

Abstract
The published papers on the effects of increased cardiac expression of adenylyl cyclase type 6 (AC6) are reviewed. These include the effects of AC on normal and failing left ventricle in several pathophysiological models in mice and pigs. In addition, the effects of increased expression of AC6 in cultured neonatal and adult rat cardiac myocytes are discussed in the context of attempting to establish mechanisms for the unanticipated beneficial effects of AC6 on the failing heart.
Keywords: Gene Therapy, Congestive Heart Failure, Adenovirus Vector, Animal Models, Nitroprusside, Cyclic AMP, Adrenergic Signaling
1. Introduction
Our purpose is to review a very specific topic: mechanisms for the favorable effects of increased cardiac expression of adenylyl cyclase type 6 (AC6) on normal and failing hearts. Unlike most reviews, where a given topic is studied by many groups of scientists, AC6 and its effects on cardiac function have, for the most part, been published by the laboratory of the authors. Citing so many of our papers was therefore unavoidable. The effect of AC in other cells and organs has been a focus of several recent reviews and original articles. For example: increased AC6 expression in cardiac fibroblasts and other cells [1-5], regulatory properties of cardiac AC6 and AC5 (the other major AC isoform expressed in cardiac myocytes) [6-8], AC5 in the heart [9-15], structure-function relationships of various AC isoforms [16-24], mechanisms for Ca2+-inhibition and stimulation of AC isoforms [25-29], and the role of AC isoforms in the brain, with a focus on memory [30-32].
2. AC Structure and Activity
Adenylyl cyclase (AC) is a transmembrane protein in cardiac myocytes and other cells and is the effector molecule for the β-adrenergic receptor (βAR) and other G-protein coupled receptors. AC regulates the conversion of adenosine triphosphate (ATP) to 3′,5′-cyclic adenosine monophosphate (cAMP), thereby, through protein kinase A (PKA), initiating a variety of intracellular signaling cascades that influence heart function. AC isoforms possess the general structure shown in Figure 1: two transmembrane regions (M1 and M2) linking a large cytosolic loop (C1) and a second cytosolic loop (C2) following the M2 region. C1 & C2 comprise the catalytic core, a primary site for regulation of AC activity [33-39]. AC activity is influenced by Gαs, Gαi, Mg2+ and ATP, and also is affected by glycosylation and phosphorylation by protein kinase A (PKA) and protein kinase C (PKC) [16-24, 38-43].
Figure 1.
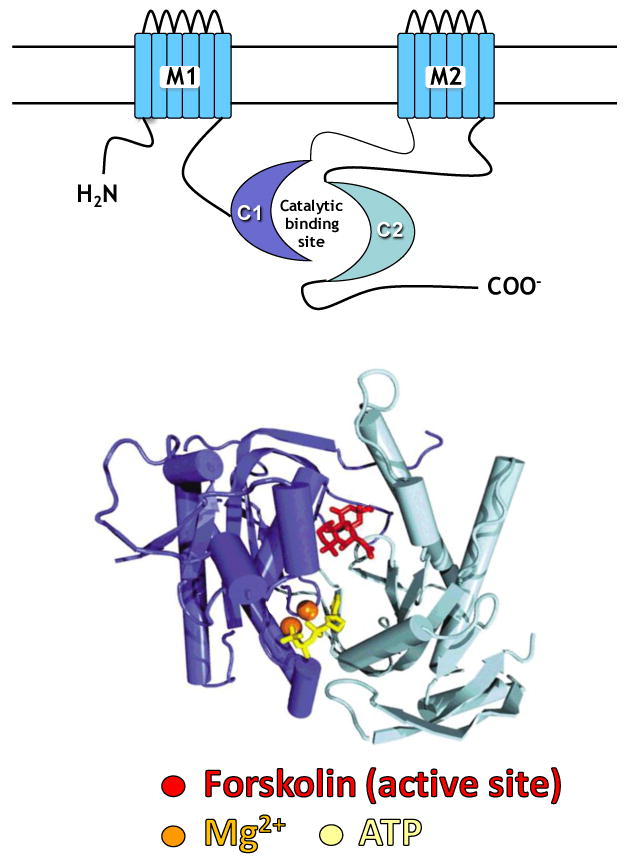
The diagram (top) shows the features of adenylyl cyclase, including the two transmembrane regions (M1 and M2), which anchor the protein to the cell membrane, and the cytoplasmic regions (C1 and C2), which form the catalytic core. Below is a representation of the C1 and C2 domains, derived from X-ray crystallography [16], displaying the structural relationships of the catalytic core. Darker blue represents the C1 region, lighter blue, the C2 region. At the interface between C1 and C2 reside binding sites for forskolinMg2+ and ATP [16].
Dynamics of the C1C2 interface is a pivotal determinant of AC activity – Gαs binds to the C2 domain and increases the affinity of C2 with C1, with consequent catalysis and generation of cAMP. In contrast, Gαi binds to the C1 domain, reduces the affinity of C1 for C2 and reduces AC activity. Forskolin alters the conformation of the C1C2 active site, thereby enabling vigorous enzyme activation [38-40]. Gilman's laboratory showed that a C1C2 fusion protein (a soluble fragment of the parent AC molecule absent its transmembrane regions) retains catalytic activity – cAMP is generated with forskolin or Gαs stimulation. However, it was unresponsive to βAR stimulation, due to its lack of association with the plasma membrane, which made ßAR coupling impossible [44-46].
X-ray crystallography of AC C1C2 fusion proteins revealed binding sites for forskolin, Gαs, Mg2+ and ATP [16]. Forskolin contacting residues are located on both C1 and C2 domains in the cleft of the C1C2 fusion protein (Figure 1). The Gαs binding site is adjacent to that of forskolin, which is the basis for their synergistic effects on catalytic activity. The specific ATP binding site is also at the interface of the C1 and C2, and residues in both C1 and C2 domains bridge the Mg2+ binding pocket. Mutagenesis studies confirm the importance of these residues in the catalytic core for AC activity [47,48]. These data have guided our generation of AC6 mutants that lack catalytic activity, but are similar otherwise to normal AC6.
3. AC6 Gene Transfer for Clinical CHF
Recent studies, which will be reviewed, indicate that increased cardiac AC type 6 (AC6), a dominant AC isoform expressed in mammalian cardiac myocytes [49] has protean beneficial effects on the left ventricle (LV) (Table). These include: 1) increased survival in mice with cardiomyopathy [50]; 2) increased survival in acute myocardial infarction (MI) [51]; 3) reduced action potential duration [52] and facilitation of atrio-ventricular (AV) conduction [53] associated with reduction of AV block [51]; 4) reductions in both LV dilation and pathological hypertrophy [54, 55]; 5) beneficial effects on Ca2+ handling through improved SERCA2a activity [56], and reduced phospholamban activity [56]; and 6) increased cardiac troponin I phosphorylation [57]. Based on these results and additional safety studies, it appeared that cardiac gene transfer of AC6 might be a rational potential therapy for clinical congestive heart failure (CHF).
AC6 and Cardiac Function.
| Model | Findings | Ref | |
|---|---|---|---|
| TG Mice | AC6 : Cardiac-Directed Expression | Basal HR, cAMP, LV dP/dt unaffected; increased recruitable LV dP/dt, no adverse effects at 24 months | 59 |
| AC6 × Gq (CHF) | AC6 expressed in Gq-associated cardiomyopathy: increased LV function & survival, decreased LVH | 50, 54 | |
| AC6 TG × Gq TG (CHF) | AC6 expressed in Gq-associated cardiomyopathy: increased LV SERCA2a activity and PLN phosphorylation | 56 | |
| AC6 : Regulated and Cardiac-Directed | Increased LV function in normal adult mice upon activation of LV AC6 expression | 69 | |
| AC6 : Regulated and Cardiac-Directed (CHF) | Activation of LV AC6 expression increased function of failing heart | 57 | |
| AC6 & Acute MI | Mortality >50% reduced in mice with increased cardiac AC6 content - similar to propranolol treatment | 51 | |
| AC6 × Gq (CHF) | AC6 expressed in Gq-associated cardiomyopathy: corrects prolonged action potential duration | 52 | |
| AC6 : Cardiac-Directed Expression | AC6 expression is associated with facilitation of AV conduction in normal mice | 53 | |
| AC6 : Cardiac-Directed Expression | AC6 expression does not affect mean HR, HR variability, vagal responsiveness, or activity levels (telemetry) | 97 | |
| AC6 Deletion | Adverse effects on LV Ca2+ signaling; reduced LV function | 93 | |
| Ad. AC6 Gene Transfer | Neonatal Rat Cardiac Myocytes | AC6 amount determines cardiac myocyte response to βAR stimulation | 58 |
| Neonatal Rat Cardiac Myocytes | AC6 increases ATF3 expression with consequent reduced PLN expression (cAMP independent) | 70 | |
| Neonatal Rat Cardiac Myocytes | AC6 activates Akt with consequent PLN phosphorylation (cAMP independent) | 71 | |
| Neonatal Rat Cardiac Myocytes | AC6 associated with dynamic regulation of Akt via PHLPP:AC6 interaction (cAMP-independent) | 72 | |
| Neonatal Rat Cardiac Myocytes | Ad. AC6 Ad. vs β1AR: differences in intracellular distribution of transgene contribute to differences in outcome | 81 | |
| Adult Rat Cardiac Myocytes | AC6 & AC6 mutant (catalytically inactive) have similar benefits: mechanism is cAMP-independent | 83 | |
| CHF, Mouse | AC6 gene transfer (indirect intracoronary) in Gq-associated cardiomyopathy: increased function of failing LV | 98 | |
| Normal Mouse | AC6 gene transfer (indirect intracoronary) increases LV function in response to βAR stimulation | 99 | |
| Normal Pig | AC6 gene transfer (intracoronary + histamine) increases recruitable LV dP/dt | 64 | |
| CHF, Pig | AC6 gene transfer (intracoronary + nitroprusside) increases function of failing LV | 55,68 | |
| Normal Pig | IND App: AC6 gene transfer (intracoronary + nitroprusside) not toxic; LV AC6 expression persists 10 weeks | * | |
| CHF, Human | Intracoronary Ad. hAC6 in patients with severe CHF (Phase 1 /Phase 2 randomized double-blind clinical trial) | * |
see ClinicalTrials.gov #NCT00787059, enrollment for this clinical trial was initiated in May 2010
A clinical trial of cardiac AC6 gene transfer in patients with CHF began enrollment in May 2010 (ClinicalTrials.gov, NCT00787059). This trial is a randomized, double blinded, placebo controlled study to evaluate the safety and clinical effectiveness of ascending doses of human adenovirus-5 (E1/E3-deleted, replication incompetent) encoding human AC6 (Ad5.hAC6) in patients with stable but severe CHF. The vector will be delivered by intracoronary injection simultaneously with intracoronary nitroprusside, used to increase gene transfer efficiency. Seventy-two patients will be enrolled in a 3:1 randomization ratio, so that 54 patients will receive increasing doses of Ad5.hAC6, and 18 will receive placebo.
This review will focus on studies of the effects of increased expression of AC6 in cultured cardiac myocytes, and in a variety of animal models of heart failure. In reviewing these studies we will adress potential mechanisms, and also adress the paradoxical finding that an agent closely linked with cAMP production has favorable effects, when other agents that promote cAMP production have deliterious consequences.
4. Preclinical Data
AC Content and cAMP Generation
Using recombinant adenovirus to increase AC6 expression in neonatal cardiac myocytes, it was found that cells with increased AC6 responded to agonist stimulation with marked increases in cAMP production in proportion to protein expressed: AC protein expression was amplified six-fold and ßAR-stimulated cAMP production was increased seven-fold (vs Ad5. lacZ control) [58]. Basal cAMP was unchanged by AC6 gene transfer. No changes in ßAR number, or in the expression of Gαs or Gαi2 were found. In other experiments using a different construct that produced a 2-fold increase in AC6 protein, isoproterenol and forskolin-stimulated cAMP generation were increased 2-fold, showing that the maximal cAMP generation is proportional to the amount of AC6 in the cell [58]. These data indicated that βAR responsiveness can be influenced by increasing the effector (AC) without changing ßAR number.
Increased Cardiac AC Expression
Transgenic mice with cardiac-directed AC6 expression have normal LV size and basal function. However, when stimulated through the ßAR, cardiac function is increased through a wide range of isoproterenol-doses (p<0.0001) and cardiac myocytes show a 2.6 fold increase in cAMP production (p<0.009). In contrast, basal cAMP and cardiac function are not changed, and long-term AC6 expression is not associated with abnormal histological findings or deleterious changes in cardiac function [59]. Increased cardiac AC6 content appears not to alter transmembrane signaling except when receptors are activated, in contrast to increased expression of cardiac ßAR or Gαs, which yields continuous activation and detrimental consequences [60-62].
Cardiomyopathy Treated by AC6
Cardiac-directed expression of Gαq is associated with LV dilation, reduced heart function, and impaired cAMP production, mimicking important aspects of clinical CHF [63]. Transgenic mice with cardiac-directed expression of AC6 were crossbred with mice with Gαq cardiomyopathy. Cardiac-directed expression of AC6 in this cardiomyopathic background increased basal LV function, and dobutamine-stimulated LV function was increased up to 40% (p<0.0005). Finally, AC6 expression prevented Gαq-associated myocardial hypertrophy and resulted in markedly increased survival (p<0.0001) [54]. In subsequent patch-clamp studies performed on isolated cardiac myocytes, Gαq expression was associated with prolonged action potential duration (APD), an effect that was abrogated by co-expression of AC6 [52]. Prolonged APD is observed in patients with CHF. If these data were extrapolated to clinical settings, one would anticipate that AC6 gene transfer may shorten APD, and thereby decrease the likelihood of automaticity and reentrant pathways leading to ventricular tachycardia and ventricular fibrillation.
Intracoronary Ad5.AC6 and Nitroprusside
Intracoronary delivery of Ad5.AC6 increases the contractile responsiveness of the heart in normal pigs [64], with no increase in arrhythmias or mean heart rate. Intracoronary delivery was preceded by intracoronary infusion of histamine in these studies, which increases gene transfer efficiency [64]. However, histamine is not an approved product for human use so other agents that might increase transvascular transport of adenovirus were explored. Nitroprusside was selected because it had been used safely, by intracoronary infusion, in patients with heart disease [65-67]. Experiments were conducted using intracoronary delivery of adenovirus encoding lacZ or AC6 in pigs with and without simultaneous infusion of nitroprusside. Nitroprusside was associated with up to a 4-fold increase in the extent of cardiac gene transfer [68].
AC6 Gene Transfer in Heart Failure
To test whether intracoronary AC6 gene transfer could improve function of the failing heart, the pacing model of CHF was used in pigs [55]. Contractile function (LV dP/dt) was measured in conscious pigs before and after twenty-one days of continuous LV pacing – used to induce severe dilated heart failure. On day seven, when substantial CHF was present, pigs received intracoronary Ad5.AC6 (1. 4 × 1012 vp + nitroprusside) or intracoronary saline (PBS). Saline-treated animals showed progressively worsening CHF associated with marked reduction in LV contractility. The fall in LV dP/dt was up to 50% less in pigs that had received AC6 gene transfer (p=0.0014). Serial echocardiography showed that Ad5.AC6 treatment was associated with reductions in LV end-diastolic (p<0.043) and end-systolic (p<0.009) diameters. AC-stimulated cAMP production was increased 1.7-fold (p=0.006) in LV samples from Ad5.AC6 treated pigs and LV gene transfer was confirmed by PCR. These data indicated that AC6 gene transfer increases function of the failing heart.
Cardiac AC6 and Acute Myocardial Infarction
To determine the consequences of increased AC6 in the setting of myocardial ischemia transgenic mice with cardiac-directed expression of AC6 [59], and transgene negative siblings underwent proximal left coronary artery occlusion, yielding large transmural infarction of the LV free wall. There was a 2-fold survival advantage of mice with cardiac-directed AC6 expression (p=0.004) [51]. Infarct size and response to global ischemia showed no group differences. Previously implanted telemetry devices allowed continuous recording of electrocardiograms after infarction, revealing that bradycardia and progressive atrio-ventricular block consistently was the fatal arrhythmia, which was less prevalent in AC6 mice. Electrophysiological measurements confirmed that AV node conduction is facilitated (up to a 30% reduction in AV-interval) in mice with increased cardiac AC6, through a wide range of heart rates (p=0.01) [53].
Activation of AC6 Expression in Severe CHF
Mice with cardiac-directed and regulated (tet-off) expression of AC6 [69] underwent left coronary artery ligation to induce CHF. Activation of cardiac AC6 expression – in the presence of severe heart failure – was associated with increases in LV ejection fraction (EF), LV +dP/dt, cardiac output, and slope of the end-systolic pressure-volume relationship. End-diastolic pressure, Tau and LV −dP/dt all decreased, documenting improved diastolic function, indicating marked increases in both systolic and diastolic function of the failing heart conferred by activation of cardiac AC6 expression (Figure 2) [57].
Figure 2.
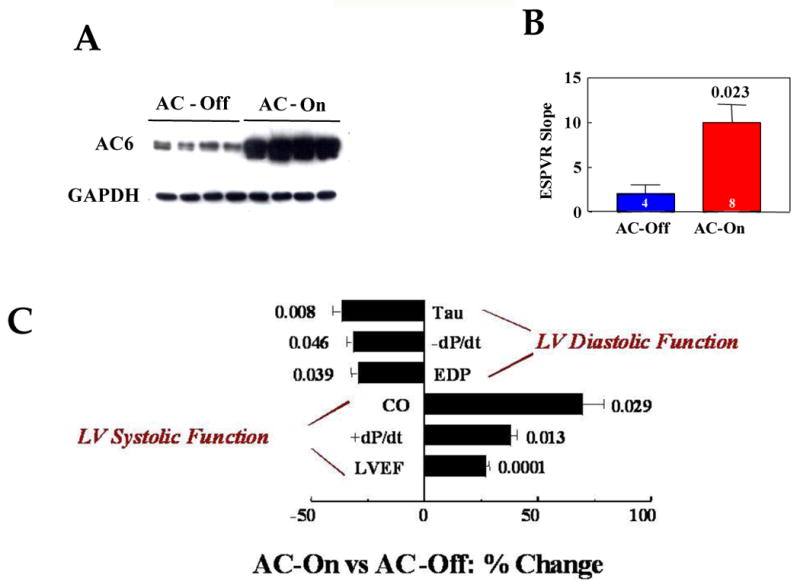
A. LV AC6 expression. The Western blot shows marked increase (p<0.0001) in AC6 protein in LV samples from mice 5 weeks after activation of AC6 transgene expression (n=8 for both groups).
B. Effect of AC6 Transgene Expression on LV Contractility. The end-systolic pressure-volume relationship (ESPVR) was measured using conductance catheters and high fidelity pressure transducers in intact anesthetized mice 5 weeks after activation of AC6 expression in animals with severe heart failure. Increased cardiac AC6 expression was associated with substantial increases in LV contractility as reflected in increased slope of the ESPVR. The graph summarizes data from all animals (AC-Off, Blue bar; AC-On, Red bar); bars represent mean values, error bars denote 1 SEM. Number above bar denotes p value comparing the two group means by Student's t-test (two tails); number in each bar denotes number of animals studied in each group.
C. Effect of AC6 Transgene Expression on LV Function. Measures of LV diastolic and systolic function were made 5 weeks after activation of AC6 expression in mice with infarct-induced CHF. The percent change in each measurement between groups is shown. Activation of cardiac AC6 expression was associated with substantial increases in measures of both systolic and diastolic function. Numbers adjacent to bars denote p values from Student's t-test (two-tails). AC-On (n=12), AC-Off (n=9). Tau, time constant of relaxation; -dP/dt, maximal rate of decline in LV pressure; EDP, end-diastolic pressure; CO, cardiac output; +dP/dt, maximal rate of rise in LV pressure; LVEF, left ventricular ejection fraction.
AC6 Paradox
The beneficial effects of AC6, so consistent in a variety of species and pathophysiological models, must be reconciled with the dire consequences on the heart of ßAR stimulation and elevations in intracellular cAMP. These unexpected beneficial effects of increased AC6 expression have been referred to as the “AC6 Paradox.” Why does AC6 have beneficial rather than deleterious effects on the failing heart expected with agents that increase intracellular cAMP? Logic would dictate that either: a) cAMP is not bad for the heart after all – that it is something else which leads to poor outcomes when cAMP levels are increased in the failing heart; or b) increased AC6 has beneficial cardiac effects independent of cAMP, which outweigh its expected deleterious effects. Using pharmacological inhibitors and other approaches, data suggest that many of the beneficial effects of increased cardiac AC6 expression do not require increased cAMP generation [70-72]. This may reflect differences in intracellular distribution, a possibility that will be discussed in the next section. We have excluded the possibility that AC6 activates cAMP-EPAC signaling [71]. As a prelude to what will be later discussed in more detail, it appears that many of the beneficial effects of AC6 occur independently of cAMP generation.
5. Mechanism for Beneficial Effects of AC6 Expression
Intracellular Distribution of AC6
Adenylyl cyclase is predominantly found in the cell membrane. However, using high-resolution electron microscopy after immunohistochemical staining, endogenous AC was also detected in the sarcoplasmic reticulum (SR), nuclear envelope, and perinuclear region in cardiac myocytes [73]. In nerve cells, endogenous AC is detected in endoplasmic reticulum and within the cytoplasm of terminal endings of nerve fibers [74]. Studies using cardiac myocyte homogenates followed by sucrose gradient centrifugation showed that AC6 is associated with caveolin proteins in lipid raft fractions [1,4,75-77] that represent caveolae, a specialized lipid raft that forms flask-shaped invaginations of the plasma membrane, involved in lipid storage, endocytosis and compartmentalization of signaling molecules [78-80]. Although the biological function of intracellular AC is unknown, organelle-targeted expression of soluble AC (a fusion protein of C1 from AC1 and C2 from AC2) suggests that AC compartmentation enables signaling specificity [46].
AC6 gene transfer increases the amount of AC6 in a variety of intracellular compartments. For example, after gene transfer, transgene was detected in the same caveolin fraction as endogenous AC6 in cardiac myocytes [1,4]. Using transgene-specific antibody and immunofluorescence staining indicated that transgene AC6 (recognized by an AU tag) was associated caveolin 3, a major component of caveolae [81]. Transgene AC6 was also detected in sarcoplasmic reticulum (SR), mitochondria and nuclear envelope (Figure 2). Although AC6 gene transfer is associated with wide intracellular distribution, ß1AR gene transfer is not. High level β1AR expression in cardiac myocytes results in transgene expression limited to the plasma membrane [81]. The mechanism explaining the disparity of distribution of β1AR vs AC6 is not known, but likely reflects differences in structure. Using AC5 and AC6, Thangavel and colleagues showed that the C1 and C2 domains of AC, but not the N-terminus, are responsible for caveolae localization [82].
AC6 Signaling
The broad intracellular distribution of AC6 seen after gene transfer provides an opportunity for AC6 to interact with previously inaccessible intracellular proteins and thereby influence signaling in unique ways. Gene transfer of AC6 into cultured cardiac myocytes was associated with: 1) increased ATF3 expression, which suppresses the phospholamban (PLB) promoter and reduces PLB transcription [70]; 2) increased phosphorylation of PLB [71]; 3) increased PI3K-Akt activation [71]; 4) increased expression of Bcl-2 protein [81]; 5) reduced cardiac ankyrin repeat protein (CARP) expression [83]; and 6) reduced phenylephrine-induced cardiac myocyte hypertrophy [83]. These effects of AC6 occurred in the absence of isoproterenol or forskolin stimulation, and in the presence of PKA inhibition, suggesting that these events were cAMP independent.
AC6 Interacting Proteins
Protein-protein interaction is required for regulation of AC activity, Gαs and Gαi being well known examples [40-45]. Additional proteins that influence AC activity include the regulator of G protein signaling (RGS2) [84-86], the protein associated with Myc (PAM) [87,88], A-kinase-anchoring protein (AKAP79) [89], Ric 8a [90], and snapin [91], to name a few. Interaction of AC6 with intracellular proteins is also important for directing AC localization and AC-associated signaling independent of receptor, G-proteins or cAMP. For example, snapin links the N-terminus of AC6 [91,92] with snapin-binding proteins. More than twenty snapin-binding proteins have been identified, each with specific intracellular locations and function [92]. Intracellular AC6, the result of increased AC6 expression, would promote its interaction with snapin-binding proteins, which may influence AC6 compartmentation, thereby leading to additional protein interactions in which AC6 might influence signaling pathways – independent of cAMP generation.
The C1 domain of AC6 is required for its interaction with PH-domain leucine-rich protein phosphatase 2 (PHLPP2), a phosphatase that acts on Akt and PKC [72]. Increased expression of AC6 was associated with increased Akt phosphorylation and activity in cardiac myocytes. However, AC6-associated Akt phosphorylation was rapidly dephosphorylated upon agonist stimulation. The mechanism appears to involve close association of transgene AC6 and PHLPP2, which may inhibit PHLPP2 activity, resulted in increased Akt phosphorylation at Ser473. Through unknown mechanisms, agonist stimulation disturbs the AC6-PHLPP2 interaction, enabling rapid and reversible Akt dephosphorylation at Ser473. Activation of PHLPP2 was cAMP and PKA-independent but required an intact catalytic domain of AC6 for the conformational change of AC6 during agonist stimulation. A single amino acid replacement that renders AC6 catalytically inactive (cannot generate cAMP) also works through AC6: PHLPP2 [72] interaction indicating that AC6-associated Akt activation was not dependent upon cAMP generation.
Do the intracellular signaling events associated with increased expression of AC6 also occur in the setting of endogenous levels of AC6? As alluded to earlier, endogenous AC6 can be detected in sarcoplasmic reticulum, nuclear envelope, and perinuclear region in cardiac myocytes [73]. Does native AC6, which is expressed at low levels, play an important role in intracellular signaling, or does this require high levels of AC6 expression? Native AC6 forms complexes with AKAPs in cardiac myocytes and other cells [24]. One source of evidence that endogenous intracellular AC6 : AKAP complexes have an important physiological role stems from the similarities in phenotype between deletion of AC6 [93] and deletion of the PKA binding site of AKAPs. For example, in heart, AKAP150Δ36 (PKA binding site-deleted) induces abnormalities in Ca2+ signaling [24] that are similar to those associated with AC6 deletion [93].
A Catalytically Inactive AC6
As previously stated, AC6 evokes many intracellular events in the absence of stimulation with isoproterenol or forskolin, suggesting that these events are cAMP independent. Pharmacological inhibitors of PKA do not block the beneficial effects of AC6 gene transfer. However, to show rigorously that these effects truly are cAMP independent requires experiments using a catalytically inactive AC6 molecule.
To achieve this goal, an AC6 mutant molecule was generated (AC6mut) by replacing aspartic acid (426) with alanine in the Mg2+ binding pocket [16] in the C1 domain of AC6 (Figure 1). Gene transfer of AC6mut in cardiac myocytes (both neonatal and adult rat) resulted in similar expression levels and intracellular distribution compared to normal (catalytically active) AC6, but showed marked impairment of cAMP generation in response to stimulation of AC (forskolin) or βAR (isoproterenol) [72,83]. Despite marked reduction in cAMP generation, AC6mut influenced intracellular signaling events similarly to what was observed following expression of catalytically intact normal AC6 [72,83]. For example, both AC6 and AC6mut increased Akt phosphorylation and activity in neonatal rat cardiac myocytes [72], reduced phenylephrine (PE)-induced cardiac myocyte hypertrophy, cell death, and expression of CARP [83]. Both AC6 and AC6mut increased ATF3 expression and reduced phospholamban expression [70,83]. In the presence of βAR activation (isoproterenol), AC6 expression was associated with increased cytoplasmic [Ca2+], results that were replicated by AC6mut gene transfer. These data confirmed that these effects of AC6 do not require cAMP.
Cardiac-directed AC5 expression
The two ACs most abundantly expressed in cardiac myocytes are types 5 (AC5) and 6 (AC6), which have 65% amino acid homology. It has been speculated that coexpression of these AC types in cardiac myocytes represents redundancy, but the specific role of AC6 in cardiac physiology and its differences from AC5 have begun to be appreciated, largely through experiments using cardiac-directed expression and targeted deletion. Unlike the case with cardiac-directed AC6 [54-57], transgenic lines with cardiac-directed expression of AC5 do not show major changes in cardiac function [94,95]. In the setting of Gαq cardiomyopathy, cardiac-directed AC5 expression, unlike AC6 expression, does not reduce hypertrophy or fetal gene expression [94]. Finally, the marked differences on LV function observed with AC5 vs AC6 deletion confirm that AC5 and AC6 have different biological roles [11-14,93].
6. Conclusion
The surprising beneficial effects of increased AC6 expression on cardiac function may reflect the effects of intracellular transgene AC6 and its interactions with key signaling molecules, kinases, phosphatases, and transcription factors. Many of these altered pathways have favorable effects on cardiac function, through abrogation of hypertrophy, increased cell survival, and improved calcium handling – effects that appear to be cAMP-independent. Whether these mechanisms are relevant or attainable in failing human myocardium remains to be seen.
There are many candidate genes proposed to treat patients with clinical heart failure. For example, SERCA2a which has recently been tested in clinical trials [94], and S100A1, a calcium regulating protein that is being developed for the treatment of clinical heart failure [95]. There is no shortage of transgenes that have favorable effects in cultured cardiac myocytes, transgenic mice, and other animal models [96]. However, the key impediment to successful cardiac gene therapy is obtaining sufficient expression in the heart to have an effect. Vectors and vector delivery methods have not advanced sufficiently, in many cases, to allow a real test of whether a candidate gene fails because it is inefficacious, or simply because of inadequate expression. Even so, gene transfer has engendered increasing interest in recent years, and holds great promise for the treatment of the failing heart. To modify the words of Tulkington in Dickens' Bleak House: “The wheels of [science] grind slow, but they grind exceedingly fine.”
Figure 3. Location of AC6 and ß1AR transgene proteins.
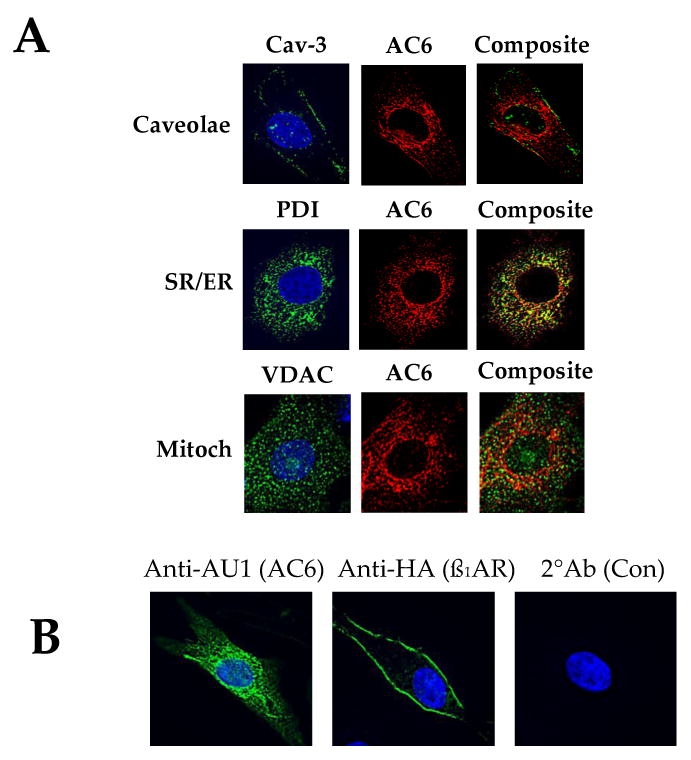
A. Double immunofluorescence staining of AC6 transgene by anti-AU1 antibody (red), anti-caveolin 3 (Cav-3) antibody (green, for caveolae); anti-voltage dependent anion selective channel protein (VDAC) antibody (green, for mitochondria); and with anti-protein disulphide-isomerase (PDI) antibody (green, for sarcoplasmic reticulum). AC6 transgene was detected in caveolae, mitochondria and SR (40×).
B. Immunofluorescence staining and deconvolution analysis of cardiac myocytes after gene transfer of Ad.AC6 and Ad.ß1AR. Uninfected cardiac myocytes served as a control (Con). Anti-AU1 antibody was used to detect AC6 transgene (green, left panel), anti-HA for ß1AR transgene (green, middle panel), and Hoechst dye to identify the nucleus (blue). AC6 transgene was evenly distributed in the plasma membrane and cytoplasm. In contrast, ß1AR transgene was limited primarily to the plasma membrane (40×).
Acknowledgments
Supported by grants from the NIH (5P01HL066941, HL081741, HL088426–01), the Department of Veterans Affairs (Merit grant) and the American Heart Association (Beginning Grant-In-Aid Awards 0765064Y and 0865147F).
Footnotes
Disclosures: Dr Hammond is founder of and consultant to Renova Therapeutics, Incorporated.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostrom RS, Violin JD, Coleman S, Insel PA. Selective enhancement of beta-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes. Mol Pharmacol. 2000;57:1075–9. [PubMed] [Google Scholar]
- 2.Ostrom RS, Naugle JE, Hase M, Gregorian C, Swaney JS, Insel PA, Brunton LL, Meszaros JG. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk redulates collagen production in cardiac fibroblasts. J Biol Chem. 2003;278:24461–8. doi: 10.1074/jbc.M212659200. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol. 2004;286:C1089–99. doi: 10.1152/ajpcell.00461.2003. [DOI] [PubMed] [Google Scholar]
- 4.Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem. 2006;281:26391–9. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- 5.Swaney JS, Patel HH, Yokoyama U, Lai NC, Spellman M, Insel PA, Roth DM. Adenylyl cyclase activity and function are decreased in rat cardiac fibroblasts after myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H3216–20. doi: 10.1152/ajpheart.00739.2007. [DOI] [PubMed] [Google Scholar]
- 6.Beazely MA, Alan JK, Watts VJ. Protein kinase C and epidermal growth factor stimulation of Raf1 potentiates adenylyl cyclase type 6 activation in intact cells. Mol Pharmacol. 2005;67:250–9. doi: 10.1124/mol.104.001370. [DOI] [PubMed] [Google Scholar]
- 7.Beazely MA, Watts VJ. Galphaq-coupled receptor signaling enhances adenylate cyclase type 6 activation. Biochem Pharmacol. 2005;70:113–20. doi: 10.1016/j.bcp.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur J Pharmacol. 2006;535:1–12. doi: 10.1016/j.ejphar.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa Y, Sorota S, Kiuchi K, Shannon RP, Komamura K, Katsushika S, Vatner DE, Vatner SF, Homcy CJ. Downregulation of adenylylcyclase types V and VI mRNA levels in pacing-induced heart failure in dogs. J Clin Invest. 1994;93:2224–9. doi: 10.1172/JCI117219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okumura S, Kawabe J, Yatani A, Takagi G, Lee MC, Hong C, Liu J, Takagi I, Sadoshima J, Vatner DE, Vatner SF, Ishikawa Y. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res. 2003;93:364–71. doi: 10.1161/01.RES.0000086986.35568.63. [DOI] [PubMed] [Google Scholar]
- 11.Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A. 2003;100:9986–90. doi: 10.1073/pnas.1733772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang T, Lai NC, Roth DM, Drumm J, Guo T, Lee KW, Han PL, Dalton N, Gao MH. Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to beta-adrenergic stimulation. Basic Res Cardiol. 2006;101:117–26. doi: 10.1007/s00395-005-0559-y. [DOI] [PubMed] [Google Scholar]
- 13.Okumura S, Vatner DE, Kurotani R, Bai Y, Gao S, Yuan Z, Iwatsubo K, Ulucan C, Kawabe J, Ghosh K, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation. 2007;116:1776–83. doi: 10.1161/CIRCULATIONAHA.107.698662. [DOI] [PubMed] [Google Scholar]
- 14.Vatner SF, Yan L, Ishikawa Y, Vatner DE, Sadoshima J. Adenylyl cyclase type 5 disruption prolongs longevity and protects the heart against stress. Circ J. 2009;73:195–200. doi: 10.1253/circj.cj-08-0957. [DOI] [PubMed] [Google Scholar]
- 15.Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, Depre C, Iwatsubo K, Ishikawa Y, Sadoshima J, Vatner SF, Vatner DE. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol. 2009;297:H1776–82. doi: 10.1152/ajpheart.00050.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha. GTPgammaS Science. 1997;278:1907–16. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 17.Whisnant RE, Gilman AG, Dessauer CW. Interaction of the two cytosolic domains of mammalian adenylyl cyclase. Proc Natl Acad Sci U S A. 1996;93:6621–5. doi: 10.1073/pnas.93.13.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sunahara RK, Dessauer CW, Whisnant RE, Kleuss C, Gilman AG. Interaction of Gsalpha with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem. 1997;272:22265–71. doi: 10.1074/jbc.272.35.22265. [DOI] [PubMed] [Google Scholar]
- 19.Dessauer CW, Scully TT, Gilman AG. Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem. 1997;272:22272–7. doi: 10.1074/jbc.272.35.22272. [DOI] [PubMed] [Google Scholar]
- 20.Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, Sprang SR. Two-metal-Ion catalysis in adenylyl cyclase. Science. 1999;285:756–60. doi: 10.1126/science.285.5428.756. [DOI] [PubMed] [Google Scholar]
- 21.Chen-Goodspeed M, Lukan AN, Dessauer CW. Modeling of Galpha(s) and Galpha(i) regulation of human type V and VI adenylyl cyclase. J Biol Chem. 2005;280:1808–16. doi: 10.1074/jbc.M409172200. [DOI] [PubMed] [Google Scholar]
- 22.Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009;284:23540–6. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–41. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper DM, Mons N, Karpen JW. Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature. 1995;374:421–4. doi: 10.1038/374421a0. [DOI] [PubMed] [Google Scholar]
- 26.Mons N, Decorte L, Jaffard R, Cooper DM. Ca2+-sensitive adenylyl cyclases, key integrators of cellular signalling. Life Sci. 1998;62:1647–52. doi: 10.1016/s0024-3205(98)00122-2. [DOI] [PubMed] [Google Scholar]
- 27.Cooper DM. Regulation and organization of adenylyl cyclases and cAMP. Biochem J. 2003;375:517–29. doi: 10.1042/BJ20031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper DM. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem Soc Trans. 2005;33:1319–22. doi: 10.1042/BST0331319. [DOI] [PubMed] [Google Scholar]
- 29.Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 30.Choi EJ, Xia Z, Villacres EC, Storm DR. The regulatory diversity of the mammalian adenylyl cyclases. Curr Opin Cell Biol. 1993;5:269–73. doi: 10.1016/0955-0674(93)90115-7. [DOI] [PubMed] [Google Scholar]
- 31.Xia Z, Storm DR. Calmodulin-regulated adenylyl cyclases and neuromodulation. Curr Opin Neurobiol. 1997;7:391–6. doi: 10.1016/s0959-4388(97)80068-2. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson GD, Storm DR. Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda) 2004;19:271–6. doi: 10.1152/physiol.00010.2004. [DOI] [PubMed] [Google Scholar]
- 33.Tang WJ, Gilman AG. Adenylyl cyclases. Cell. 1992;70:869–72. doi: 10.1016/0092-8674(92)90236-6. [DOI] [PubMed] [Google Scholar]
- 34.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–80. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 35.Hanoune J, Pouille Y, Tzavara E, Shen T, Lipskaya L, Miyamoto N, Suzuki Y, Defer N. Adenylyl cyclases: structure, regulation and function in an enzyme superfamily. Mol Cell Endocrinol. 1997;128:179–94. doi: 10.1016/s0303-7207(97)04013-6. [DOI] [PubMed] [Google Scholar]
- 36.Tesmer JJ, Sprang SR. The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr Opin Struct Biol. 1998;8:713–9. doi: 10.1016/s0959-440x(98)80090-0. [DOI] [PubMed] [Google Scholar]
- 37.Smit MJ, Iyengar R. Mammalian adenylyl cyclases. Adv Second Messenger Phosphoprotein Res. 1998;32:1–21. doi: 10.1016/s1040-7952(98)80003-7. [DOI] [PubMed] [Google Scholar]
- 38.Hurley JH. Structure, mechanism, and regulation of mammalian adenylyl cyclase. J Biol Chem. 1999;274:7599–602. doi: 10.1074/jbc.274.12.7599. [DOI] [PubMed] [Google Scholar]
- 39.Whisnant RE, Gilman AG, Dessauer CW. Interaction of the two cytosolic domains of mammalian adenylyl cyclase. Proc Natl Acad Sci U S A. 1996;93:6621–5. doi: 10.1073/pnas.93.13.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen-Goodspeed M, Lukan AN, Dessauer CW. Modeling of Galpha(s) and Galpha(i) regulation of human type V and VI adenylyl cyclase. J Biol Chem. 2005;280:1808–16. doi: 10.1074/jbc.M409172200. [DOI] [PubMed] [Google Scholar]
- 41.Wu GC, Lai HL, Lin YW, Chu YT, Chern Y. N-glycosylation and residues Asn805 and Asn890 are involved in the functional properties of type VI adenylyl cyclase. J Biol Chem. 2001;276:35450–7. doi: 10.1074/jbc.M009704200. [DOI] [PubMed] [Google Scholar]
- 42.Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y. Regulation of adenylyl cyclase by protein kinase A. J Biol Chem. 1995;270:12481–4. doi: 10.1074/jbc.270.21.12481. [DOI] [PubMed] [Google Scholar]
- 43.Lin TH, Lai HL, Kao YY, Sun CN, Hwang MJ, Chern Y. Protein kinase C inhibits type VI adenylyl cyclase by phosphorylating the regulatory N domain and two catalytic C1 and C2 domains. J Biol Chem. 2002;277:15721–8. doi: 10.1074/jbc.M111537200. [DOI] [PubMed] [Google Scholar]
- 44.Tang WJ, Gilman AG. Construction of a soluble adenylyl cyclase activated by Gs alpha and forskolin. Science. 1995;268:1769–72. doi: 10.1126/science.7792604. [DOI] [PubMed] [Google Scholar]
- 45.Yan SZ, Hahn D, Huang ZH, Tang WJ. Two cytoplasmic domains of mammalian adenylyl cyclase form a Gs alpha- and forskolin-activated enzyme in vitro. J Biol Chem. 1996;271:10941–5. doi: 10.1074/jbc.271.18.10941. [DOI] [PubMed] [Google Scholar]
- 46.Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res. 2006;98:675–681. doi: 10.1161/01.RES.0000209516.84815.3e. [DOI] [PubMed] [Google Scholar]
- 47.Tang WJ, Stanzel M, Gilman AG. Truncation and alanine-scanning mutants of type I adenylyl cyclase. Biochemistry. 1995;34:14563–72. doi: 10.1021/bi00044a035. [DOI] [PubMed] [Google Scholar]
- 48.Yan SZ, Huang ZH, Shaw RS, Tang WJ. The conserved asparagine and arginine are essential for catalysis of mammalian adenylyl cyclase. J Biol Chem. 1997;272:12342–9. doi: 10.1074/jbc.272.19.12342. [DOI] [PubMed] [Google Scholar]
- 49.Ping P, Anzai T, Gao M, Hammond HK. Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol Heart Circ Physiol. 1997;273:H707–17. doi: 10.1152/ajpheart.1997.273.2.H707. [DOI] [PubMed] [Google Scholar]
- 50.Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989–94. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–96. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 52.Timofeyev V, He Y, Tuteja D, Zhang Q, Roth DM, Hammond HK, Chiamvimonvat N. Cardiac-directed expression of adenylyl cyclase reverses electrical remodeling in cardiomyopathy. J Mol Cell Cardiol. 2006;41:170–81. doi: 10.1016/j.yjmcc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 53.Sastry A, Arnold E, Gurji H, Iwasa A, Hassankhani A, Lai NC, Roth DM, Patel HH, Hammond HK, Narayan SM. Adenylylcyclase VI overexpression facilitates atrioventricular nodal conduction in mice. J Am Coll Cardiol. 2006;48:559–565. doi: 10.1016/j.jacc.2006.01.082. [DOI] [PubMed] [Google Scholar]
- 54.Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou JY, Zhu J, Entrikin D, Hammond HK. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99:3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 55.Lai NC, Roth DM, Gao MH, Tang T, Dalton D, Lai YY, Spellman M, Clopton P, Hammond HK. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in CHF. Circulation. 2004;110:330–336. doi: 10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- 56.Tang T, Gao MH, Roth DM, Guo T, Hammond HK. Adenylyl cyclase type VI corrects cardiac sarcoplasmic reticulum Ca2+ uptake defects in cardiomyopathy. Am J Physiol Heart Circ Physiol. 2004;287:H1906–12. doi: 10.1152/ajpheart.00356.2004. [DOI] [PubMed] [Google Scholar]
- 57.Lai NC, Tang T, Gao MH, Saito M, Takahashi T, Roth DM, Hammond HK. Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J Am Coll Cardiol. 2008;51:1490–7. doi: 10.1016/j.jacc.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK. Increased expression of adenylylcyclase type VI proportionately increases ß-adrenergic receptor-stimulated cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A. 1998;95:1038–43. doi: 10.1073/pnas.95.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao M, Lai NC, Roth DM, Zhou J, Anzai T, Dalton N, Hammond HK. Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation. 1999;99:1618–22. doi: 10.1161/01.cir.99.12.1618. [DOI] [PubMed] [Google Scholar]
- 60.Geng YJ, Ishikawa Y, Vatner DE, Wagner TE, Bishop SP, Vatner SF, Homcy CJ. Apoptosis of cardiac myocytes in Gsα transgenic mice. Circ Res. 1999;84:34–42. doi: 10.1161/01.res.84.1.34. [DOI] [PubMed] [Google Scholar]
- 61.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in ß1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–64. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW., II Early and delayed consequences of ß2-adrenergic receptor overexpression in mouse hearts, Critical role for expression level. Circulation. 2000;101:1707–14. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 63.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:121–6. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lai NC, Roth DM, Gao MH, Fine S, Head BP, Zhu J, McKirnan MD, Kwong C, Dalton N, Urasawa K, Roth DA, Hammond HK. Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP-generating capacity. Circulation. 2000;102:2396–401. doi: 10.1161/01.cir.102.19.2396. [DOI] [PubMed] [Google Scholar]
- 65.Quyyumi AA, Dakak N, Mulcahy D, Andrews NP, Husain S, Panza JA, Cannon RO. Nitric oxide activity in the atherosclerotic human coronary circulation. J Am Coll Cardiol. 1997;29:308–17. doi: 10.1016/s0735-1097(96)00472-x. [DOI] [PubMed] [Google Scholar]
- 66.Hillegass WB, Dean NA, Liao L, Rhinehart RG, Myers PR. Treatment of no-reflow and impaired flow with the nitric oxide donor nitroprusside following percutaneous coronary interventions: initial human clinical experience. J Amer Coll Cardiol. 2001;37:1335–43. doi: 10.1016/s0735-1097(01)01138-x. [DOI] [PubMed] [Google Scholar]
- 67.Resnic FS, Wainstein M, Lee MK, Behrendt D, Wainstein RV, Ohno-Machado L, Kirshenbaum JM, Rogers CD, Popma JJ, Piana R. No-reflow is an independent predictor of death and myocardial infarction after percutaneous coronary intervention. Am Heart J. 2003;145:42–46. doi: 10.1067/mhj.2003.36. [DOI] [PubMed] [Google Scholar]
- 68.Roth DM, Lai NC, Gao MH, Fine S, McKirnan MD, Roth DA, Hammond HK. Nitroprusside increases gene transfer associated with intracoronary delivery of adenovirus. Human Gene Ther. 2004;15:989–94. doi: 10.1089/hum.2004.15.989. [DOI] [PubMed] [Google Scholar]
- 69.Gao MH, Bayat H, Zhou JY, Roth DM, Drumm JD, Burhan J, Hammond HK. Controlled expression of cardiac-directed adenylylcyclase type IV provided increased contractile function. Cardiovasc Res. 2002;56:197–204. doi: 10.1016/s0008-6363(02)00539-4. [DOI] [PubMed] [Google Scholar]
- 70.Gao MH, Tang T, Guo SQ, Sun JR, Feramisco JF, Hammond HK. Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J Biol Chem. 2004;279:38797–802. doi: 10.1074/jbc.M405701200. [DOI] [PubMed] [Google Scholar]
- 71.Gao MH, Tang T, Guo T, Pestonjamasp K, Feramisco JR, Hammond HK. Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. J Biol Chem. 2008;283:33527–35. doi: 10.1074/jbc.M805825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao MH, Miyanohara A, Feramisco JR, Tang T. Activation of PH-domain leucine-rich protein phosphatase 2 (PHLPP2) by agonist stimulation in cardiac myocytes expressing adenylyl cyclase type 6. Biochem Biophys Res Commun. 2009;384:193–8. doi: 10.1016/j.bbrc.2009.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamamoto S, Kawamura K, James TN. Intracellular distribution of adenylate cyclase in human cardiocytes determined by electron microscopic cytochemistry. Microsc Res Tech. 1998;40:479–87. doi: 10.1002/(SICI)1097-0029(19980301)40:6<479::AID-JEMT8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 74.Drescher MJ, Kern RC, Hatfield JS, Drescher DG. Cytochemical localization of adenylyl cyclase activity within the sensory epithelium of the trout saccule. Neurosci Lett. 1995;196:145–8. doi: 10.1016/0304-3940(95)11764-n. [DOI] [PubMed] [Google Scholar]
- 75.Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol Pharmacol. 2002;62:983–92. doi: 10.1124/mol.62.5.983. [DOI] [PubMed] [Google Scholar]
- 76.Head BP, Patel BP, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem. 2005;280:31036–44. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 77.Insel PA, Head BP, Patel HH, Roth DM, Bundey RA, Swaney JS. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Biochem Soc Trans. 2005;33:1131–4. doi: 10.1042/BST20051131. [DOI] [PubMed] [Google Scholar]
- 78.Palade G. Fine structure of blood capillaries. J Appl Physics. 1953;24:1424. Abstract. [Google Scholar]
- 79.Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol. 1955;1:445–8. doi: 10.1083/jcb.1.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–67. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 81.Gao MH, Tang T, Miyanohara A, Feramisco JR, Hammond HK. Beta1-adrenergic receptor vs adenylyl cyclase 6 expression in cardiac myocytes: differences in transgene localization and intracellular signaling. Cell Signal. 2010;22:584–9. doi: 10.1016/j.cellsig.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thangavel M, Liu X, Sun SQ, Kaminsky J, Ostrom RS. The C1 and C2 domains target human type 6 adenylyl cyclase to lipid rafts and caveolae. Cell Signal. 2009;21:301–8. doi: 10.1016/j.cellsig.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao MH, Tang T, Miyanohara A, Guo A, Tang R, Roth DM, Firth AL, Yuan JX, Hammond HK. Cyclic AMP-dependent and cAMP-independent effects of adenylyl cyclase 6: Insights from a catalytically inactive mutant. Molecular Pharmacology Fast Forward. doi: 10.1124/mol.110.067298. Published on December 2, 2010 as. [DOI] [Google Scholar]
- 84.Sinnarajah S, Dessauer CW, Srikumar D, Chen J, Yuen J, Yilma S, Dennis JC, Morrison EE, Vodyanoy V, Kehrl JH. RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature. 2001;409:1051–5. doi: 10.1038/35059104. [DOI] [PubMed] [Google Scholar]
- 85.Salim S, Sinnarajah S, Kehrl JH, Dessauer CW. Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem. 2003;278:15842–9. doi: 10.1074/jbc.M210663200. [DOI] [PubMed] [Google Scholar]
- 86.Roy AA, Baragli A, Bernstein LS, Hepler JR, Hébert TE, Chidiac P. RGS2 interacts with Gs and adenylyl cyclase in living cells. Cell Signal. 2006;18:336–48. doi: 10.1016/j.cellsig.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 87.Scholich K, Pierre S, Patel TB. Protein associated with Myc (PAM) is a potent inhibitor of adenylyl cyclases. J Biol Chem. 2001;276:47583–9. doi: 10.1074/jbc.M107816200. [DOI] [PubMed] [Google Scholar]
- 88.Gao X, Patel TB. Histidine residues 912 and 913 in protein associated with Myc are necessary for the inhibition of adenylyl cyclase activity. Mol Pharmacol. 2005;67:42–9. doi: 10.1124/mol.104.005355. [DOI] [PubMed] [Google Scholar]
- 89.Piggott LA, Bauman AL, Scott JD, Dessauer CW. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci U S A. 2008;105:13835–40. doi: 10.1073/pnas.0712100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang SC, Lai HL, Chiu YT, Ou R, Huang CL, Chern Y. Regulation of type V adenylate cyclase by Ric8a, a guanine nucleotide exchange factor. Biochem J. 2007;406:383–8. doi: 10.1042/BJ20070512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chou JL, Huang CL, Lai HL, Hung AC, Chien CL, Kao YY, Chern Y. Regulation of type VI adenylyl cyclase by Snapin, a SNAP25-binding protein. J Biol Chem. 2004;279:46271–9. doi: 10.1074/jbc.M407206200. [DOI] [PubMed] [Google Scholar]
- 92.Wang SC, Lin JT, Chern Y. Novel regulation of adenylyl cyclases by direct protein-protein interactions: insights from snapin and ric8a. Neurosignals. 2009;17:169–80. doi: 10.1159/000200076. [DOI] [PubMed] [Google Scholar]
- 93.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, Yuan JX, Roth DM, Hammond HK. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired Ca2+ handling. Circulation. 2008;117:61–9. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 94.Tepe NM, Liggett SB. Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alphαq overexpressing mouse. FEBS Lett. 1999;458:236–40. doi: 10.1016/s0014-5793(99)01147-3. [DOI] [PubMed] [Google Scholar]
- 95.Esposito G, Perrino C, Ozaki T, Takaoka H, Defer N, Petretta MP, De Angelis MC, Mao L, Hanoune J, Rockman HA, Chiariello M. Increased myocardial contractility and enhanced exercise function in transgenic mice overexpressing either adenylyl cyclase 5 or 8. Basic Res Cardiol. 2008;103:22–30. doi: 10.1007/s00395-007-0688-6. [DOI] [PubMed] [Google Scholar]
- 96.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–81. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–15. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 98.Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ Res. 2008;102:1458–70. doi: 10.1161/CIRCRESAHA.108.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roth DM, Drumm JD, Bhargava V, Swaney JS, Gao MH, Hammond HK. Cardiac-directed expression of adenylyl cyclase and heart rate regulation. Basic Res Cardiol. 2003;98:380–7. doi: 10.1007/s00395-003-0429-4. [DOI] [PubMed] [Google Scholar]
- 100.Rebolledo B, Lai NC, Gao MH, Takahashi T, Roth DM, Baird SM, Hammond HK. Adenylylcyclase gene transfer increases function of the failing heart. Hum Gene Ther. 2006;17:1043–8. doi: 10.1089/hum.2006.17.1043. [DOI] [PubMed] [Google Scholar]
- 101.Roth DM, Lai NC, Gao MH, Drumm JD, Jimenez J, Feramisco JR, Hammond HK. Indirect intracoronary delivery of adenovirus encoding adenylyl cyclase increases left ventricular contractile function in mice. Am J Physiol. 2004;287:H172–7. doi: 10.1152/ajpheart.01009.2003. [DOI] [PubMed] [Google Scholar]