volume density), proliferation was significantly reduced as compared to macroscale cultures of the same density (A). Glucose consumption rates in media with a dilution of starting glucose concentration were assayed (B). ICWs for AMPK phosphorylation at 24 and 48 h after seeding for macro- and microscale cultures with the dilution of glucose showed no significant differences between any of the conditions (C), though proliferation was significantly reduced in microchannels, except for the 0 g L−1 condition (D). Significant differences between microchannel cultures and the corresponding macroscale culture at the same time points were seen regardless of media serum concentrations (E), though proliferation was significantly lower in microchannel cultures regardless of media serum concentration (F). All proliferation data listed here compare the fold increase in cell number at 48 h versus that at 3 h post-seeding.
Meanwhile, cells cultured in dilutions of starting media glucose levels showed that as glucose concentrations increase, the differences in glucose consumption between microchannel and macroscale culture becomes more pronounced (Fig. 3B), though changes in AMPK signaling at 24 or 48 h were not significantly different regardless of culture scale or media glucose concentration (Fig. 3C). Despite relatively high glucose consumption rates, and similar AMPK phosphorylation, the proliferation of cells in microchannel cultures failed to be restored as compared to macroscale rates, and in all except the 0 g L−1 condition (in which proliferation was largely inhibited in all cultures, see ESI for additional data†) were still significantly lower than the corresponding macroscale culture (Fig. 3D).
When cells were cultured in dilutions of serum components, similar trends as demonstrated previously were seen regardless of serum concentration for phosphorylation of S6 (Fig. 3E). Microchannel levels were significantly higher than macroscale at 24 h regardless of serum concentration, and these levels reduced drastically by 48 h, returning to closer to macroscale levels. However, none of the serum concentrations resulted in a significant increase in proliferation in microchannels (Fig. 3F), though macroscale cultures seemed to be sensitive to serum concentration as expected for cells of this type (many fibroblastic cells tend to be very serum-dependent). These data suggest that not only is proliferation in microchannel cultures insensitive to volume density changes, but also that glucose and serum supplementation are not sufficient to significantly change the outcome of the cultures either with respect to AMPK and S6 phosphorylation or proliferation. However, microscale cultures did seem sensitive to the changes in glucose concentration with respect to their glucose consumption, beyond the sensitivity seen in macroscale cultures, though it is not clear why this effect would be seen. Thus, the changes in volume density and media component availability are likely not dominant factors in why microchannel cultures are inhibited from proliferating as compared to macroscale cultures.
Surface area to volume ratios and polymer interactions
Although microfluidic devices can provide the benefits of low reagent volumes, as the scale of the culture is reduced, the susceptibility of the culture volume to evaporative losses increases as well as the total SA/V increases. For example, in these conditions, 1 μL of evaporative water loss in a macroscale culture results in a 0.5% shift in osmolarity, while the same loss in a microchannel culture will result in a 33% increase. While most incubators are humidified, often humidity levels are approximately 80% relative humidity, with significant variability (e.g. when the door is opened); while this may be sufficient to limit significant concentration of macroscale cultures, it often is not sufficient for microdevices.27,28 Recently, evidence of changes in cell behavior and morphology as a result of evaporative losses and subsequent concentration of the media in a micro-device as compared to a macroscale culture was shown.29 Because of this, we have optimized our culture conditions to minimize evaporation via the use of sacrificial water pools and measured media volume over time to ensure that our microscale cultures are not experiencing osmotic shock (data not shown).
While the issue of evaporation from a cellular perspective is beginning to be addressed in the literature for microfluidic cell culture systems (despite it being a very common challenge), the influence on cell behavior of the increased SA/V is less well understood. Conceivably, surface interactions with media components either via leaching of media components into the polymer bulk or vice versa, and the interactions of proteins with surfaces may become an important factor in controlling the microenvironment cells are exposed to in these devices. Most macroscale cultures are performed in polysytrene (or glass bottomed) tissue culture flasks, dishes and plates. While many microfluidic cultures are performed with similar substrates as macroscale cultures by adding micropatterned channel materials35 onto tissue culture substrates, newer materials are used to fabricate the body of the devices. As new materials are integrated into microfluidic devices for cell based assays, the limitations of these materials are also being evaluated. Often the materials that ells interact with are considered to be “inert” with respect to their effects on cellular behavior and are largely ignored unless they are designed specifically to be bioactive.
Recent work has shown for a common polymer used for microfabrication, PDMS, that the partitioning of hydrophobic molecules into the polymer bulk can result in significant changes in the solution concentrations.30 This issue becomes particularly important when compounds used to stimulate or block cellular processes or pathways are either small and hydrophobic, such as many small molecule inhibitors, steroid hormones, cell secreted factors or other compounds used in drug screening, but also may be important for basic cell culture itself. Additionally, titrations of compounds used for screening or controls that may potentially interact with the materials used can be done to determine whether or not this might be a significant issue for the molecules/materials of interest. However, if PDMS does significantly interact with the basal media components in a culture, it will be challenging to determine specifically which components are being affected and even more so to determine the extent of the effects on the range of cellular processes occurring in cultures.
Experiments which give insight into the potential effect of SA/V ratios and the influence of PDMS were performed to begin to understand if these may be limiting factors in microfluidic cell based assays. These results are an examination of the specific effects of microculture on cellular behavior, with a focus on metabolism, proliferation and cell cycle progression. For all subsequent analyses, a third culture type was used to illustrate how SA/V ratios and PDMS contact influence cell behavior, microwells, (mWells, described in Fig. 1). The total and PDMS-only SA/V ratios of the three conditions are shown in Fig. 4A. The macroscale and microwell cultures have very similar total surface area to volume ratios (SA/Vtotal), and are analogous to the differences between a 6 well plate and a 96 well plate (which typically provide indistinguishable results), with the exception of a low amount of contact with PDMS (SA/VPDMS of ~0.5 mm−1) in the microwell cultures. Additionally, microwell cultures serve as a good comparison to microchannels because they are a control for both small volumes (they are equally as sensitive to evaporation as microchannels) and volume densities (densities are similar to those in microchannels).
Fig. 4.

Effects of total and PDMS SA/V ratios on glucose metabolism and proliferation in normal and supplemented medium. Total and PDMS-only SA/V ratios for microchannel, macroscale, and microwell cultures vary significantly (A). Volume density changes in the three culture types did prove to affect proliferation, but both microchannels and microwell proliferation were significantly reduced as compared to the macroscale culture of the corresponding volume density (though the differences were not as significant for microwell cultures, B). In dilutions of media glucose concentrations, the average per cell consumption rates were closer to macroscale levels in all conditions in microwell cultures than in microchannel cultures (C), though proliferation was consistently reduced in both microscale culture types (E). Similar reduction in proliferation in microscale cultures were seen when serum concentrations were varied (D), suggesting that neither volume density nor supplementation could rescue proliferation in cultures in contact with PDMS.
If SA/Vtotal is critical, then cultures in microwells and macroscale cultures with the same volume densities would be expected to proliferate similarly, while microchannels would be much more affected because they have nearly double the total SA/V. However, if SA/VPDMS was more significant, it would be likely that despite the equal total SA/V of microwells and macroscale, that the proliferation and behavior of cells in microwells would fall either intermediate to the other two conditions, or be more similar to microchannels. To elucidate these issues with respect to proliferation, cells were seeded in macrocultures, microwells and microchannels with a range of volume densities (Fig. 4B) and also in the same media supplementation conditions used previously (both glucose and serum dilutions) and the glucose consumption (Fig. 4C), and proliferation (serum dilutions, Fig. 4D; glucose dilutions, Fig. 4E) were measured.
While microwell cultures do show more controlled glucose consumption rates that fall closer to those seen in macroscale cultures than microchannels, proliferation is still significantly inhibited in these cultures regardless of volume density and media supplementation. A statistically significant increase in proliferation in response to a decrease in volume density or increase in serum concentration does not occur in microwells or microchannels while these changes are significant in macroscale cultures. For both 2.5× and 5× densities, proliferation rates were higher in microwells than those in microchannels (but still significantly lower than macroscale cultures, though less so) and when glucose and serum were supplemented microwell proliferation rates were consistently the same or higher than in microchannels.
These data suggest that contact with PDMS is a dominating factor over volume density, media supplementation or SA/Vtotal for these cells. Microchannel cultures seemed to consistently show a maximum of nearly one population doubling over 2 days, typically in the first 24 h after seeding, while macrocultures go through an average of 2 population doublings (for a total of 4+ fold increase in cell number). Also, microwell cultures tended to show slightly more proliferation than microchannel cultures, suggesting that the mechanisms for how PDMS may be influencing these cultures may be dependent on how much PDMS is available to interact with the medium (with respect to SA/VPDMS). Because no experimental condition showed significant rescue of proliferation in microcultures, further investigation of the dynamics of any cell proliferation was warranted to identify if cells in microchannels and microwells were successfully entering the cell cycle and if there was any consistent cell cycle block (such as arrest at specific parts of the cell cycle resulting in the halt in proliferation seen after 24 h).
Nuclear size and cell division
To determine if cells in all of the culture types were capable of entering the cell cycle rather than remaining quiescent, the presence of cells in mitosis via microscopy was verified (Fig. 5). Cells in all three culture types were seeded in the standard media (4.5 g L−1 glucose and 10% serum), and after 48 h fixed and stained for nuclei and imaged via microscopy. Qualitatively, the frequency of cells actively dividing (cells with condensed chromosomes) was lower in microchannels vs. microwells and 96 well plates, but was evidence of cells entering the cell cycle in all culture types.
Fig. 5.
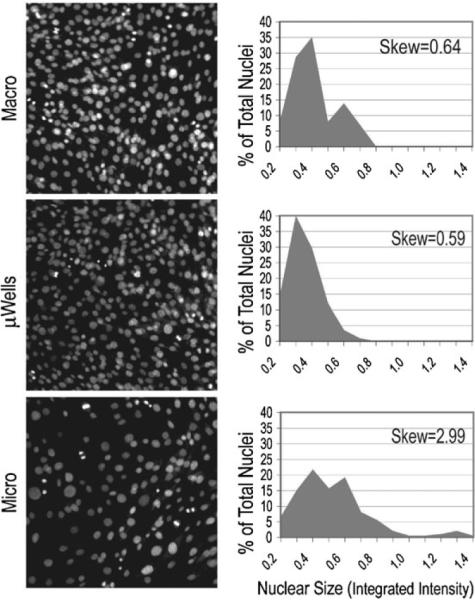
Representative images of the nuclei of macroscale cultures (top, macro), microwell cultures (middle, μwells) and microchannel cultures (bottom, micro) taken with the same exposure time and magnification. When images were analyzed by determining the integrated intensity of a sample of nuclei (minimum of 250), distributions of the nuclear size as a percentage of the total nuclei were determined, shown to the right of the images for each culture type.
Because cells in microchannels were clearly entering the cell cycle, whether the cells were successfully dividing was then considered. Qualitatively, microchannels frequently contained more nuclei with very large area (images in Fig. 5 are of the same magnification). To begin to quantify this observation, the center of 3 wells, microchannels or microwells were imaged with the same excitation intensity and magnification and the integrated intensity of the nuclei in each image was quantified using NIH ImageJ (sample sizes were a minimum of 250 nuclei per culture type per well or channel). Nuclei that were actively dividing and visibly in some phase of mitosis were not included (determined by the presence of condensed chromosomes, chromosomes aligned for mitosis or actively separating cells).
The distributions of the integrated intensity of the nuclei in each scale are plotted in Fig. 5 beside representative images of nuclei in each culture type at 48 h after seeding. In both macroscale and microwell cultures, the distributions were closer to expected cell cycle distributions, with very low skew (the degree of asymmetry of a distribution around its mean, zero being a symmetric, normal distribution), near 0.6, and with a predominating (presumably 2n) peak and in the case of macroscale (since microwell cultures are inhibited from proliferating long before 48 h), a 4n peak from cells in S/G2. However, in microchannel cultures the skew was nearly 3, and positive, indicating a large degree of skew towards the right, or nuclei with more DNA. The skew in microchannels was beyond the expected values for 4n cells presumably in G2/S (~0.8 fluorescence units maximum), and thus these high DNA content cells could not be explained by simply a G2/S arrest. Upon further observation, images from microchannels showed several different cell cycle progression problems occurring that could not be found in cells growing in either of the other two culture types. An image showing several cell division defects commonly found in microchannel cultures at 48 h after seeding is shown in Fig. S7.† These included multinucleated cells, those with far more than the expected 2n or 4n amount of nuclear DNA, those with disorganized chromosomes during mitosis among other defects.
These data suggest that in microchannel cultures, these cells can successfully enter the cell cycle, but have a much higher frequency of either arrest in the S/G2 phases of the cell cycle, unsuccessful division and/or accumulation of nuclear DNA. The wide distribution of nuclear sizes is not seen in microwell cultures, or macroscale cultures. If cells in microchannels were inhibited from finishing a round of cell division, they would generally remain quiescent afterwards (as perhaps for these mouse mammary fibroblasts (MMFs) like many cell types, 4n cells are inhibited from initiating another round of cell division or cells are arrested in S/G2), which would be consistent with the proliferation kinetics seen in these cultures. One round of cell division may be initiated in the first 24 h when proliferation is generally seen in these cultures, but after this point, very little further division occurs because the first round was unsuccessful in so many cells.
In these experiments, the cells used were mammary fibroblasts isolated from p16INK4a knockout mice. p16INK4a functions to regulate the entrance of cells into the S phase of the cell cycle, and over or under expressing it results in errant cell cycle control or arrest.36 This defect is likely why these cells did not undergo apoptosis when faced with severe cell cycle inhibition as cells with functional p16INK4a likely would have (as we have observed for other cell types in our lab). While for these cells, the effects of microfluidic channel culture have resulted in the accumulation of nuclear DNA likely due to the lack of p16INK4a, other effects, potentially more or less disruptive to cellular functions may be present for other cell types in other microfluidic devices. Also, it seems that as SA/VPDMS increases, cell proliferation and changes in baseline functions become more pronounced. To begin to better understand what upstream events occur that result in these changes, we began to study how the expression and baseline activation levels of many proteins that play crucial roles in basic cell survival change in response to microchannel culture.
Baseline status of other signaling pathways
In these experiments, assays for the levels of each protein, or protein modification of interest were done to determine if there are significant differences in activation levels of various stress related signaling pathways in macroscale and microchannel cultures. Biologically relevant readouts were chosen that relate to metabolic and growth factor signaling processes, heat shock protein/ER stress, important cellular signaling pathways (mitogen activated protein kinases (MAPKs)), and DNA damage (summarized in Fig. 6). This panel of stress responses provides an overview of the basic types of cellular functions that are nearly universal, and could be applied to any mammalian cell type of interest to begin to understand what types of effects a specific microfluidic device may have on the cellular baseline functions.
Fig. 6.

Summary of the readouts tested in microscale cultures. From left to right, columns show which stimuli result in the changes in phosphorylation or increases in expression (described in the center column) and what the subsequent activity of the protein is. Arrows indicate that the protein activates the processes listed, while blunt arrows indicate inhibition.
Ideally, in order to compare how microchannel culture alone influences cell behavior, other differences such as cell surface density should be minimized between the culture types, while maintaining an exposure time long enough for differences (if any) to be seen, and enough cell density for ICWs to pick up smaller changes in signal. For these experiments, cells were seeded at the same surface densities (higher than for normal culture, see Methods) in microchannels and in macroscale cultures, and assayed after 24 h, before large differences in confluency were obtained, and in a predominately non-proliferative microenvironment (thus minimizing the effects of PDMS-induced cell cycle inhibition on cellular responses in microchannels). Validation of the ICW technique for these cultures, this cell type and all the readouts included has been included in the supplementary information along with all supporting validation work (positive and negative controls, etc.).†
This panel of stress assays aimed to provide insight into how microfluidic cultures are different, from a cellular perspective than the corresponding macroscale cultures (results summarized in Fig. 7). From a metabolic and growth factor signaling perspective, microchannel culture reduces the total nutrient availability per cell due to the increase in volume density. In addition, the total amount of growth factors per cell (typically from FBS used in many media) is reduced as volume density increases. Either of these factors may cause a significant change in cellular behavior and proliferative decisions in microcultures when in these conditions (equal surface density vs. normal culture conditions shown previously). AMP kinase and S6 (previously discussed) are readouts sensitive to these kinds of stressors. Statistically significant differences in AMP kinase and S6 phosphorylation indicate that perhaps the reduced media volumes result in nutrient depletion (resulting in increased phosphorylation of AMP kinase) or growth factor depletion (resulting in reduced activation of S6) when cultures are compared in this way. Because cells in both culture types are somewhat inhibited from proliferating due to surface density inhibition in this condition, volume density (and thus media component availability) may become a dominant factor, previously clouded by PDMS-induced responses seen in longer term cultures.
Fig. 7.
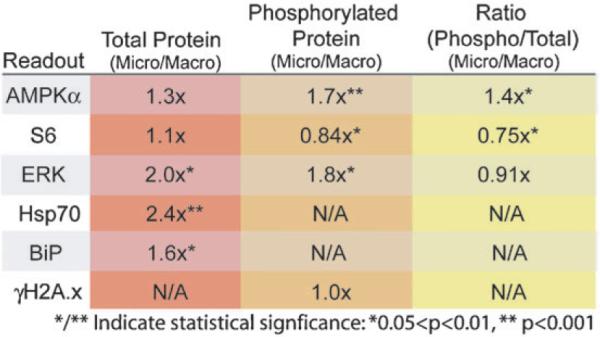
Summary of readout results comparing macro- and microscale cultures for total protein, the phosphorylated protein (if applicable) and the ratio of phosphorylated to total for each applicable readout. Several of these proteins exhibit approximately 2 fold (or more) changes in expression in only 24 h of microculture, indicating that the influences of microculture are significant and relatively rapid. Statistical significance via Student's t-test is listed when p values fell within the limits denoted.
Other readouts showed statistically significant differences in microchannel cultures compared to macrocultures such as the up regulation of ERK1/2, BiP and HSP70 (Fig. 7). These proteins are sensitive to a wide variety of potential stressors, thus it is unclear what specific characteristic of microfluidic culture is causing each of the responses seen. However, these differences do reflect that the microenvironment in microcultures is truly different and results in different levels of activation and expression of key proteins involved in basic cell functions including attachment, growth, and protein folding/production, even without any experimental differences such as growth factor stimulation or other treatments. The functions of these proteins that may be responsive to microchannel culture conditions are summarized here:
The members of mitogen activated protein kinases (MAPKs) mitigate a wide variety of cellular responses to stimuli.37–39 These kinases are part of a complex system of signaling cascades, which ultimately serve to translate signals from cell surface receptors into a cellular response. Extracellular signal-regulated kinases 1 and 2 (ERK1/2) are MAPKs involved in cell attachment, migration, proliferation, and differentiation (all may be influenced by microchannel culture).37 ERK1/2 are found in a variety of locations in the cell, resulting in a variety of different functions. ERK1/2 are found at adherens junctions and focal adhesions and as much as half the total cellular ERK1/2 are bound to microtubules throughout the cell altering their polymerization, and when activated via phosphorylation, activate a wide variety of target genes.37
Environmental changes that result in protein misfolding include glucose starvation, exposure to reducing agents, low pH, hypoxia, which, if present in microcultures, could result in changes in heat shock protein (HSP) expression or ER stress. The HSP70 family serves to fold nascent proteins and respond to a wide range of cellular stressors that adversely affect protein function (e.g., thermal, oxidative, or metabolic stressors). HSP70 binds hydrophobic patches of unfolded or incorrectly folded proteins and, via an ATP dependent mechanism, refolds the proteins or assists in targeting them for degradation.40,41 Increased levels of HSP70 tend to be anti-apoptotic.
Meanwhile, the endoplasmic reticulum (ER) is the cell's main protein and lipid production organelle and is responsible for protein folding, targeting and quality control processes. ER stress can occur as a result of large amounts of unfolded proteins (due to overexpression of proteins, or blockage of their exit from the ER), reductive environments, low glucose, or lowered pH.42,43 When unfolded protein levels increase in the ER, higher levels of BiP are required in order to properly fold or target for degradation the backlog of protein. As a cell survival protein, the loss of or underexpression of BiP in response to stress induces apoptosis.44 Interestingly, BiP was originally known as glucose regulated protein 78 (GRP78) since it was discovered in cells grown in vitro after glucose starvation,43 which may be the case in microcultures depending on the conditions and cell type. Thus, there are several potential sources of ER stress and glucose restriction in microculture. The responses of both heat shock proteins and resident endoplasmic reticulum proteins to stress are critical for cell survival and apoptosis.
Variations in microenvironments and behavior of cells in macro- and microscale cultures could potentially cause significant differences in levels of DNA damage (although it is not readily apparent which culture type is at higher risk). To determine if DNA damage is occurring at a higher level in microcultures versus macrocultures, we stained for a marker for DNA damage and activation of the ATM/ATR pathways, phosphorylation of histone H2A.x. H2A.x represents from 2–25% of total H2A and is phosphorylated (γH2A.x) as a result of double strand breaks by ATM and ATR.45 γH2A.x foci appear in the nucleus at sites of DNA damage. However, differences in levels of γH2A.x were not seen, indicating that significant differences in rates of DNA damage between the scales are not likely. This also suggests that reductions in proliferation seen in microcultures are not due to delays for DNA repair, nor that widespread apoptosis is occurring in these cultures.
Conclusions
We have demonstrated that even in a relatively simple, microfluidic culture channel array, significant differences in cell behavior from glucose consumption to proliferation to markers of stress as compared to macroscale techniques are seen. Though the fold changes in protein expression and activation levels demonstrated via stress assays are not large, they are statistically significant differences, and may be large enough to cause misinterpretation of cellular behavior in microchannels. While the data presented suggest that PDMS may be causing artifacts in microfluidic cell culture results depending on the level of interaction with the cells/culture medium, it is still unclear whether this may be due to molecules leaching into or out of the PDMS or via some other mechanism. One possibility is that uncrosslinked low molecular weight polymers may leach from the PDMS into the medium. If this were the case, the monomers could potentially partition into the hydrophobic portions of the cells such as the plasma membrane, ER or nuclear envelope or otherwise function to disrupt signaling or metabolism via an unknown mechanism. Likewise, it is possible that hydrophobic growth factors or lipids from the cell culture medium and serum are being depleted by diffusion into the PDMS bulk. If lipid metabolism was an important source of energy for these cells, the loss of lipids to the PDMS in microcultures might explain why their use of glucose as an energy source increases by several fold over the rate in macrocultures or microwells. Either of these potential explanations would explain why cultures with extra medium (lower volume densities) proliferate more than those with less (either the concentration of the low molecular weight polymers in the medium is less, or larger amounts of lipid/growth factors are provided replacing some lost to the bulk PDMS). Also, these factors would be consistent with the increase in growth in microwell cultures with respect to microchannel cultures, but not equal to that of macrocultures, as they have an intermediate SA/VPDMS. Regardless of which effect is primarily responsible for the differences seen in microchannel cultures, these data illustrate a significant change in phenotype depending on the type of microenvironment the cells are exposed to, which is likely to be both cell type- and device-specific.
With the integration of ICWs to high throughput microfluidic assays the panel of stress assays could be reproduced for a wide variety of cell types and could be expanded to include more aspects of cellular function important for the assays and cell types of interest. This technique could be used to validate and troubleshoot microfluidic cultures for cell based assays to better understand the cellular baseline for specific cell types of interest prior to large assays being run. Also, the ability to do quantitative studies of signaling cascades in situ in microfluidic devices expands the available readouts for microfluidic assays allowing for more variety of assays to be performed in them. The activation or inhibition of signaling pathways in response to drugs or other stimuli can now be screened using microfluidic devices, with all of the resource benefits that they provide (in reduced cell sample sizes and reagent costs).
Microfluidic devices for cell based assays have provided new types of engineered microenvironments and new methods for controlling and observing the cellular responses to them. The field has begun to analyze the biological effects of the physical differences of microfluidic devices for cell based assays, ranging from evaporation in static microfluidic cultures to flow induced artifacts in gradient generation devices. Nonetheless, the relative lack of quantitative biological analysis techniques that have been interfaced with microfluidic devices has prevented more facets of cellular function beyond viability or proliferation to be analyzed in them in a simple way. The results shown here indicate that, from a cellular perspective, the microenvironment in microfluidic cultures can be significantly different from those in traditional macroscale cultures. However, the results we've shown are likely unique to the specific conditions we tested, which is an important issue regarding how microfluidic devices should be validated in the future. The responses presented are likely specific to this cell type, given these seeding densities/culture protocols, the time points chosen for assay, and the devices themselves.
There will be a wide range of physical phenomena and device characteristics specific to every microfluidic device and set of working protocols that may influence cell behavior. Fig. 8 summarizes phenomena known to be of interest even in relatively simple microfluidic culture devices, some of which we have discussed the influences of in this manuscript and some which may be more applicable to other devices or protocols. The identification of the most relevant and influential sources of artifacts from microfluidic devices will be critical for better validation of the devices from a cellular perspective. A variety of conditions testing the influence of these phenomena will be important to evaluate at some level for every cell type, protocol and device used for cell based assays because of how sensitive and variable the responses to all aspects of the microenvironment can be. In addition, signaling pathways and cell behavior characteristics particularly crucial for the success of the specific assay being run will be important to validate in the specific assay condition prior to large scale experiments. Finally, studies of the cellular baseline should be performed using targets of interest relevant for the specific assay, for example using PCR based assays if mRNA expression changes are the final readout of an assay, versus the protein-based approach we have demonstrated here.
Fig. 8.

Summary of likely phenomena and characteristics commonly found in microfluidic devices that may influence cell behavior. These examples are likely only a subset of potential sources of variation and the specific validation steps required will vary depending on specific device designs.
It is possible that when more complex functions such as flow, gradient introduction, growth factor or drug stimulation, are incorporated into microfluidic devices that perhaps these too may affect the cellular baseline. A better understanding of how the microenvironment in microfluidic devices for cell based assays affects basic cellular functions will be critical for future work. Also, understanding the unique limitations and benefits of the microfluidic systems in use for biological assays will provide insight into what controls will be necessary to more fully validate the results in context of current techniques. These differences might also be leveraged to provide new ways to assay cellular responses by comparing macro- and microscale assays. Future studies integrating cell biological assays with microfluidic cultures will rely upon well designed studies with correct and thorough positive and negative controls for validation purposes.
Methods
All statistical analyses were conducted using the Student's t-test and p values cited when levels fell below either 0.05 or 0.01, as labeled. All error bars are one standard deviation.
Microscale and macroscale cultures
Simple microfluidic channels were used for this study to illustrate how even simple devices can influence cell behavior. Dimensions were 750 μm wide, 250 μm tall and 5 mm long, with 750 μm diameter ports at either end. PDMS channel bodies were placed on tissue culture treated Petri dishes and placed in larger Petri dishes with sacrificial water to prevent significant evaporation (the effectiveness of which was tested by measuring the volume remaining in the channels over the 2 day culture period). These channels were used with 4 μL of total culture volume each, and compared to 96 well plates with 200 μL of medium per well.
Cell culture
Mouse mammary fibroblasts (MMFs) isolated from p16/INK4a knockout mice were cultured in DMEM with 10% serum, and 1% P/S (passage numbers ranged from 20–35). When not specified, high glucose medium was used, containing 4.5 g L−1 glucose, otherwise no glucose DMEM was used and glucose was added to the specified concentration. Cells were passaged every 2 days with approximately a 1 : 5 dilution, (initial confluence was approximately 20–30% and at 2 days was approximately 70–80%). For stress assays MMFs were seeded at the same surface density (approx. 90–100 K cm−2)in microchannels and in 96 well plates and allowed to plate and proliferate for 24 h (initial seeding densities of approx. 50–60% reaching 70–80% at 24 h in macroscale cultures). For proliferation assays and glucose measurements, cells were seeded at a normal passage density (20–30%) and allowed to proliferate for 48 h which is the normal passage protocol for cells grown in flasks for this cell type. This results in a 5 fold increase in volume density in microchannels versus a typical macroscale culture with the same surface density. At 24 h after seeding, positive and negative control treatments were performed as described for each readout and then cells were fixed and stained for ICWs.
Proliferation assays
For proliferation assays, channels and wells were sacrificed at each time point (5 or more channels or microwells for microcultures, 3–5 wells for macrocultures), and fixed and stained for nuclei with ToPro3. Cells were washed briefly with PBS, then fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, then permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. ToPro3 (Molecular Probes) was diluted 1 : 500 in PBS and incubated for 10 min at room temperature, then washed twice with PBS and dried prior to scanning.
Glucose concentration
Glucose concentration was determined by taking media samples from cultures prior to fixing, adding 500 μL of the glucose assay reagent (BioAssay Systems, QuantiChrom Glucose Assay Kit) and boiling for 8 min. The cooled samples were transferred to a 96 well plate, analyzed via plate reader colorimetric assay at 630 nm and compared to a standard curve.
In Cell Westerns
For ICWs, cells were fixed and stained for either phosphorylated and/or total protein. Cells were washed briefly with PBS, then fixed with 4% PFA in PBS on ice for 20 min with the addition of phosphatase inhibitor cocktail at 1 : 100 dilution (Pierce, Halt Phosphatase inhibitor cocktail, #78420). To permeabilize, two washes with 0.1% Triton X-100 in PBS, 7 min each were done then cells were blocked in Licor blocking buffer (Licor Biosciences, #927-40000) for 90 min at room temperature. Primary antibodies were added to Licor blocking buffer with 0.1% Tween-20 and incubated overnight at 4 °C.
Primary antibodies were used at the following dilutions: 1 : 50 dilution for phospho-AMPKα (Cell Signaling, #2535, rabbit monoclonal), AMPKα (Cell Signaling, #2603, rabbit monoclonal), and phospho-S6 ribosomal protein (Cell Signaling, #2211, rabbit monoclonal), 1 : 100 for S6 ribosomal protein (Cell Signaling, #2217, rabbit monoclonal), 1 : 250 for Ki67 (Transduction Labs, #610968 mouse), 1 : 200 for phospho-ERK1/2, (Cell Signaling, #4370, rabbit monoclonal), 1 : 100 for total ERK1/2 (Cell Signaling #4695, rabbit monoclonal), 1 : 100 for BiP (Cell Signaling, #3177 rabbit monoclonal), 1 : 50 for HSP70 (Cell Signaling, #4876 rabbit polyclonal), and 1 : 500 for γH2A.x (abcam #ab2893 rabbit polyclonal). Cells were then washed 3 times with PBS with 0.1% Tween-20 for at least 7 min each wash at room temperature. IR dye conjugated secondary antibody was then added to Licor blocking buffer with 0.1% Tween-20 at 1 : 200 dilution (Rockland Inc., #611-731-127, IRDye 800CW conjugated donkey anti rabbit) for 45 min at room temperature in the dark. Secondary antibody was washed out with two washes with PBS with 0.1% Tween-20, 7 min each wash, then cells were incubated with ToPro3 (Molecular Probes) at 1 : 500 dilution for 10 min in PBS at room temperature, in the dark. Cells were then washed twice with PBS and allowed to dry prior to scanning on an infrared laser scanner (Odyssey, Licor Biosciences).
Western blotting
Cells were seeded at the same density as used for the corresponding ICWs (approx. 90–100 K cm−2) in 6 well plates, the same positive and negative control conditions performed at 24 h before cells were lysed for Western blots. After positive and negative control treatments, cells were washed briefly with PBS on ice, then lysed in RIPA buffer with a protease inhibitor cocktail (Roche, Complete Mini tablets, #11836153001), and for phosphorylated proteins, a phosphatase inhibitor cocktail at 1 : 100 dilution (Pierce, Halt Phosphatase inhibitor cocktail, #78420). Cells were homogenized via sonication, then tris-glycine SDS sample buffer was added with 4% β-mercaptoethanol, and boiled for 5 min. Lysates were loaded onto either 12% or 8% tris-glycine gels depending on the molecular weight of the protein of interest (Invitrogen), and run in tris-glycine SDS running buffer (Invitrogen) with molecular weight markers suitable for infrared detection (Licor Biosciences, #928-40000). Protein was transferred to nitrocellulose membranes and subsequently blocked in Licor blocking buffer for at least 1 h at room temperature, in the dark. Primary antibodies to the proteins of interest were diluted into Licor blocking buffer with 0.1% Tween-20 (all at 1 : 1000, antibody specifications given above), along with a primary antibody to actin (either mouse monoclonal to α-actin from MP Biomedicals, #69100 at 1 : 10 000, or rabbit monoclonal to β-actin from Cell Signaling, #4970 at 1 : 1000).
Blots were incubated with the primary antibodies overnight at 4 °C in the dark. Membranes were then washed 3 times with PBS with 0.1% Tween-20 for 10–12 min each wash, then were incubated with secondary antibodies in Licor blocking buffer with 0.1% Tween-20 for 45 min at room temperature, in the dark, with shaking. Secondary antibodies were used at 1 : 20 000 and were from either Rockland Inc., (#611-731-127-IRDye 800CW conjugated donkey anti-rabbit, #610-131-121-IRDye 800CW conjugated goat anti-mouse, #611-130-122-IRDye 700DX conjugated goat anti-rabbit), or Licor Biosciences (#926-32220, IRDye 680 conjugated goat anti-mouse). Blots were then washed 3 times with PBS with 0.1% Tween-20 for 10–12 min each wash and allowed to dry prior to scanning. Blots were scanned using the Odyssey laser scanner, and integrated intensities of the bands of interest were normalized to the actin signal as a loading control.
Immunocytochemistry
For immunocytochemistry (ICC), cells were seeded into glass chamber slides at 90–100 K cm−2, and after 24 h positive and negative controls for each readout were performed. The cells were fixed and stained using the same protocol as described above for ICWs, with the exception of using a secondary antibody labeled with Alexa 488 (Molecular Probes, either goat anti-rabbit or anti-mouse), and the cells were mounted instead of dried. Cells were then imaged via microscopy. All images of paired positive and negative controls were taken with the same exposure length, intensity and objective to ensure a quantitative relationship between them. Additionally, controls without primary antibodies were imaged with the same exposure length to determine the levels of background due to non-specific staining of the secondary antibody. Any image processing (e.g. exporting in formats suitable for publication) was done exactly the same for these paired images as well, to maintain image consistency and validity.
Supplementary Material
Acknowledgements
Thanks to Dr Caroline Alexander and Young Chul Kim for providing the p16/INK4a KO mouse mammary fibroblasts. Funding sources: DJB, NIH grants R21CA122672 and K25CA104162, and ALP, DOD/BRCP W81XWH-06-1-0487.
Footnotes
Electronic supplementary information (ESI) available: Additional validation and background information, and supporting data.
References
- 1.Folch A, Toner M. Microengineering of Cellular Interactions. Annu. Rev. Biomed. Eng. 2000;2(1):227–256. doi: 10.1146/annurev.bioeng.2.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Masuda S, Washizu M, Nanba T. Novel method of cell fusion in field constriction area in fluid integrated circuit. IEEE Trans. Ind. Appl. 1989;25:732–737. [Google Scholar]
- 3.Sims CE, Allbritton NL. Analysis of single mammalian cells on-chip. Lab Chip. 2007;7(4):423–440. doi: 10.1039/b615235j. [DOI] [PubMed] [Google Scholar]
- 4.Keenan TM, Folch A. Biomolecular gradients in cell culture systems. Lab Chip. 2008;8(1):34–57. doi: 10.1039/b711887b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyvantsson I, Warrick JW, Hayes S, Skoien A, Beebe DJ. Automated cell culture in high density tubeless microfluidic device arrays. Lab Chip. 2008;8(5):717–724. doi: 10.1039/b715375a. [DOI] [PubMed] [Google Scholar]
- 6.Tan W, Desai TA. Microfluidic patterning of cells in extracellular matrix biopolymers: effects of channel size, cell type, and matrix composition on pattern integrity. Tissue Eng. 2003;9(2):255–267. doi: 10.1089/107632703764664729. [DOI] [PubMed] [Google Scholar]
- 7.Folch A, Toner M. Microengineering of cellular interactions. Annu. Rev. Biomed. Eng. 2000;2:227–256. doi: 10.1146/annurev.bioeng.2.1.227. [DOI] [PubMed] [Google Scholar]
- 8.Fisher RJ, Peattie RA. Controlling tissue microenvironments: biomimetics, transport phenomena, and reacting systems. Adv. Biochem. Eng. Biotechnol. 2007;103:1–73. doi: 10.1007/10_018. [DOI] [PubMed] [Google Scholar]
- 9.Kim L, Toh YC, Voldman J, Yu H. A practical guide to microfluidic perfusion culture of adherent mammalian cells. Lab Chip. 2007;7(6):681–694. doi: 10.1039/b704602b. [DOI] [PubMed] [Google Scholar]
- 10.Walker GM, Ozers MS, Beebe DJ. Insect cell culture in microfluidic channels. Biomed. Microdevices. 2002;4(3):161–166. [Google Scholar]
- 11.Yu H, Alexander CM, Beebe DJ. A plate reader-compatible microchannel array for cell biology assays. Lab Chip. 2007;7(3):388–391. doi: 10.1039/b612358a. [DOI] [PubMed] [Google Scholar]
- 12.Yu HM, Alexander CM, Beebe DJ. Understanding microchannel culture: parameters involved in soluble factor signaling. Lab Chip. 2007;7(6):726–730. doi: 10.1039/b618793e. [DOI] [PubMed] [Google Scholar]
- 13.Sawano A, Takayama S, Matsuda M, Miyawaki A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev. Cell. 2002;3(2):245–257. doi: 10.1016/s1534-5807(02)00224-1. [DOI] [PubMed] [Google Scholar]
- 14.Lucchetta EM, Munson MS, Ismagilov RF. Characterization of the local temperature in space and time around a developing Drosophila embryo in a microfluidic device. Lab Chip. 2006;6(2):185–190. doi: 10.1039/b516119c. [DOI] [PubMed] [Google Scholar]
- 15.Thery M, Racine V, Pepin A, Piel M, Chen Y, Sibarita JB, Bornens M. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 2005;7(10):947–953. doi: 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- 16.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276(5317):1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 17.Zigmond S. Ability of polymorphonuclear leukocytes to orient in gradients of chemotactic factors. J. Cell Biol. 1977;75(2 Pt 1):606–616. doi: 10.1083/jcb.75.2.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yap B, Kamm RD. Mechanical deformation of neutrophils into narrow channels induces pseudopod projection and changes in biomechanical properties. J. Appl. Physiol. 2005;98(5):1930–1939. doi: 10.1152/japplphysiol.01226.2004. [DOI] [PubMed] [Google Scholar]
- 19.Walker GM, Sai JQ, Richmond A, Stremler M, Chung CY, Wikswo JP. Effects of flow and diffusion on chemotaxis studies in a microfabricated gradient generator. Lab Chip. 2005;5(6):611–618. doi: 10.1039/b417245k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Zhong JF. Microfluidic devices for high-throughput gene expression profiling of single hESC-derived neural stem cells. Methods Mol. Biol. (Totowa, N. J.) 2008;438:293–303. doi: 10.1007/978-1-59745-133-8_22. [DOI] [PubMed] [Google Scholar]
- 21.El-Ali J, Sorger PK, Jensen KF. Cells on chips. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 22.Stangegaard M, Petronis S, Jorgensen AM, Christensen CBV, Dufva M. A biocompatible micro cell culture chamber (mu CCC) for the culturing and on-line monitoring of eukaryote cells. Lab Chip. 2006;6(8):1045–1051. doi: 10.1039/b603379b. [DOI] [PubMed] [Google Scholar]
- 23.Stangegaard M, Wang Z, Kutter J, Dufva M, Wolff A. Whole genome expression profiling using DNA microarray for determining biocompatibility of polymeric surfaces. Mol. Biosyst. 2006;2:421–428. doi: 10.1039/b608239d. [DOI] [PubMed] [Google Scholar]
- 24.Yu HM, Meyvantsson I, Shkel IA, Beebe DJ. Diffusion dependent cell behavior in microenvironments. Lab Chip. 2005;5(10):1089–1095. doi: 10.1039/b504403k. [DOI] [PubMed] [Google Scholar]
- 25.Raty S, Walters EM, Davis J, Zeringue H, Beebe DJ, Rodriguez-Zas SL, Wheeler MB. Embryonic development in the mouse is enhanced via microchannel culture. Lab Chip. 2004;4(3):186–190. doi: 10.1039/b316437c. [DOI] [PubMed] [Google Scholar]
- 26.Paguirigan A, Puccinelli J, Su X, Beebe DJ. unpublished data.
- 27.Berthier E, Warrick J, Beebe DJ. Managing evaporation for more robust microscale assays. Part 2: Characterization of convection and diffusion for cell biology. Lab Chip. 2008;8(6):860–864. doi: 10.1039/b717423c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berthier E, Warrick J, Yu H, Beebe DJ. Managing evaporation for more robust microscale assays. Part 1: Volume loss in droplet based assays. Lab Chip. 2008;8(6):852–859. doi: 10.1039/b717422e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heo YS, Cabrera LM, Song JW, Futai N, Tung YC, Smith GD, Takayama S. Characterization and resolution of evaporation-mediated osmolality shifts that constrain microfluidic cell culture in poly(dimethylsiloxane) devices. Anal. Chem. 2007;79(3):1126–34. doi: 10.1021/ac061990v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toepke MW, Beebe DJ. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip. 2006;6(12):1484–1486. doi: 10.1039/b612140c. [DOI] [PubMed] [Google Scholar]
- 31.Paguirigan A, Puccinelli J, Su X, Beebe DJ. unpublished data.
- 32.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144(12):5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 33.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007;100(3):328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 34.Averous J, Proud CG. When translation meets transformation: the mTOR story. Oncogene. 2006;25(48):6423–6435. doi: 10.1038/sj.onc.1209887. [DOI] [PubMed] [Google Scholar]
- 35.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001;3(1):335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 36.Berger A, Bardeesy N. Modeling INK4/ARF tumor suppression in the mouse. Curr. Mol. Med. 2007;7(1):63–75. doi: 10.2174/156652407779940477. [DOI] [PubMed] [Google Scholar]
- 37.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 38.Shi Y, Gaestel M. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol. Chem. 2002;383(10):1519–1536. doi: 10.1515/BC.2002.173. [DOI] [PubMed] [Google Scholar]
- 39.Strniskova M, Barancik M, Ravingerova T. Mitogen-activated protein kinases and their role in regulation of cellular processes. Gen. Physiol. Biophys. 2002;21(3):231–55. [PubMed] [Google Scholar]
- 40.Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation) Biochem. J. 2007;404(3):353–363. doi: 10.1042/BJ20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hohfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2(10):885–90. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35(4):373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 43.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 2001;26(8):504. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med. 2006;6(1):45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka T, Kajstura M, Halicka HD, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation are strongly amplified during mitogenic stimulation of lymphocytes. Cell Proliferation. 2007;40(1):1–13. doi: 10.1111/j.1365-2184.2007.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.