

Abstract
Objectives
This study sought to evaluate whether increased left ventricular (LV) adenylyl cyclase VI (ACVI) expression, at a time when severe congestive heart failure (CHF) was present, would increase function of the actively failing heart.
Background
Increased LV ACVI content markedly reduces mortality and increases LV function after acute myocardial infarction (MI) in mice. However, the effects of increased cardiac ACVI content in the setting of severe heart failure caused by ischemic cardiomyopathy are unknown.
Methods
Mice with cardiac-directed and regulated expression of ACVI underwent coronary artery ligation to induce severe CHF 5 weeks later. ACVI expression was then activated in 1 group (AC-On) but not the other (AC-Off). Multiple measures of LV systolic and diastolic function were obtained 5 weeks later, and LV samples were assessed for alterations in calcium and beta-adrenergic receptor signaling, apoptosis, and cardiac troponin I phosphorylation.
Results
The LV systolic and diastolic function was increased 5 weeks after activation of ACVI expression. Improved LV function was associated with normalization of cardiac troponin I phosphorylation and reduced apoptosis.
Conclusions
Activation of cardiac ACVI expression in mice with ischemic cardiomyopathy and severe CHF improves function of the failing heart.
Increased adenylyl cyclase VI (ACVI) content reduces mortality and increases left ventricular (LV) function after acute myocardial infarction (MI) (1), and also has beneficial effects in congestive heart failure (CHF) (2–4). Cardiac gene transfer of ACVI attenuates the decline of cardiac function in pacing-induced CHF (2). Increased LV ACVI content prevents CHF from occurring in a genetic model of cardiomyopathy using a crossbreeding strategy (3,4). Other genes have been shown to slow the decline of LV function (5), or in genetic models, to prevent heart failure from occurring (6–8). However, studies that have documented increased function of an actively failing heart by increased expression of a putatively therapeutic gene are remarkably rare (9). Demonstration that a candidate gene’s expression leads to increased function of the failing heart in a relevant animal model is an essential criterion for identifying potentially useful genes for the treatment of clinical CHF. In the present study we ask whether activation of ACVI expression—in the presence of severe CHF caused by MI—would have beneficial effects.
We used a transgenic line that provides cardiac-specific expression of ACVI under tet-regulation (10). This enabled rapid activation of cardiac transgene ACVI expression at any desired point in time. Left coronary artery occlusion was used to induce severe CHF in mice (11), providing a suitable model of clinical ischemic cardiomyopathy, a common etiology for clinical CHF. When evidence of severe CHF was present (5 weeks after MI), we activated transgene cardiac ACVI expression in 1 group; transgene suppression was maintained in the other group. Five weeks after activation of ACVI expression, physiological and molecular studies were conducted and the 2 groups were compared (Fig. 1). Our hypothesis was that increased cardiac expression of ACVI, in the setting of severe CHF, would be associated with increased function of the failing heart. An additional goal was to determine mechanisms for differences in LV function.
Figure 1. Experimental Design.
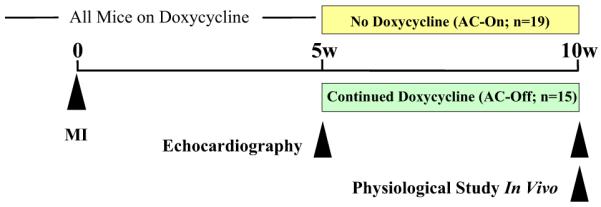
See the text for a full description.
AC = adenylyl cyclase; MI = myocardial infarction.
Methods
Animals
Animal use and care were in accordance with institutional and National Institutes of Health guidelines.
Transgenic mice with cardiac-directed and regulated (tet-off) expression of ACVI (congenic C57BL/6 background) were generated by our laboratory and previously described in detail (10). Suppression is complete until doxycycline is removed from the water supply (10).
A total of 234 mice were used in the present studies: 174 for the primary comparison and 60 for additional controls (see later text). Of the 174 mice for the AC therapy comparison, 50 mice survived infarction and met the entry criterion: fractional area change (FAC) <30% (normal is >50%). Tetracycline suppression was continued in 1 subgroup (AC-Off), but removed in the other (AC-On) (10). To test the influence of doxycycline on LV size and function, 52 nontransgenic C57BL/6 mice were infarcted. Doxycycline was given in the water supply (as in the previous text) from 10 days before until 5 weeks after MI. For the doxycycline group, 7 met entry criterion; for the no doxycycline group, 11 met entry criterion. We also used 8 nontransgenic C57BL/6 mice as normal control subjects (no MI, no doxycycline treatment).
Heart failure model
We used coronary occlusion to induce large anterior wall MI and CHF as described in detail previously (11). Because MI size deliberately was large, this model is associated with a high initial mortality (>30%); 50% of surviving mice have severe CHF, as has been reported previously (11). The primary end point of the study was LV function 5 weeks after initiation of cardiac-directed ACVI expression in the failing heart (Fig. 1). Group assignment occurred 5 weeks before enrollment. Because we could not predict the number in each group that would meet enrollment criteria 5 weeks later, group sizes were not identical. At enrollment, groups were randomly assigned to continue on doxycycline (AC-Off) or to have it withdrawn (AC-On). Data were acquired and analyzed without knowledge of group identity.
Echocardiography
Echocardiography was performed as previously described (1,2).
In vivo physiology
Mice were anesthetized with sodium pentobarbital (100 mg/kg intraperitoneally), and a 1.4-F conductance-micromanometer catheter (SPR 716, Millar Instruments, Houston, Texas) was inserted via the right carotid artery across the aortic valve and into the LV chamber. After LV pressures were recorded, bilateral vagotomy was performed to minimize confounding effects of reflex activation. End-systolic pressure (ESP) and LV pressure development (+dP/dt) and relaxation (−dP/dt) were obtained. Inferior vena cava occlusion was performed to reduce LV volume, and end-systolic pressure–volume relationship (ESPVR) was obtained. Stroke volume was determined by subtracting the LV end-systolic from the LV end-diastolic volume, measured by conductance catheter; stroke volume × heart rate provided cardiac output.
Calcium uptake
The initial rate of adenosine triphosphate-dependent sarcoplasmic reticulum (SR) calcium uptake in viable LV samples was measured as previously described (12).
LV cyclic adenosine monophosphate (cAMP) generation
Cyclic adenosine monophosphate production in viable LV samples was measured as previously described (1).
Protein kinase and phosphatase activity
Akt was immunoprecipitated from 500-μg LV samples and assayed (Akt Kinase Assay Kit, Cell Signaling Technology, Danvers, Massachusetts). Protein kinase A (PKA) activity (cAMP-dependent) (12) and activities of protein phosphatases 1 (PP1) and 2A (PP2A) were measured.
Western blotting
Viable LV samples were homogenized and underwent Western blotting as described previously (12). Antibodies to cTnI and phospho-Ser23/24-cTnI were obtained from Cell Signaling Technology (Danvers, Massachusetts).
Matrix metalloproteinase (MMP) expression
Quantitative real-time reverse-transcriptase polymerase chain reaction was conducted to compare MMP-2 and -8 messenger ribonucleic acid (mRNA) contents. Primer pairs were MMP2: forward 5′-GAGTTGCAACCTCTTTGTGC-3′ and reverse 5′-CAGGTGTGTAACCAATGATCC-3′; MMP8: forward 5′-CCCAGCACCTATTCACTACC-3′ and reverse 5′-CTGTTCTCAGCTGAGGATGC-3′. Specificity of each reverse-transcriptase polymerase chain reaction was checked by its dissociation curve and agarose gel electrophoresis.
Apoptosis
Terminal 2′-deoxyuridine 5′-triphosphate nick end-labeling (TUNEL) assays were performed as described previously (1).
Caspase 3/7 activity
Caspase 3/7 activity in LV homogenates was measured using the Caspase-Glo 3/7 Assay (Promega, Madison, Wisconsin).
Necropsy and histology
Body and LV weights were recorded. Transmural LV samples from regions bordering and remote from the infarction were formalin-fixed; hematoxylin-and-eosin and Masson trichrome stainings were used to assess inflammation and fibrosis. A pathologist scored LV samples for the presence of inflammation and fibrosis (blinded analysis).
Statistical analysis
Results are shown as mean ± SE. Group comparisons were made using the Student t test (2-tailed) or 2-way analysis of variance with Bonferroni t testing. For histological studies, the degree of inflammation and fibrosis were rated (0, none; 1, mild; 2, moderate; 3, severe), and the Mann-Whitney U test was used to test for difference between groups. The null hypothesis was rejected when p < 0.05. Analyses were performed using GraphPad Prism (GraphPad Software, Inc., San Diego, California).
Results
Animal model
In previous studies using the same MI procedures, we have shown that infarct size averages 49 ± 3% of the total LV (11), comprising most of the LV free wall. Such large infarctions result in high mortality and severe CHF in survivors. In the present study, 29% of the animals survived infarction, developed CHF, and entered into the study, a typical outcome with this model. Data were not adversely affected by this because we had strict criteria for enrollment into the study: surviving animals had to have substantial LV dysfunction. There were no group differences in body weight (AC-Off: 27.2 ± 0.7 g, n = 11; AC-On: 28.8 ± 1.5 g, n = 8; p = 0.30), LV weight (AC-Off: 138 ± 6 mg, n = 11; AC-On: 130 ± 4 mg, n = 8; p = 0.37) or LV/body weight ratio (AC-Off: 5.1 ± 0.2 mg/g, n = 11; AC-On: 4.6 ± 0.3 mg/g, n = 8; p = 0.17). However, there was LV hypertrophy in both groups compared with uninfarcted mouse hearts where the LV/body weight ratio is 3.5 ± 0.1 mg/g (11). We previously found that the lung weight/tibial length ratio is increased in this model of CHF (p = 0.037) (11). Activation of transgene provided a 16-fold increase in LV ACVI (p < 0.0001) (Fig. 2), confirming the efficiency of the tet-regulated transgene (10).
Figure 2. LV ACVI Content.
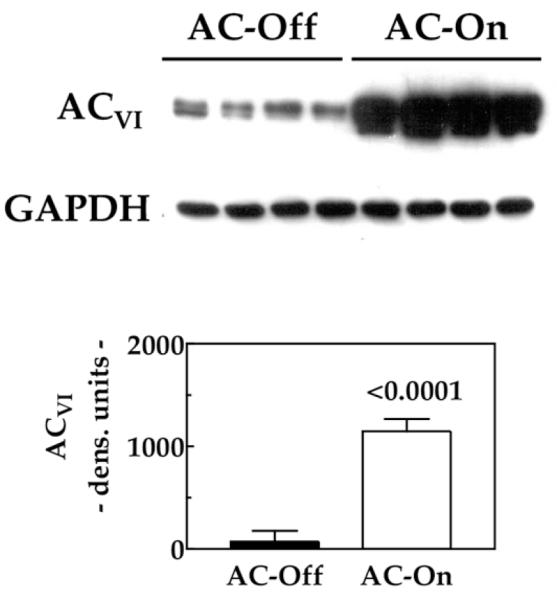
The Western blot shows a marked increase in ACVI protein in LV samples from mice 5 weeks after activation of ACVI transgene expression (n = 8 for both groups). The graph summarizes data from Western blotting. Bars = mean values; error bars = 1 SE; number above bars = probability value (Student unpaired t test, 2-tailed). ACVI = adenylyl cyclase VI; LV= left ventricular.
Echocardiography
Basal heart rates were similar in both groups both before and after treatment (Table 1). The LV end-diastolic and end-systolic dimensions were increased 5 weeks after MI, and activation of transgene ACVI expression had no effect on LV dimensions, which did not change from 5 weeks through 10 weeks after MI and CHF. Thicknesses of the posterior and septal walls were reduced by MI, but did not change during treatment. However, there were increases in the LV fractional area change (p = 0.0001) and ejection fraction (EF) (p = 0.0001) (Table 1). Pre-treatment EF was the same in both groups: 24%. Post-treatment EF in the AC-On group was 6 percentage units greater than that in the AC-Off group, a 27% relative increase (AC-Off: 22 ± 2%, n = 15; AC-On: 28 ± 2%, n = 19; p = 0.02).
Table 1.
Echocardiography: Effects of Increased ACVI Expression
| AC-Off (n = 15) | AC-On (n = 19) | ||||||
|---|---|---|---|---|---|---|---|
| Before Rx | 5 Weeks After Rx | Change | Before Rx | 5 Weeks After Rx | Change | p Value | |
| HR (beats/min) | 394 ± 14 | 412 ± 15 | 18 ± 13 | 408 ± 11 | 430 ± 12 | 21 ± 17 | 0.88 |
| EDD (mm) | 5.5 ± 0.1 | 5.6 ± 0.1 | 0.1 ± 0.1 | 5.9 ± 0.2 | 5.9 ± 0.2 | 0.03 ± 0.1 | 0.70 |
| ESD (mm) | 4.8 ± 0.1 | 5.0 ± 0.2 | 0.2 ± 0.1 | 5.1 ± 0.2 | 5.0 ± 0.3 | 0.1 ± 0.1 | 0.17 |
| PWd (mm) | 0.66 ± 0.03 | 0.57 ± 0.02 | −0.09 ± 0.03 | 0.64 ± 0.02 | 0.61 ± 0.02 | −0.03 ± 0.02 | 0.14 |
| IVSd (mm) | 0.48 ± 0.02 | 0.47 ± 0.02 | −0.01 ± 0.02 | 0.49 ± 0.03 | 0.48 ± 0.02 | −0.02 ± 0.02 | 0.89 |
| FAC (%) | 13 ± 1 | 12 ± 1 | −1 ± 1 | 13 ± 1 | 16 ± 1 | 3 ± 1 | 0.0001 |
| LVEF (%) | 24 ± 1 | 22 ± 1 | −2 ± 1 | 24 ± 2 | 28 ± 2 | 5 ± 1 | 0.0001 |
Values represent mean ± SE. The p values are from the Student t test comparing changes in each parameter between groups (2-tailed, unpaired).
ACVI = adenylyl cyclase VI; EDD = end-diastolic diameter; ESD = end-systolic diameter; FAC = fractional area change; HR = heart rate; IVSd = interventricular septal diastolic wall thickness; LVEF = left ventricular ejection fraction; PWd = posterior diastolic wall thickness; Rx = treatment.
LV contractile function
To assess the effect of ACVI transgene expression on cardiac contractile function, we measured LV pressure and volume to assess ESPVR. Activation of ACVI expression was associated with increases in basal LV +dP/dt (p = 0.013) and cardiac output (70% increase; p = 0.029) (Table 2). The ESPVR, a relatively load-independent measure of contractile function, was increased 4-fold (p = 0.014) by activation of ACVI expression (Table 2, Fig. 3). Because of difficulties with catheter engagement, sample sizes differ in Table 2 compared with Table 1.
Table 2.
Basal Left Ventricular Function: Effects of Increased ACVI Expression
| AC-Off (n = 9) | AC-On (n = 12) | p Value | |
|---|---|---|---|
| HR (beats/min) | 424 ± 34 | 452 ± 20 | 0.46 |
| CO (ml/min) | 4.0 ± 0.4 | 6.8 ± 1.0 | 0.029 |
| ESPVR (mm Hg/μl) | 2.4 ± 0.7* | 10.1 ± 1.8* | 0.014 |
| LV +dP/dt (mm Hg/s) | 5,178 ± 375 | 7,149 ± 551 | 0.013 |
| LV −dP/dt (mm Hg/s) | −4,901 ± 459 | −6,408 ± 505 | 0.046 |
| EDP (mm Hg) | 17 ± 2 | 12 ± 1 | 0.039 |
| Tau (ms) | 11 ± 1 | 7 ± 1 | 0.008 |
Values represent mean ± SE. p value is from Student t test comparing changes in each parameter between groups (2-tailed, unpaired).
For ESPVR, AC-Off, n = 5; AC-On, n = 10.
CO = cardiac output; EDP = end-diastolic pressure; ESPVR = end-systolic pressure–volume relationship; HR = heart rate; LV = left ventricular.
Figure 3. Contractile Function.
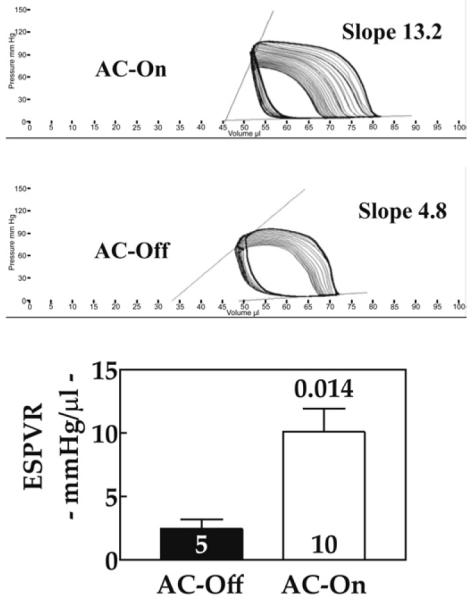
The ESPVR was measured in intact mice 5 weeks after activation of ACVI expression in animals with severe CHF. Increased cardiac ACVI expression was associated with substantial increases in LV contractility as reflected in increased slope of the ESPVR. Cardiac loops, generated by altering loading conditions of the LV, are shown with the slope of the end-systolic pressure-volume point depicted. The graph below summarizes data from all animals. Numbers in bars denote animals studied. Bars = mean values; error bars = 1 SE; number above bars = probability value (Student unpaired t test, 2-tailed). CHF = congestive heart failure; ESPVR = end-systolic pressure-volume relationship; other abbreviations as in Figure 2.
LV diastolic function
Activation of ACVI expression had favorable effects on LV end-diastolic pressure (p = 0.039), basal LV −dP/dt (p = 0.046), and tau (p = 0.0008), indicating increased cardiac relaxation (Table 2).
Effects of doxycycline
We included 2 additional control groups: 1) doxycycline: these nontransgenic C57BL/6 mice received doxycycline continuously from 10 days before until 5 weeks after MI, a protocol similar to the AC-On group; and 2) no doxycycline: these nontransgenic C57BL/6 mice were included as a control for the doxycycline group. There were no differences in any measure of LV size or function 10 weeks after MI in these 2 groups (Table 3), and these values closely matched those in the AC-Off group (unpaired Student t test) (Table 1).
Table 3.
Echocardiography: Nontransgenic C57BL/6 Mice ± Doxycycline
| Doxycycline (n = 7) | No Doxycycline (n = 11) | ||||||
|---|---|---|---|---|---|---|---|
| 5 Weeks After MI | 10 Weeks After MI | Change | 5 Weeks After MI | 10 Weeks After MI | Change | p Value | |
| HR (beats/min) | 443 ± 20 | 459 ± 30 | 16 ± 31 | 464 ± 10 | 453 ± 24 | −11 ± 18 | 0.43 |
| EDD (mm) | 4.9 ± 0.4 | 5.3 ± 0.5 | 0.5 ± 0.2 | 5.0 ± 0.3 | 5.3 ± 0.4 | 0.3 ± 0.2 | 0.46 |
| ESD (mm) | 4.3 ± 0.4 | 4.8 ± 0.6 | 0.6 ± 0.2 | 4.3 ± 0.4 | 4.7 ± 0.5 | 0.3 ± 0.2 | 0.52 |
| PWd (mm) | 0.6 ± 0.1 | 0.5 ± 0.1 | −0.1 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.05 | −0.01 ± 0.04 | 0.81 |
| IVSd (mm) | 0.6 ± 0.1 | 0.6 ± 0.04 | 0.01 ± 0.04 | 0.6 ± 0.03 | 0.6 ± 0.02 | −0.02 ± 0.03 | 0.61 |
| FAC (%) | 12 ± 2 | 11 ± 2 | −0.6 ± 1.0 | 14 ± 2 | 14 ± 2 | −0.1 ± 1.0 | 0.74 |
| LVEF (%) | 19 ± 4 | 20 ± 4 | 1 ± 2 | 22 ± 4 | 21 ± 4 | −1.1 ± 1.5 | 0.32 |
Values represent mean ± SE. p value is from Student t test comparing changes in each parameter between groups (2-tailed, unpaired). Data from nontransgenic C57BL/6 mice (strain-matched). Experimental protocols were identical. Doxycycline was given continuously from 10 days before to 5 weeks after MI, or was not given (No Doxycycline group).
Abbreviations as in Table 1.
LV Ca2+ handling
Activation of ACVI expression did not alter the maximal velocity of Ca2+ uptake (AC-Off: 268 ± 31 nmol/mg/min, n = 9; AC-On: 292 ± 8 nmol/mg/min, n = 7; p = 0.47) or the affinity constant of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) for Ca2+ (KCa) (AC-Off: 1.32 ± 0.51 μM, n = 9; AC-On: 1.25 ± 0.50 μM, n = 7; p = 0.87), although the values for KCa were increased compared with normal mice (12). There were no group differences in the following proteins: SERCA2a, Ser16 phosphorylated phospholamban (PLN), Thr17 phosphorylated PLN, calsequestrin, RyR2, and Ser2809 phosphorylated RyR2.
LV cAMP generation
Activation of transgene ACVI was associated with a 2-fold increase in NKH477-stimulated cAMP production in LV membranes (p = 0.0001), basal cAMP was unchanged, and PKA activity was increased (Fig. 4). The PKA catalytic subunit expression was unchanged (data not shown). Stimulation by NKH477 of LV samples from age-matched normal mice show 121 ± 10 pmol/mg/min cAMP generation (n = 8), indicating that LV cAMP-generating capacity is reduced by 28% in untreated CHF (p = 0.02) and increased to 45% above normal levels after activation of ACVI expression (p = 0.006). There were no group differences in protein contents of β1AR, β2AR, or Gαs.
Figure 4. LV cAMP Production and PKA Activity.

(Left) Increased ACVI expression was associated with increased cAMP generation in response to NKH477 (10 μM) stimulation of LV samples. Basal cAMP generation was not changed by ACVI expression. (Right) Increased cardiac ACVI expression was associated with increased PKA activity in LV samples. Numbers in bars = animals studied; bars = mean values; error bars = 1 SE; number above bars = probability value (Student unpaired t test, 2-tailed). cAMP = cyclic adenosine monophosphate; PKA = protein kinase A; other abbreviations as in Figure 2.
Phosphorylation of cTnI
Activation of cardiac expression of ACVI in the setting of CHF was associated with a 2.7-fold increase in cTnI phosphorylation at Ser23/24 (p = 0.01) (Fig. 5). The LV samples from normal mice have levels of cTnI phosphorylation at Ser23/24 of 361 ± 34 densitometric unit (du) (n = 8), indicating that severe CHF is associated with a 70% reduction in cTnI phosphorylation at Ser23/24 (p < 0.0001). Activation of ACVI expression in failing hearts restored cTnI phosphorylation at Ser23/24 to normal (p = 0.33). The LV samples showed no differences in protein phosphatase PP1 or PP2A content (PP1: AC-Off: 2,516 ± 62 du, n = 8; AC-On: 2,431 ± 83 du, n = 8; p = 0.43; PP2A: AC-Off: 770 ± 55 du, n = 8; AC-On: 728 ± 123 du, n = 8; p = 0.76) or activity (PP1: AC-Off: 1.01 ± 0.12 nmol/mg/min, n = 8; AC-On: 1.24 ± 0.08 nmol/mg/min, n = 8; p = 0.43; PP2A: AC-Off: 1.04 ± 0.06 nmol/mg/min, n = 8; AC-On: 1.22 ± 0.10 nmol/mg/min, n = 8; p = 0.12).
Figure 5. LV cTnI Phosphorylation.
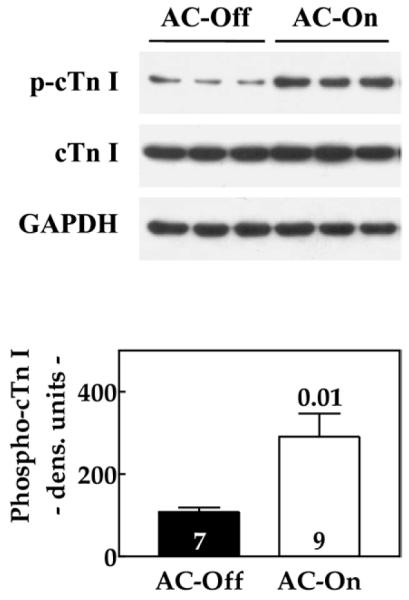
The Western blot shows a marked increase in cTnI phosphorylation in LV samples from mice 5 weeks after activation of ACVI transgene expression. Summary of data from Western blotting is shown in the lower panel. Numbers in bars = animals studied; bars = mean values; error bars = 1 SE; number above bars = probability value (Student unpaired t test, 2-tailed). Abbreviations as in Figure 2.
MMP expression
There were no group differences in LV expression of MMP2 mRNA (AC-Off: 100 ± 15%, n = 5; AC-On: 110 ± 17%, n = 7; p = 0.68) or MMP8 mRNA (AC-Off: 100 ± 31%, n = 5; AC-On: 85 ± 52%, n = 7; p = 0.83).
Apoptosis
TUNEL staining was used to quantify apoptosis in the border and remote zones 10 weeks after MI. The LV samples from AC-Off mice showed a 7.3-fold increase in apoptosis in the border zone and a 3.9-fold increase in the remote zone compared with baseline LV apoptosis (1). Activation of ACVI expression was associated with a 59% reduction of apoptosis in the border zone and a 46% reduction in the remote zone (p = 0.024) (Fig. 6). There was no group difference in LV Bcl-2 protein content (AC-Off: 205 ± 26 du, n = 8; AC-On: 201 ± 53 du, n = 8; p = 0.94) or caspase 3/7 activity (AC-Off: 828 ± 92 relative light units (RLU)/μg, n = 9; AC-On: 654 ± 71 RLU/μg, n = 7; p = 0.18), and LV Akt activity was reduced by activation of ACVI expression (AC-Off: 1,199 ± 93 du, n = 8; AC-On: 722 ± 74 du, n = 8; p = 0.001).
Figure 6. Apoptosis.
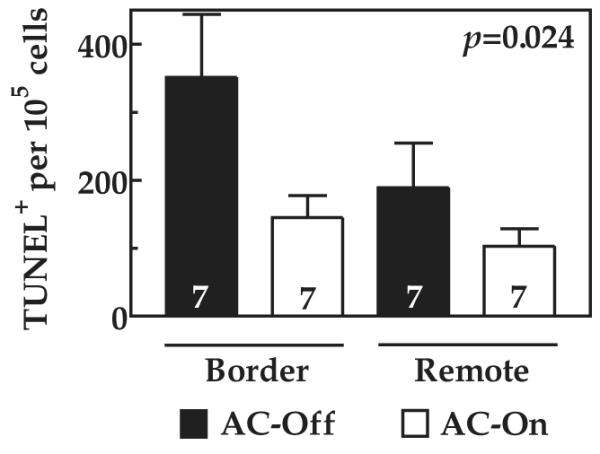
Rates of LV apoptosis (TUNEL staining) were increased in both border and remote zones compared with the normal rate in murine LV of 50 TUNEL-positive nuclei per 100,000 cells (1). Activation of ACVI expression reduced the rate of apoptosis by about 50% in both regions. Numbers in bars = animals studied; bars = mean values; error bars = 1 SE; p value = probability value for gene effect (2-way analysis of variance). TUNEL = terminal 2′-deoxyuridine 5′-triphosphate nick-end labeling; other abbreviations as in Figure 2.
Histology
There was evidence of mild to moderate inflammation and fibrosis, which were more apparent in areas adjacent to the infarction than in the remote regions. There were no group differences in these findings.
Discussion
A key finding of the study is improved systolic and diastolic LV function 5 weeks after activation of cardiac ACVI expression in the setting of severe CHF because of ischemic cardiomyopathy. Animals in the AC-Off group showed a decline in LV ejection fraction between weeks 5 and 10. In contrast, after ACVI expression was activated, LV ejection fraction increased during the same interval, substantiating a true treatment effect. LV +dP/dt, a measure of LV systolic function, was increased by activation of cardiac ACVI expression. In addition, the slope of the ESPVR—perhaps the best measurement of contractile function of the intact heart—was increased 4-fold. This confirms a marked ACVI effect on contractile function of the failing heart, and likely explains the increase in cardiac output. Furthermore, 3 measures of diastolic function—LV end-diastolic pressure, LV −dP/dt, and tau, were all decreased by activation of cardiac ACVI expression, documenting improved diastolic function.
About 70% of patients with severe CHF have previous MI as the cause, thus coronary occlusion with subsequent LV chamber dilation and CHF provided a suitable model. Our methods result in infarction of 49 ± 3% of the LV and septum, with a transmural scar comprising the majority of the LV free wall (11). Pre-treatment LV ejection fraction was 24% in both groups, the result of large MI. Our model was relevant to clinical CHF and provided a substantial challenge for therapeutic intervention. An additional critical feature of this study is that transgene expression was activated only after severe CHF was present, thus providing a stringent test for efficacy. This was achieved using a cardiac-directed and tet-regulated transgene. We previously have shown that transgene expression is undetectable during doxycycline suppression (10). Once severe CHF was documented, 1 group continued to receive doxycycline and thus transgene suppression (AC-Off), whereas the other group no longer received doxycycline, which activated cardiac ACVI expression (AC-On). Indeed, we found a 16-fold increase in ACVI protein in LV samples from the AC-On versus AC-Off animals, documenting substantial expression in response to transgene activation.
Tetracyclines, which may attenuate MMP expression and activity, can influence LV remodeling when administered in the first few days after MI (13). Therefore, it was important that all animals received identical treatment during the healing phase of MI. We maintained this inhibition similarly in animals before and 5 weeks after MI. This ensured that scar development, adverse LV remodeling, and degree of heart failure were similar between groups. The LV samples from the 2 groups showed no differences in MMP2 or MMP8 expression, suggesting that doxycycline treatment, at the dose required for suppression of ACVI expression, did not influence MMP in this model. This was confirmed in additional nontransgenic control animals, which showed similar severe abnormalities in LV size and function 10 weeks after MI with or without doxycycline treatment (Table 3).
We also sought specific mechanisms by which increased expression of ACVI improved LV systolic and diastolic function. We found no change in several signaling proteins that can influence cardiac function—β1AR, β2AR, Gαs, SERCA2a, calsequestrin, PLN (and its phosphorylation), and RyR2 (and its phosphorylation) were similar in both groups. Furthermore, no group differences in SR calcium uptake were observed. In 2 previous reports, increased cardiac ACVI content was associated with improved SR calcium uptake (1,12). However, 1 of these studies was conducted in the acute phase of MI (1), and the other used a crossbreeding strategy in which heart failure was prevented rather than treated (12). Our data indicate that increased cardiac expression of ACVI in this setting does not affect LV SR calcium uptake.
We found that cAMP-generating capacity, which is substantially decreased in the failing heart, was returned to normal levels by increased ACVI expression. These data suggest that improved cardiac function was attributable, at least in part, to increased cardiac ACVI expression and associated increases in cAMP-generating capacity. It is important to point out, however, that increased cardiac expression of other βAR signaling elements that increase cAMP (βAR, Gs) have disastrous effects in both normal and failing hearts (6,14,15), so the favorable outcome with ACVI expression is unique, and likely reflects effects of ACVI not solely attributable to cAMP amplification.
Increases in LV contractile function in the absence of alterations in calcium handling suggested that distal changes, perhaps in contractile proteins, were evoked by activation of ACVI expression. We focused on cTnI phosphorylation because, in human failing hearts, dephosphorylation is specific to cTnI, and is thought not to occur among other contractile proteins such as cardiac troponin T, myosin light chain, or myosin binding protein C (16). Activation of cardiac ACVI expression in the setting of CHF was associated 5 weeks later with a 2.7-fold increase in cTnI phosphorylation at Ser23/24. Such an increase would be anticipated to have a positive effect on cardiac function. Transgenic mice with pseudophosphorylated cTnI at Ser23/24 have increased LV contractility (17). Furthermore, phosphorylation of cTnI at Ser23/24 increases the off-rate for Ca2+ exchange with cTnC (18), which increases relaxation rate (19). Phosphorylation of cTnI regulates maximal tension development, crossbridge kinetics, and systolic power production in skinned myocardial fiber bundles (19). Reduced phosphorylation of cTnI at Ser23/24 is associated with marked abnormalities in diastolic and systolic function in failing human hearts, and may account for reduced LV contractile function even when calcium handling is relatively normal (16,20).
Untreated heart failure (AC-Off) was associated with a 70% reduction in cTnI phosphorylation at Ser23/24 compared with normal murine LV, and activation of ACVI restored cTnI phosphorylation to normal levels. We found increased phosphorylation of cTnI but not PLN or RyR2—even though activation of ACVI expression increased cAMP-generating capacity and PKA activity. Phosphorylation of cTnI but not PLN has previously been reported in the setting of increased cardiac cAMP content (21). Selective phosphorylation of cTnI cannot easily be explained by ACVI activation of PKA, and indicates a novel association between cardiac ACVI content and cTnI that may reflect differences in compartmentation or association with A-kinase anchoring protein (22). How increased ACVI expression enables selective phosphorylation of cTnI is a focus of ongoing studies in our laboratory. We have found no previous examples in which a treatment for heart failure increased cTnI phosphorylation. Phosphorylation of cTnI alone, in the absence of alterations in calcium handling, seems to be sufficient for substantial improvement in function of the failing heart (16).
Increased apoptosis in both border (7.3-fold increase) and remote (3.9-fold increase) zones were found in failing hearts 10 weeks after MI (AC-Off). Increased ACVI expression was associated with a 46% to 59% reduction in apoptosis rates (p = 0.024). These results are in contrast to cardiac-directed expression of β1AR or Gαs, which are associated with increased cardiac apoptosis (14,23). A 50% reduction in apoptosis would be anticipated to preserve viable LV mass over time, and thus have implications in longer-term studies and in clinical settings.
LV Bcl-2 protein content and caspase 3/7 activities were not altered by ACVI expression. However, increased LV Akt activity, seen in untreated heart failure (AC-Off), was reduced by activation of ACVI expression. Although expression of constitutively active Akt1 seems to be protective, prolonged Akt activation is associated with contractile dysfunction and CHF (24). The precise molecular mechanism for reduced apoptosis conferred by activation of ACVI expression in CHF will require additional studies in isolated cardiac myocytes.
Despite striking improvement in LV systolic and diastolic function, we did not find changes in LV dimensions 5 weeks after treatment. Increased ACVI expression attenuates adverse remodeling in acute MI (1). However, all animals in the current study had the same low level of ACVI in the heart 5 weeks after large MI, and extensive adverse LV remodeling occurred before treatment was initiated. Sustained ACVI expression in genetic cardiomyopathy prevents deleterious LV remodeling and increases survival (4), and intracoronary delivery of an adenovirus vector encoding ACVI reduces LV enlargement in pacing-induced CHF (2). Whether longer or earlier ACVI expression also will diminish adverse LV remodeling in this model, as it has in other models (1,2,4), is currently being addressed. Pleger et al. (9), using S100A1 gene transfer in MI-induced CHF in rats, found attenuation of LV dilation, unlike our study. Differences on LV remodeling may be caused by smaller MIs in their study. We infarcted 49% of the LV (pre-treatment EF 24%); Pleger et al. (9) infarcted 25% of LV (pre-treatment EF 40%). Their study confirms that gene transfer strategies can effectively treat CHF in pre-clinical models.
Study limitations
The current study was designed to determine whether activation of cardiac ACVI expression could increase function of the failing heart, but was not adequately powered to determine whether mortality was reduced by the intervention. The death rates were similar between groups during the 5-week treatment phase in the current study (AC-Off: 2 deaths; AC-On: 1 death). However, we have shown a mortality advantage of ACVI in acute MI (1) and in murine genetic cardiomyopathy (4), an effect associated with favorable electrical remodeling conferred by ACVI expression (25). The present study shows that global cardiac expression of ACVI has beneficial effects on the failing heart. It remains to be seen whether exogenous gene transfer of ACVI can achieve similar results. However, we recently showed a reduced rate of decline in LV function in pacing-induced CHF after intracoronary delivery of an adenovirus encoding ACVI (2).
Clinical implications
Increased cardiac ACVI expression has favorable effects on function of the failing heart. In previous studies, we have shown that increased cardiac ACVI also has favorable effects on survival in acute MI (1) and on electrical remodeling in CHF (25). These data provide a rationale for increasing expression of cardiac ACVI in clinical CHF.
Conclusions
In conclusion, our data show that, in an animal model of ischemic cardiomyopathy, increased cardiac expression of ACVI improves function of the failing heart and increases phosphorylation of cTnI.
Acknowledgments
The authors thank Matthew Spellman and Yuan Yang for technical assistance and Dr. Glenn I. Fishman for providing the tTA mice.
This work was supported by National Institutes of Health grants 5P01HL066941, HL088426, and HL081741 (Dr. Hammond), Merit Review Awards from the Department of Veteran’s Affairs (Drs. Roth and Hammond), a Grant-in-Aid from the American Heart Association Western States Affiliate (Dr. Gao), and a fellowship from the Banyu Life Science Foundation International (Dr. Takahashi).
Abbreviations and Acronyms
- ACVI
adenylyl cyclase VI
- cAMP
cyclic adenosine monophosphate
- CHF
congestive heart failure
- +dP/dt
pressure development
- EF
ejection fraction
- ESP
end-systolic pressure
- ESPVR
end-systolic pressure–volume relationship
- FAC
fractional area change
- LV
left ventricular
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- PKA
protein kinase A
- PP1
protein phosphatases 1
- PP2A
protein phosphatases 2A
- RLU
relative light units
- SR
sarcoplasmic reticulum
Footnotes
Lynne Warner Stevenson, MD, served as Guest Editor for this article.
REFERENCES
- 1.Takahashi T, Tang T, Lai NC, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–96. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 2.Lai NC, Roth DM, Gao MH, et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110:330–6. doi: 10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- 3.Roth DM, Gao MH, Lai NC, et al. Cardiac-directed adenylylcyclase expression improves heart function in cardiomyopathy. Circulation. 1999;99:3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 4.Roth DM, Bayat H, Drumm D, et al. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105:1989–94. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- 5.Hoshijima M, Ikeda Y, Iwanaga Y, et al. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med. 2002;8:864–71. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 6.Engelhardt S, Hein L, Dyachenkow V, et al. Altered calcium handling is critically involved in the cardiotoxic effects of chronic β-adrenergic stimulation. Circulation. 2004;109:1154–60. doi: 10.1161/01.CIR.0000117254.68497.39. [DOI] [PubMed] [Google Scholar]
- 7.Harding VB, Jones LR, Lefkowitz RJ, et al. Cardiac βARK1 inhibition prolongs survival and augments β-blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci U S A. 2001;98:5809–14. doi: 10.1073/pnas.091102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rockman HA, Chien KR, Choi DJ, et al. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:7000–5. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pleger ST, Most P, Boucher M, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–15. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 10.Gao MH, Bayat H, Zhou JY, et al. Controlled expression of cardiac-directed adenylylcyclase type VI provided increased contractile function. Cardiovasc Res. 2002;56:197–204. doi: 10.1016/s0008-6363(02)00539-4. [DOI] [PubMed] [Google Scholar]
- 11.Bayat H, Swaney J, Ander A, et al. Progressive heart failure after myocardial infarction in mice. Basic Res Cardiol. 2002;97:206–13. doi: 10.1007/s003950200013. [DOI] [PubMed] [Google Scholar]
- 12.Tang T, Gao MH, Roth DM, et al. Adenylyl cyclase type VI corrects cardiac sarcoplasmic reticulum calcium uptake defects in cardiomyopathy. Am J Physiol Heart Circ Physiol. 2004;287:H1906–12. doi: 10.1152/ajpheart.00356.2004. [DOI] [PubMed] [Google Scholar]
- 13.Villarreal FJ, Griffin M, Omens J, et al. Early short-term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation. 2003;108:1487–92. doi: 10.1161/01.CIR.0000089090.05757.34. [DOI] [PubMed] [Google Scholar]
- 14.Geng YJ, Ishikawa Y, Vatner DE, et al. Apoptosis of cardiac myocytes in Gsα transgenic mice. Circ Res. 1999;84:34–42. doi: 10.1161/01.res.84.1.34. [DOI] [PubMed] [Google Scholar]
- 15.Dorn GW, Tepe NM, Lorenz JN, et al. Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gαq-overexpressing mice. Proc Natl Acad Sci U S A. 1999;96:640–5. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–59. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Takimoto E, Soergel DG, Janssen PM, et al. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 18.Zhang R, Zhao J, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease Ca2+ affinity of cardiac troponin C. J Biol Chem. 1995;270:30773–80. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
- 19.Solaro RJ. Modulation of cardiac myofilament activity by protein phosphorylation. In: Fozzarad H, Solaro RJ, editors. Handbook of Physiology. Volume 1, The Heart. Oxford University Press; New York, NY: 2002. pp. 264–300. [Google Scholar]
- 20.Zakhary DR, Moravec CS, Stewart RW, et al. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–10. doi: 10.1161/01.cir.99.4.505. [DOI] [PubMed] [Google Scholar]
- 21.Karczeski P, Bartel S, Krause EG. Differential sensitivity to isoprenaline of troponin I and phospholamban phosphorylation in isolated rat hearts. Biochem J. 1990;266:115–22. doi: 10.1042/bj2660115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruehr ML, Russell MA, Bond M. A-kinase anchoring protein targeting of protein kinase A in the heart. J Mol Cell Cardiol. 2004;37:653–65. doi: 10.1016/j.yjmcc.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Bisognano JD, Weinberger HD, Bohlmeyer TJ, et al. Myocardial-directed overexpression of the human beta1-adrenergic receptor in transgenic mice. J Mol Cell Cardiol. 2000;32:817–30. doi: 10.1006/jmcc.2000.1123. [DOI] [PubMed] [Google Scholar]
- 24.Shiojima I, Walsh K. Regulation of cardiac growth and coronary aniogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–65. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 25.Timofeyev V, He Y, Tuteja D, et al. Cardiac-directed expression of adenylyl cyclase reverses electrical remodeling in cardiomyopathy. J Mol Cell Cardiol. 2006;41:170–81. doi: 10.1016/j.yjmcc.2006.04.008. [DOI] [PubMed] [Google Scholar]